

Abstract
Maximizing the scientific impact of NMR-based structure determination requires robust and statistically sound methods for assessing the precision of NMR-derived structures. In particular, a method to define a core atom set for calculating superimpositions and validating structure predictions is critical to the use of NMR-derived structures as targets in the CASP competition. FindCore (D.A. Snyder and G.T. Montelione PROTEINS 2005;59:673–686) is a superimposition independent method for identifying a core atom set, and partitioning that set into domains. However, as FindCore optimizes superimposition by sensitively excluding not-well-defined atoms, the FindCore core may not comprise all atoms suitable for use in certain applications of NMR structures, including the CASP assessment process. Adapting the FindCore approach to assess predicted models against experimental NMR structures in CASP10 required modification of the FindCore method. This paper describes conventions and a standard protocol to calculate an “Expanded FindCore” atom set suitable for validation and application in biological and biophysical contexts. A key application of the Expanded FindCore method is to identify a core set of atoms in the experimental NMR structure for which it makes sense to validate predicted protein structure models. We demonstrate the application of this Expanded FindCore method in characterizing well-defined regions of 18 NMR-derived CASP10 target structures. The Expanded FindCore protocol defines “expanded core atom sets” that match an expert’s intuition of which parts of the structure are sufficiently well-defined to use in assessing CASP model predictions. We also illustrate the impact of this analysis on the CASP GDT assessment scores.
Keywords: CASP, Protein Structure, Superimposition, RMSD, NMR
Introduction
Protein structure determination by NMR continues to expand its impact in biological and biophysical research. Traditionally, the producers of NMR structure models of proteins and nucleic acids have used an ensemble representation to document the precision of these experimental models. Assessment of the precision of structural information is critical in its validation, application and prediction. Regions of structural models that are less precise (i.e. less converged) are more limited in their application than more precise (i.e. better converged) regions. In particular, users of NMR-derived structural information require annotations indicating which regions of the structural model are well-defined, and which regions require special consideration to avoid misinterpretation.1–4 By generating several (typically 10 – 40) models of the protein structure, each of which is considered to fit the NMR data about equally well, the experimentalist conveys to users of the NMR model information about which regions of the structure are well-defined and suitable for applications requiring a significant level of structural precision, and which are less well-defined. Structural comparisons across the ensemble provide information about the precision of different regions of the model, which users of NMR structures need to consider with respect to their specific applications.
One of the many uses of experimental NMR structures is for assessment of model prediction methods in projects like the Critical Assessment of Protein Structure Prediction (CASP).5,6 In this application, protein structures determined by NMR or X-ray crystallography are used to benchmark the accuracy of various methods of comparative modeling and protein structure prediction. In such projects, it is critical to identify those regions of the target NMR structure that are sufficiently well-defined to use for assessment of the accuracy of model predictions. Predictions of a protein’s structure in regions where the experimental structure is not well-defined cannot be evaluated with the same stringency as predictions in regions where experimental data provides precise structural information. Similarly, the evaluation of the quality of a structural model should not penalize the model for fine-scale deviations from acceptable validation statistics in regions where the model provides only a rough idea of the true protein structure. In each cycle of CASP, this problem has been addressed in some way or another.7–10 However, no standard has been defined for annotating biomolecular NMR structure coordinates with the information derived from the ensemble, directing assessors to which parts of the structure are sufficiently precise for use in the assessment analysis.
This paper presents an extension of the FindCore1 method, using an iterated process to establish a core set of well-defined atoms with atomic position variances within three standard deviations of the range observed among the initial FindCore core atom set. This expanded set of well-defined atoms includes all atoms for which it is reasonable to assess the quality of the NMR structure using knowledge-based methods, including evaluation of dihedral angles and hydrogen bonds. This expanded core atom set can also be used evaluate structure prediction methods on their ability to accurately determine the precise coordinates of the structure predicted. Statistics associated with this set also provide benchmarks to evaluate the quality of the prediction. This iterated FindCore process was used in CASP10 to identify the atoms to use in evaluating predictions of 18 NMR-derived structures. It is anticipated that the well-defined atom set construction procedure described here and similar extensions of the FindCore method will find more general utility for prediction, application and validation methods that utilize biomolecular NMR structures.
Methods
In this study, we utilized 18 ensembles of NMR structures, experimentally-determined targets contributed to CASP10 by several laboratories. Table I lists the CASP Target and PDB IDs for these targets.
Table I.
Coordinate Uncertainties for CASP NMR Targets through the Expanded FindCore Process
| Protein ID | Find Core | Initial Iteration | Expanded Core | Edited Core | |||||
|---|---|---|---|---|---|---|---|---|---|
| CASP | PDB | Mean | Crit Val | Mean | Crit Val | Mean | Std Dev | Mean | Std Dev |
| T0655 | 2LUZ | 0.560 | 1.305 | 0.642 | 1.763 | 0.718 | 1.517 | 0.718 | 1.521 |
|
| |||||||||
| T0657 | 2LUL | 0.504 | 1.490 | 0.599 | 2.327 | 0.717 | 1.757 | 0.704 | 1.739 |
|
| |||||||||
| T0662 | 2LTE | 0.706 | 1.839 | 0.782 | 2.532 | 0.871 | 1.604 | 0.870 | 1.601 |
|
| |||||||||
| T0665 | 2LR8 | 0.506 | 1.564 | 0.574 | 2.234 | 0.675 | 1.696 | 0.677 | 1.701 |
|
| |||||||||
| T0668 | 0.557 | 1.810 | 0.719 | 3.235 | 0.939 | 1.813 | 0.939 | 1.813 | |
|
| |||||||||
| T0669 | 2LTL | 0.521 | 1.402 | 0.592 | 1.909 | 0.667 | 1.586 | 0.664 | 1.585 |
|
| |||||||||
| T0675 | 2LV2 | 1.010 | 2.855 | 1.100 | 3.551 | 1.150 | 1.530 | 1.143 | 1.521 |
|
| |||||||||
| T0677_A | 0.486 | 1.201 | 0.527 | 1.494 | 0.568 | 1.514 | 0.563 | 1.507 | |
|
| |||||||||
| T0677_B | 0.456 | 1.228 | 0.525 | 1.756 | 0.610 | 1.614 | 0.601 | 1.602 | |
|
| |||||||||
| T0709 | 0.430 | 1.252 | 0.458 | 1.550 | 0.527 | 1.617 | 0.514 | 1.596 | |
|
| |||||||||
| T0711 | 0.488 | 1.497 | 0.579 | 2.288 | 0.679 | 1.699 | 0.680 | 1.708 | |
|
| |||||||||
| T0714 | 2LVC | 0.581 | 1.357 | 0.685 | 1.975 | 0.764 | 1.538 | 0.768 | 1.547 |
|
| |||||||||
| T0716 | 2LY9 | 0.562 | 1.948 | 0.647 | 2.732 | 0.735 | 1.746 | 0.732 | 1.743 |
|
| |||||||||
| T0727 | 2LTM | 0.359 | 1.000 | 0.425 | 1.365 | 0.454 | 1.556 | 0.450 | 1.545 |
|
| |||||||||
| T0729 | 2LU7 | 0.433 | 1.139 | 0.525 | 1.717 | 0.624 | 1.626 | 0.631 | 1.643 |
|
| |||||||||
| T0731 | 0.884 | 3.010 | 0.957 | 3.806 | 1.030 | 1.665 | 1.030 | 1.667 | |
|
| |||||||||
| T0751 | 2LVA | 0.823 | 5.899 | 0.806 | 5.805 | 0.806 | 1.932 | 0.801 | 1.920 |
|
| |||||||||
| T0754 | 2LV9 | 0.480 | 1.225 | 0.563 | 1.672 | 0.633 | 1.534 | 0.628 | 1.527 |
Coordinate uncertainties (<u2> values, converted to Å) and their means, standard deviations and critical values are calculated, as described in the main text, from each core atom set (and the associated superimposition) produced in the iterated, editing process.
Construction of a well-defined atom set begins with the identification of an initial core atom set via the previously described FindCore algorithm.1 The FindCore method can identify core atom sets and identify multiple “domains” of core atoms sets that are not well-defined with respect to one another, from any set of atoms of interest: e.g. Cα atoms, backbone heavy atoms, heavy atoms, protons or all atoms. Construction of well-defined atom sets for CASP10 NMR target structures selected atoms from the set of all heavy (N, C, S, and O) atoms, including both backbone and sidechain atoms.
Briefly, the FindCore core algorithm represents the uncertainty in an NMR-derived ensemble of structural models using the inter-atomic variance matrix, reduces this matrix to a binary array using an internally and automatically calibrated threshold, sums the row of the binary array to obtain an atom-specific “order parameter” and uses these order parameters to cluster atoms into a core and a non-core set. The core set may be further partitioned into “domains”, i.e. multiple core atom sets that are not well defined with respect to one another, using the inter-atomic variance matrix. The core atom set (or each “domain” found) can be used to calculate RMSD minimizing superimpositions of the models in the ensemble.
Following superimposition using the core atom set identified by FindCore, we often observe that the variations in atomic positions for some atoms not included in the core atom set are not significantly higher than for some of the core atoms. This suggests that the FindCore algorithm is overly stringent in selecting for atoms whose relative positions are well defined in the ensemble.
Iterative editing of the FindCore core overcomes the stringency with which FindCore defines the core atom set. First, the superimposition allows for the meaningful calculation for each atom of mean squared displacements from the mean atomic coordinates, averaged across all models in the ensemble,
Where the brackets indicate taking an average across all models and [x y z] indicates the x, y and z coordinates (as superimposed) of the current atom. Note that if NMR-ensembles were actually Boltzmann ensembles representing the actual conformational variability in the protein structure, then the <u2> value defined above would be the same <u2> used in interpreting the Debye-Waller B-factor as 8π2<u2>/3.11
A subtlety involved in the superimposition process is the selection of a representative model onto which to superimpose the other structures. In the Expanded FindCore method, pairwise superimpositions (defined using the initial core atom set) are used to identify the medoid12 model. The superimposition of all other models onto the medoid is then used to calculate average coordinates, and all models are re-superimposed onto this average. This superimposition is then used to calculate mean squared displacements from mean atomic coordinates, <u2>.
It is found that the <u2> values for the core atom set follow a log-normal distribution rather than a normal distribution (cf. Figure 1A and 1B). This is not surprising given the previous observation of log-normal distributions of square deviations in atomic positions across NMR-derived ensembles in related work seeking to compare crystallographic structures with NMR ensembles.13 The sample mean mu and standard deviation su of log(<u2>) are thus used to define a critical <u2> = emu+3*su corresponding to a Z-score of 3 (approximately the 99.9th percentile) for the (log) normal distribution. Atoms characterized by <u2> values less than the critical <u2> value are then ruled into the core atom set, while core atoms characterized by a <u2> value greater than the critical value are removed. Typically, this results in an expansion of the core atom set by about 17%, while only a small number (1–2) of core atoms are removed as a Z-value of 3 corresponds to a 0.13% probability of a core atom being censored (Figure 1C). The resulting initial expanded core atom set provides a basis for re-superimposition of the NMR-derived ensemble, and <u2> values are re-calculated using this new superimposition. The sample mean mu and standard deviation su are recalculated from the log(<u2>) values for the edited core. These statistics are again used to define a critical <u2> and to guide the construction of an expanded core set of atoms with recalculated <u2> values less than the new critical value (Figure 1D). Ideally, the resulting ensemble of conformers, annotated with respect to “expanded core” vs. “non-core” atoms based on Expanded FindCore would then be used for assessment of predicted models. However, for the CASP10 prediction assessment, only the single medoid model14 defined from the “expanded core atoms” and this superimposition was used by the prediction center as the representative model of the experimental NMR structure.
Figure 1. Lognormal distribution of core atom <u2> values.

All plots were made using the heavy atoms of NMR ensemble target T0655 (2LUZ) as an example. (A) Normal QQ plot of <u2> values (calculating using the FindCore superimposition) for FindCore core atoms in T0655 demonstrating a lack of normality in the <u2> statistic. (B) Normal QQ plot of log(<u2>) values for the same core atom set demonstrating the lognormal distribution of the <u2> statistic. (C,D) Histograms (with a logarithmic scale for the abscissa) of <u2> values for heavy atoms in T0655, with <u2> calculated from (C) the original FindCore superimposition and (D) the superimposition calculated in the first iteration of the Expanded FindCore method. Blue bars correspond to atoms in the original FindCore core, cyan bars to atoms added to the core in the iteration process, magenta bars to atoms removed from the core during the editing process, which ensures all atoms in the final core belong to residues for which all backbone atoms are in the core. Yellow bars correspond to carbonyl oxygens added to the core in the editing process. The orange, green and blue lines mark Z score cut-offs of 2, 3 and 4 respectively. Note that the Z score = 3 cut-off (green lines) is (B) a tight upper boundary for the distribution of <u2> values for the core atom set and (D) is a tight lower boundary for the second (non-core) mode of the distribution of <u2> values calculated using the final Expanded FindCore superimposition.
At this point, the expanded core atoms set is suitable for various applications requiring the distinction of well-defined from ill-defined atoms in the protein NMR structure model. However, certain software tools used in CASP10 assessment (e.g. Ramachandran analysis) require complete backbone atom information. These applications may require editing of the expanded core atoms set. In particular, for CASP10 many different methods were used to compare the “representative conformer” from the target NMR structures, as defined in the previous paragraph, with each of the predicted models using only atoms in the expanded core atom set, and excluding atoms that are not-well-defined in the ensemble of coordinates. However, for some residues, some backbone atoms are included in the “core atom set” and others are not, frustrating the application of some structural comparison methods that require a complete set of backbone atoms for each residue in order to be used without significant code modifications. For the purpose of CASP10, the expanded core set was next edited to ensure that the well-defined atom set includes, for each residue, either all of the backbone heavy atoms or none of them. Thus, atoms are only retained in the core atom set if they belong to residues for which N, C′ and Cα are all in the expanded core; i.e if any one of these three atoms was in included in the expanded core atom set, then all three were removed from the core atom set. In general, backbone carbonyl oxygen atoms are less frequently observed in the expanded core atom set than backbone N, C′ and Cα atoms, perhaps due to the lack of direct NMR-derived constraints to these oxygen atoms resulting from the lack of attached protons capable of yielding NOESY cross-peaks. Accordingly, for the purpose of using the software tools required for CASP10 assessment without significant code modifications, if the N, C′ and Cα atoms of a residue are included in the edited expanded core atom set, but its carbonyl oxygen is not, the carbonyl oxygen was added to create the complete edited expanded core atoms set used for structure prediction assessment.
The expanded FindCore method described in this paper is implemented in MATLAB and will be available as part of a future release of the FindCore Toolbox available via the MATLAB Central File Exchange. Both MATLAB and Microsoft Excel were used for statistical analyses and tabulations. MATLAB generated the plots reported here, and PyMOL15 rendered structures.
Results
Tables I and II track the process, for each of the 18 CASP10 targets studied here, from initial identification of the core atom set and superimposition, to expansion of the core atom set and superimposition, to refinement of the expanded core and final editing of backbone atoms for software compatibility (as described above) resulting in a set of well-defined atoms. Table I illustrates the changes in mean (log) and critical <u2> values calculated from the FindCore core and its associated superimposition, and from the first iteration of the core expansion using its associated superimposition and additionally tabulates the mean and standard deviation of the (log) <u2> values for both the expanded and edited core atom sets. The statistics reported in this table are transformed (exponentiation followed by square root) to have units of Å. Table II tracks additions and subtractions from the core atom set through both iterated expansions as well as through the editing process. Note that both iterations of the expansion process add potentially large numbers of atoms to the core while only slightly increasing the mean coordinate variances of core atoms. The set of atoms added to the core by the expansion, summarized in Table I, ranges from ranges from no net change for T0751 to 271 atoms (72%) for T0668 with a median expansion of 36%. Since the critical Z value used corresponds to a probability of 0.13%, only very few if any atoms are removed from the core in the iterative superimposition process.
Table II.
Changes in the Size of the Core Atom Set through the Iterated, Editing Process
| Protein ID | FindCore | Initial Iteration | Second Iteration | Editing | |||||
|---|---|---|---|---|---|---|---|---|---|
| CASP | PDB | Core | Non-Core | Added | Removed | Added | Removed | Added | Removed |
| T0655 | 2LUZ | 726 | 686 | 203 | 0 | 135 | 0 | 9 | 5 |
|
| |||||||||
| T0657 | 2LUL | 723 | 649 | 181 | 0 | 158 | 0 | 21 | 2 |
|
| |||||||||
| T0662 | 2LTE | 446 | 365 | 67 | 0 | 59 | 0 | 1 | 0 |
|
| |||||||||
| T0665 | 2LR8 | 306 | 272 | 49 | 2 | 44 | 0 | 0 | 1 |
|
| |||||||||
| T0668 | 379 | 358 | 149 | 0 | 122 | 0 | 0 | 0 | |
|
| |||||||||
| T0669 | 2LTL | 534 | 421 | 106 | 1 | 77 | 0 | 9 | 4 |
|
| |||||||||
| T0675 | 2LV2 | 380 | 266 | 42 | 0 | 17 | 0 | 3 | 0 |
|
| |||||||||
| T0677_A | 266 | 198 | 30 | 0 | 25 | 0 | 6 | 1 | |
|
| |||||||||
| T0677_B | 408 | 337 | 81 | 0 | 69 | 0 | 12 | 2 | |
|
| |||||||||
| T0709 | 113 | 111 | 9 | 0 | 14 | 0 | 5 | 1 | |
|
| |||||||||
| T0711 | 133 | 88 | 27 | 0 | 21 | 0 | 2 | 2 | |
|
| |||||||||
| T0714 | 2LVC | 386 | 325 | 143 | 0 | 74 | 0 | 0 | 3 |
|
| |||||||||
| T0716 | 2LY9 | 327 | 284 | 51 | 0 | 36 | 0 | 3 | 1 |
|
| |||||||||
| T0727 | 2LTM | 487 | 374 | 160 | 0 | 43 | 0 | 7 | 1 |
|
| |||||||||
| T0729 | 2LU7 | 328 | 296 | 114 | 0 | 87 | 0 | 1 | 6 |
|
| |||||||||
| T0731 | 389 | 358 | 31 | 0 | 21 | 0 | 1 | 1 | |
|
| |||||||||
| T0751 | 2LVA | 359 | 484 | 0 | 1 | 0 | 0 | 1 | 0 |
|
| |||||||||
| T0754 | 2LV9 | 339 | 304 | 95 | 0 | 54 | 0 | 6 | 1 |
Number of atoms classified as core vs. non-core by FindCore and the changes in the size of the core atom set occurring at each step in the iterated editing process described in the main text
Figure 2 displays superimpositions of the 18 CASP targets analyzed in this paper. As expected, many loops and tails are excluded from the well-defined atom set while the iterative editing process extends the original FindCore core to include most regions of secondary structure and even turns and bends where the “trajectory” of the backbone trace remains sufficiently well-defined to expect predictions to match experimentally determined backbone traces in these loops. Note that residues rejected in the editing process (shown in purple), which limits the expanded core atom set to atoms in those residues for which N, Cα and C′ are all in the expanded core, tend to occur on the boundaries between well-ordered secondary structural elements and disordered loops.
Figure 2. CASP Targets superimposed using the Expanded FindCore approach for defining the set of “well defined” core atoms.

Superimpositions are shown for each target and labeled by their CASP10 Target ID. Coloration of the backbone traces indicates atom classification according to FindCore and subsequent expansion of the core atom set by iterative superimpostion. Saturated blue regions indicate the original FindCore atom set, regions added to the core in the iterated expansion process are sky blue and red regions are atoms that are not included in the well-defined atom set. Residues removed during the final editing process to ensure all core atoms belong to only those residues with a complete backbone set in the core are colored magenta. Note that the Expanded FindCore core (saturated and light blue) includes the well-packed, secondary structure rich geometric cores of the protein structures while most loops are excluded from the Expanded FindCore core. Residues excluded in the editing process typically are at the edges of well-defined structural elements. The circled (core) residue in T0655 is LYS 92, which is discussed in the main body of the text.
In some cases, “islands” of core atoms appear in otherwise disordered loops and “gaps” of ill-defined atoms occur in otherwise well-defined structural elements. For example, in T0655 (2LUZ), GLY 142 is not-well-defined even though it is flanked by residues with backbone atoms in the extended core. The lack of precision of the atomic coordinates of this residue is not surprising as, being a glycine residue, it can impart flexibility to the peptide backbone. Interestingly, the PROCHECK16,17 G-score for GLY 142’s φ and ψ dihedral angles is −1.67, suggesting that the less well-defined coordinates for this residue are potentially inaccurate. Variability in this residue’s coordinates may reflect some amount of arbitrariness in the inference of these potentially inaccurate as well as imprecise atomic positions. T0665 (2LUZ) also contains an example of an island of core atoms in a loop: in particular the backbone (although not the sidechain beyond Cα) atoms of LYS 92 (orange circle), sandwiched between non-core residues GLY 91 and PRO 93, are in the expanded core. Being on the N-terminal side of a proline residue may restrain the backbone atoms of LYS 92 sufficiently to allow their admittance into the core. Interestingly, the PROCHECK G-score for LYS 92’s φ and ψ dihedral angles is −0.55, which, while not ideal, is quite reasonable for residues in a surface loop. Though islands and gaps in the core atom set are not unreasonable outcomes in assessing which atoms have converged to well-defined coordinates, such islands and gaps may confound human evaluation and even some automated validations of protein structure. Thus, for the purposes of CASP10 assessment applications, manual “smoothing” was done to eliminate such islands and gaps from the expanded core atom set.
Isolated stretches of secondary structure, such as the proto-helix in the extensive tail region of Target 751, were also excluded from the well-defined atom set for the purposes of in the CASP10 assessment, as their position relative to the molecule’s overall frame of reference is not-well-determined. While such partially formed stretches of secondary structure are often functionally important as they indicate a tendency to fold upon binding,18 such conformations often form transiently and are derived from ensemble-averaged NMR data that is not properly interpreted by standard methods of protein NMR structure determination. For this reason, our definition of well-defined atoms correctly excludes such “mini-domains”. However, the underlying FindCore method, and hence the iterated editing process reported here, can appropriately handle larger, fully formed multiple domains.1
The original FindCore method1 uses a statistical parsing of the interatomic variance matrix that maximizes the separation between two populations of atoms: those that are well-defined with respect to one another and those that have high interatomic variances. The Expanded FindCore method uses the resulting core atom set to superimpose the ensemble and then includes / excludes atoms based on their coordinate uncertainties in the superimposition. Atoms with coordinate uncertainties within three standard deviations (Z = 3) of the mean (log) coordinate uncertainty within this core atom set are added into the core atom set. This “expanded core atom set” better fits our expert, intuitive expectations of what is well-defined in the NMR ensemble. As an example, Figure 3 compares, for NMR ensemble PDB entry 2L7W, the core atom sets defined by FindCore with its internally calibrated statistical cutoff 1, Expanded FindCore using the Z=3 cutoff, and an analysis done with the program CYRANGE 2. Expanded FindCore results with the Z=3 cutoff are similar to those of the CYRANGE program. A second example is illustrated in Figure 4 for the Zn-binding sites in NMR structures of CASP10 targets T0657 and T0754. For both targets, the statistically-based cutoffs of the original FindCore algorithm are too conservative, and exclude relatively well-defined atom positions of side chains that are ligands of the Zn atoms, while the Expanded FindCore method developed for CASP10 includes these structurally-important atoms in the set of atoms to be used in prediction model quality assessment. Hence, the Expanded FindCore protocol provides “expanded core atom sets” which, upon superimposition, fit well to expert intuition of which parts, on an atom-wise and not merely residue-wise basis, of the structure are sufficiently well-defined to be used in assessing CASP model predictions.
Figure 3. Comparison of FindCore, Expanded FindCore and CYRANGE methods for identifying core atom sets.
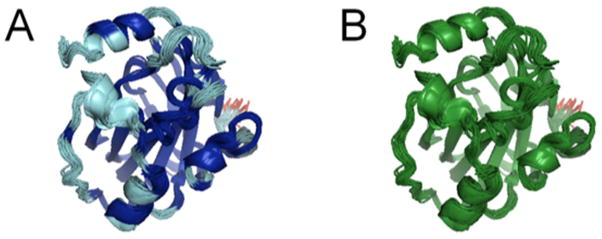
(A) FindCore (blue) and Expanded FindCore (cyan) regions of the human Raf-1 kinase inhibitor protein (PDB ID 2L7W). In this case, Expanded FindCore greatly expands upon the original core atom set identified by FindCore, only excluding the poorly converged N-terminal residue (red) from the core. (B) CYRANGE 2 produces a result similar to Expanded FindCore, except that CYRANGE defines its core (green) on a per-residue, rather than per-atom basis.
Figure 4. The Zn binding sites of CASP10 NMR targets T0657 and T0754 are fully covered in the Expanded FindCore core atom set.
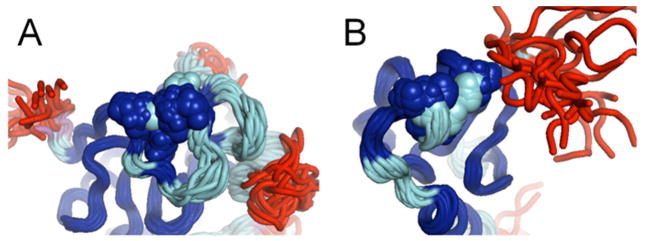
Zn binding sites in (A) CASP Target T0657 (PH domain of tyrosine protein kinase TEC, PDB ID 2LUL) and (B) CASP Target T0754 (human MLL5 PHD domain, PDB ID 2LV9) shown with individual atoms rendered as spheres. Note that the original FindCore core (blue) includes only some of the atoms in Zn-binding residues. The remaining heavy atoms (backbone and sidechain) of Zn-binding residues in these proteins are, however, included the Expanded FindCore core atom set (cyan).
A key application of the Expanded FindCore method presented here is to identify a core set of atoms for which it makes sense to validate predicted protein structure models. In order to illustrate the impact of this analysis on CASP assessment scores, we calculated GDT-TS scores for predicted models assessed against 18 experimental NMR structures from each of 5 top ranked groups, using in each case three representations of the NMR structure: (i) the untrimmed coordinates, (ii) coordinates trimmed using the original FindCore method, and (iii) coordinates trimmed using the expanded FindCore protocol outlined above. These results are summarized in Supplementary Table S1. As expected, excluding the ill-defined regions the experimental protein NMR structure significantly increases the GDT-TS score. A particularly dramatic example is target T0751, where GDT-TS scores increase from ca. 0.38 to ca. 0.90. All of the predictions have significantly higher GDT-TS scores when considering only the core atom sets indicated by FindCore. Comparing GDT-TS scores for FindCore and Expanded FindCore atom sets, the GDT-TS scores, as expected, are somewhat lower for the Expanded FindCore atom sets (Supplementary Table S1). This suggests that the additional atoms added to the core atom set by the expansion algorithm are sometimes less accurately positioned than those in the initial FindCore core atom set. Nonetheless, the original FindCore atom set is clearly too conservative as demonstrated by the examples shown Figures 3 and 4. The final cutoff values for including/excluding atoms in the Expanded FindCore atom sets are determined by the conventions outlined in this paper, and can affect the relative ranking of model predictions assessed by NMR structures. For these reasons, it is important to have a standardized and reproducible protocol for defining the atoms in the NMR structure coordinate set that are used in the assessment, as provided by this study.
The importance of trimming the NMR coordinates to well-defined regions is demonstrated graphically in Figure 5, showing a CASP10 prediction for NMR target T0731 (blue ribbon diagram) against a representative NMR-derived structure model (the medoid model12,14 from the ensemble of conformers) from either the (i) complete or (ii) Expanded FindCore experimental NMR structure coordinates. Because both the N-terminal and C-terminal regions of the experimental NMR structure are not well-defined by the NMR ensemble, it is inappropriate to use these atomic coordinates of the experimental NMR structure to assess the predicted model in these regions that are excluded by the Expanded FindCore algorithm. In particular, the predicted C-terminal helix for T0731 cannot be judged either consistent or inconsistent with the C-terminus of the NMR-derived structure: due to its high uncertainty, Expanded FindCore correctly excludes this C-terminal region from the core.
Figure 5.
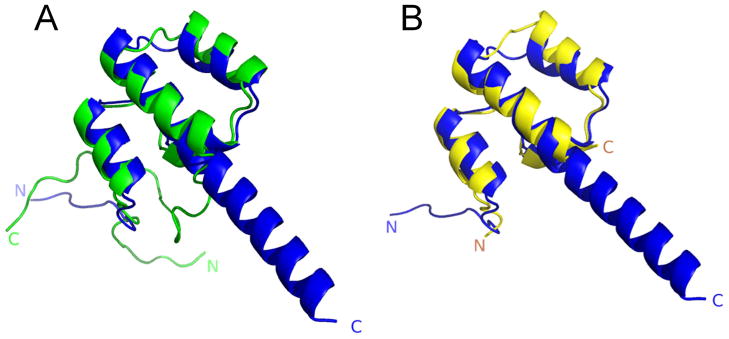
Demonstration of the importance of using a standardized protocol for trimming NMR structures to a well-defined core atom set using the NMR ensemble information prior to assessment. Comparison of CASP10 structure prediction by QUARK31 for NMR target T0731 (blue ribbon diagram) against a representative NMR-derived structure from either the untrimmed (green, left) or Expanded FindCore (yellow, right) experimental NMR structure coordinates.
As part of this analysis, we also analyzed the regularity of the structural features in expanded core vs. non-core atom sets. These results are presented in the Supplementary Materials. Regions of the structure added to the core by the Expanded FindCore algorithm are not more likely to have poor backbone and sidechain dihedral angle values (i.e. PROCHECK16,17 G scores < −1) compared with regions of the original core atom set. The Expanded FindCore method also does a better job in assigning residues having poor PROCHECK G scores to the not-well-defined class than does the original FindCore method. We also assessed the consistency of hydrogen-bonded interactions across the NMR ensemble, another measure of structural reliability. In this analysis, the more conservative FindCore definition is better than the Expanded FindCore definition in including consistent hydrogen-bonded pairs and excluding inconsistently hydrogen-bonded pairs.
Discussion
The importance of distinguishing well-defined and not-well-defined regions of protein NMR structures in CASP assessment
Issues related to use of the ensemble representation of protein NMR structures often confound the application of these structures by biologists, particularly in the field of structural bioinformatics. For this reason, conventions defining how to utilize NMR structures for assessing model predictions in CASP are an important part of the CASP experiment, both for the free-modeling (FM) and template-based modeling (TBM) assessments. The importance of distinguishing well-defined and ill-defined regions in interpreting NMR-derived protein structure models has recently been reviewed in the context of validating the accuracy of experimentally-determined protein NMR structures.3,4 The work presented in this paper provides a convention and standardized protocol for defining which parts of the experimental NMR structure are sufficiently well-defined for assessing the accuracy of a CASP model prediction, and should be useful in future CASP experiments.
GDT measures form an alternative family of metrics for comparing superimposed structures.7,19 GDT scores are the most common metrics used to evaluate predictions in the CASP competition.7–9 They are relatively insensitive to regions of disorder, but are still dependent on the quality of the superimposition of the prediction to the actual structure. Thus the use of GDT measures does not obviate the need to assess the precision of atomic coordinates (i.e. the similarity of their relative positions across the ensemble of conformers) prior to the superimposition process, in order to prevent disordered regions of the protein structure from biasing the superimposition and hence the resulting GDT score.
The ensemble representation of NMR structural uncertainty
The ensemble representation of protein NMR suffers from several shortcomings. Most significantly, NMR ensembles are rarely generated in a statistically justified fashion, considering how the uncertainties in the underlying data propagate to uncertainties in the protein model.20–23 Secondly, the structure generation process usually assumes a static underlying structure rather than an ensemble-averaged sampling of structural models from a Boltzmann distribution. This assumption requires each member of the ensemble to best fit the complete set of experimental data, which is fundamentally incorrect, as each NMR observable arises from ensemble averaging of the dynamic protein structure conformation. The approximation is acceptable when the structural variations in the underlying molecular structure are small, but can result in various artifacts when the data arising from highly variable regions of the structure (e.g. backbone loops and some side chains) are best fit to a single conformer, even when this is done multiple times to generate the ensemble representation.24 For example, NOEs arising from multiple conformations in fast exchange on the NMR time scale may result in inconsistent restraints, which “pin” the conformation in a narrow and physically unrealistic conformational space (see for example Fig. 4 of Tejero, et al., 1996 24).
In spite of these shortcomings, the ensemble representation still remains the most common approach for presenting a protein NMR structure model. In this representation, uncertainty in atomic positions is generally assessed by comparing structural features across the ensemble. The most common method for comparing two conformational models is to superimpose (translate and rotate) one model onto the other model in order to minimize the RMSD between the two models. However, disordered loops and N- or C-terminal segments, as well as multiple domains whose relative orientations are not well-defined with respect to each other, can bias the optimal rotation as well as the resulting RMSD.1, 25
Alternative methods for distinguishing well-defined from not-well-defined regions of protein NMR structures
Well-defined atom sets can also be identified by analysis of dihedral angle circular variance, or dihedral angle order parameters.2,25,26 These methods are reasonably robust and simple. Like the method outlined here, they generally require an arbitrary cutoff to determine which dihedral angles are “well defined” and which are “not well defined” across the NMR ensemble.
Theobald and Wuttke27,28 have described a method of superimposition guided by maximum likelihood. For ensembles sampled from well-behaved statistical distributions, such as the multivariate normal distributions that occur in coordinates of models sampled from a harmonic energy landscape according to the Boltzmann distribution, such maximum likelihood methods do avoid the necessity to define a core atom set for superimposition.27, 28 However, in the absence of a well-defined sampling distribution, such methods may not produce maximum likelihood superimpositions. In any case, some convention is required in utilizing the superimposed ensembles to distinguish “well defined” from “not-well-defined” atomic positions.10
FindCore1 is a superimposition independent method for isolating a core atom set, and partitioning that set into domains. Unlike dihedral angle order parameters25, which are commonly used to identify a core atom set, FindCore uses non-local data, in the form of the inter-atomic variance matrix29, to identify the core atom set. A key advantage of using such non-local data is that regions within loops and tails, for which well-defined, precise conformational information is not available, may be locally very consistent due to “pinning artifacts” 24 that arise from ensemble averaging effects, and/or to a underlying propensity for disordered regions to form secondary structure, e.g. upon binding a suitable partner. Moreover, well-defined regions of a structure may have relatively under-constrained dihedral angles due to a lack of direct constraints on heavy atoms in NMR data.
Another advantage of FindCore in ensuring superimpositions and their associated RMSD statistics are not biased by the inclusion of disordered regions in the core atom set, is that FindCore, in practice, is particularly stringent about its definition of the core: if a few well-defined atoms are not included in the core, the resulting superimposition still adequately superimposes those atoms (Snyder and Montelione, 20051: Fig 1A). Using a stringently-classified set of core atoms for the initial superimposition also gives a reasonably RMSD statistic whereas inclusion of ill-defined atoms in the core can throw off a superimposition and the associated RMSD. However, a thorough evaluation of the quality of a prediction should include a comparison of all well-defined atoms, not just the subset of such atoms sufficient to fairly calculate an RMSD.1
Defining cutoff values for discrimination well-defined and not-well defined regions
Several attempts have been made in the literature to design an objective score for defining well- vs. not-well-defined regions of a biomolecular NMR structure that does not use a preset cutoff. These are discussed in detail by Snyder and Montelione (2005) 1 in presenting the original FindCore algorithm, and in a recent review on protein NMR structure validation 4. The Z score = 3 cutoff used in the Expanded FindCore is a convention that was selected to include atoms in the expanded core atom set which, have coordinate uncertainty <u2> within 3 standard deviations (Z=3) of the distribution of coordinate variances within the initial core atom set. These results are similar to those obtained with the CYRANGE program 2 based on dihedral angle distributions. While either approach is suitable for the problem at hand, at the time of the CASP10 assessment we elected to use Expanded Find Core, as it is an atom, rather than residue, based discriminator.
We considered two ways of discriminating well-defined vs. not-well-defined atom sets; the first would be based on a hard coordinate uncertainty cutoff (e.g. 1 Å uncertainty after superimposition), the second would be based on a Z score of deviations within the core atoms of the ensemble after superimposition. The second approach results in a critical cutoff that depends on the overall convergence within the well-defined regions of each NMR ensemble. In this case, an NMR ensemble with a well-defined core would require a tighter match between a predicted model and the “NMR target”; while a NMR ensemble with a less-well converged core tolerates a looser match between prediction and “NMR target”.
The convergence within each of the 18 NMR ensembles used in CASP10 varies considerably between the ensembles. Using a “hard cutoff” would result in some ensembles including atoms that clearly have not converged given the data available and thus would unfairly penalize predictors for deviations from the experimental structure in relatively poorly defined loop regions. A hard cutoff would additionally result in other experimental NMR structures being excluded all together because the coordinate uncertainty even in “well defined” regions does not meet the criterion. Using a fixed coordinate uncertainty cutoff would also exclude some key ligand binding sites of certain targets.
While for other applications of the FindCore method a fixed coordinate uncertainty cutoff may be appropriate, we decided that the latter approach, using a looser cutoff between well-defined and not-well-defined regions for experimentally less-well defined NMR structures, was more appropriate to the CASP10 assessment application. This was done to minimize the effect of penalizing predictors for poor predictions of imprecise experimental structures while still including such less-well-converged experimental NMR structures in the CASP competition and thus rewarding methods able to predict relatively well-defined regions of these structures. This “ensemble-dependent” cutoff was implemented in our Expanded FindCore protocol, prior to any comparison between experimental and predicted structure and independent of the “difficulty” of the target, by using a coordinate uncertainty cutoff <u2> corresponding to a Z score of 3 of the distribution of atomic variances within the core atom set of each NMR structure ensemble. This resulted in an absolute cutoff that ranged from 1.0 Å (T0727) to 5.89 Å (T0751).
Identifying well-defined regions of the NMR structure that are not well-defined with respect to one another
As mentioned above, a key feature of FindCore is its ability to identify “domains” from the core atom set that are internally well defined, by not well defined with respect to one another. For the 18 CASP10 NMR target structures used in this study, FindCore did not characterize any of the ensembles as having more than a single domain. The full-length target T0677 would likely have been split into two domains by FindCore, but was provided by the experimentalists as two separate domain targets (i.e. T0677_A and T0677_B). However, extension of the methods described above to multiple domain cores is straightforward: each domain requires a separate superimposition and hence, in an n-domain ensemble, n <u2> values characterize the variation in atomic position (relative to each domain’s optimal superimposition). Iterated editing of the core would simply use the per-atom minimum of these n per-atom <u2> values rather than the one per-atom value calculated in the case of a single domain structure. Alternatively, following initial domain identification using FindCore, assignment of all (heavy) atoms (core and non-core) can proceed by identifying the superimposition minimizing an atom’s <u2> value. The iterated superimposition approach described in the Methods section would then be applied on a per-domain basis.
Conclusions
The original FindCore method stringently defines a relatively sparse core atom set, in order to ensure the best possible core atom set for estimation of the precision of NMR-derived structures. For certain applications, such as phasing of X-ray diffraction data using NMR structures 30, this stringent core is appropriate. Expanded FindCore starts with the core robustly defined by FindCore and iteratively expands it to provide an intuitive description of core atoms. The use of Expanded FindCore in defining core atom sets for evaluating CASP10 predictions of protein structures experimentally determined via NMR confirmed the utility of Expanded FindCore in characterizing protein NMR structure ensembles. As the superimpositions and validation statistics reported in this paper demonstrate, Expanded FindCore defines a set of atoms within the NMR structure ensemble suitable for evaluation of CASP10 model predictions.
Supplementary Material
Acknowledgments
The authors wish to acknowledge many fruitful and enlightening conversations with Andriy Kryshtafovych, B.K. Lee, Lucio Ferella, Antonio Rosato, James Aramini, Jeremy Block, Binchen Mao, and Todd Taylor. This work was supported by a grant from the Protein Structure Initiative of the National Institutes of Health (U54-GM094597). RT also acknowledges support from CONSOLIDER INGENIO CSD2010-00065 and Generalitat Valenciana PROMETEO 2011/008. DS also acknowledges support from the Research Corporation for Science Advancement, College Cottrell Grant, Award #19803, and that he completed this work with support with an award of Assigned Release Time (ART) from the Office of the Provost of William Paterson University of NJ.
References
- 1.Snyder DA, Montelione GT. Clustering algorithms for identifying core atom sets and for assessing the precision of protein structure ensembles. PROTEINS: Struct Funct Bioinf. 2005;59:673–686. doi: 10.1002/prot.20402. [DOI] [PubMed] [Google Scholar]
- 2.Kirchner DK, Guntert P. Objective identification of residue ranges for the superposition of protein structures. BMC Bioinformatics. 2011;12:170–180. doi: 10.1186/1471-2105-12-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Montelione GT, Nilges M, Bax A, Guntert P, Herrmann T, Richardson JS, Schwieters CD, Vranken WF, Vuister GW, Wishart DS, Berman HM, Kleywegt GJ, Markley JL. Recommendations of the wwPDB NMR Validation Task Force. Structure. 2013;21:1563–1570. doi: 10.1016/j.str.2013.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosato A, Tejero R, Montelione GT. Quality assessment of protein NMR structures. Curr Opin Struct Biol. 2013 doi: 10.1016/j.sbi.2013.08.005. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moult J, Pedersen JT, Judson R, Fidelis K. A large-scale experiment to assess protein structure prediction methods. PROTEINS: Struct Funct Bioinf. 1995;23:ii–iv. doi: 10.1002/prot.340230303. [DOI] [PubMed] [Google Scholar]
- 6.Moult J, Fidelis K, Kryshtafovych A, Tramontano A. Critical assessment of methods of protein structure prediction (CASP) Round IX. PROTEINS: Struct Funct Bioinf. 2011;79:1–5. doi: 10.1002/prot.23200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zemla A, Venclovas C, Moult J, Fidelis K. Processing and analysis of CASP3 protein structure predictions. PROTEINS: Struct Funct Bioinf. 1999;37:22–29. doi: 10.1002/(sici)1097-0134(1999)37:3+<22::aid-prot5>3.3.co;2-n. [DOI] [PubMed] [Google Scholar]
- 8.Zemla A, Venclovas C, Moult J, Fidelis K. Processing and evaluation of predictions in CASP4. PROTEINS: Struct Funct Bioinf. 2001;45:13–21. doi: 10.1002/prot.10052. [DOI] [PubMed] [Google Scholar]
- 9.Read RJ, Chavali G. Assessment of CASP7 predictions in the high accuracy template-based modeling category. PROTEINS: Struct Funct Bioinf. 2007;69:27–37. doi: 10.1002/prot.21662. [DOI] [PubMed] [Google Scholar]
- 10.Kinch LN, Shi S, Cheng H, Cong Q, Pei J, Mariani V, Schwede T, Grishin NV. CASP9 target classification. PROTEINS: Struct Funct Bioinf. 2011;79:21–36. doi: 10.1002/prot.23190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sevillano E, Meuth H, Rehr JJ. Extended x-ray absorption fine structure Debye-Waller factors. I. Monatomic crystals. Phys Rev B. 1979;20:4908–4911. [Google Scholar]
- 12.Struyf A, Hubert M, Rousseeuw P. Clustering in an Object-Oriented Environment. J Stat Softw. 1997;1:1–30. [Google Scholar]
- 13.Andrec M, Snyder DA, Zhou Z, Young J, Montelione GT, Levy RM. A large data set comparison of protein structures determined by crystallography and NMR: Statistical test for structural differences and the effect of crystal packing. PROTEINS: Struct Funct Bioinf. 2007;69:449–465. doi: 10.1002/prot.21507. [DOI] [PubMed] [Google Scholar]
- 14.Tejero R, Snyder D, Ma B, Aramini JM, Montelione GT. PDBStat: A Universal Restraint Converter and Restraint Analysis Software Package for Protein NMR. J Biomol NMR. 2013;56:337–351. doi: 10.1007/s10858-013-9753-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schrödinger L. The PyMOL Molecular Graphics System. 1.2r3pre. [Google Scholar]
- 16.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 17.Laskowski R, Rullmann JA, MacArthur M, Kaptein R, Thornton J. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 18.Dyson HJ, Wright PE. Equilibrium NMR studies of unfolded and partially folded proteins. Nat Struct Mol Biol. 1998;5:499–503. doi: 10.1038/739. [DOI] [PubMed] [Google Scholar]
- 19.Zemla A. LGA: a method for finding 3D similarities in protein structures. Nucleic Acids Res. 2003;31:3370–3374. doi: 10.1093/nar/gkg571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rieping W, Habeck M, Nilges M. Inferential Structure Determination. Science. 2005;309:303–306. doi: 10.1126/science.1110428. [DOI] [PubMed] [Google Scholar]
- 21.Rieping W, Habeck M, Bardiaux B, Bernard A, Malliavin TE, Nilges M. ARIA2: Automated NOE assignment and data integration in NMR structure calculation. Bioinformatics. 2007;23:381–382. doi: 10.1093/bioinformatics/btl589. [DOI] [PubMed] [Google Scholar]
- 22.Rieping W, Nilges M, Habeck M. ISD: a software package for Bayesian NMR structure calculation. Bioinformatics. 2008;24:1104–1105. doi: 10.1093/bioinformatics/btn062. [DOI] [PubMed] [Google Scholar]
- 23.Richter B, Gsponer J, Varnai P, Salvatella X, Vendruscolo M. The MUMO (minimal under-restraining minimal over-restraining) method for the determination of native state ensembles of proteins. J Biomol NMR. 2007;37:117–135. doi: 10.1007/s10858-006-9117-7. [DOI] [PubMed] [Google Scholar]
- 24.Tejero R, Bassolino-Klimas D, Bruccoleri RE, Montelione GT. Simulated annealing with restrained molecular dynamics using CONGEN: Energy refinement of the NMR solution structures of epidermal and type-α transforming growth factors. Protein Sci. 1996;5:578–592. doi: 10.1002/pro.5560050403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hyberts SG, Goldberg MS, Havel TF, Wagner G. The solution structure of eglin c based on measurements of many NOEs and coupling constants and its comparison with X-ray structures. Protein Sci. 1992;1:736–751. doi: 10.1002/pro.5560010606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhattacharya A, Tejero R, Montelione GT. Evaluating protein structures determined by structural genomics consortia. PROTEINS: Struct Funct Bioinf. 2007;66:778–795. doi: 10.1002/prot.21165. [DOI] [PubMed] [Google Scholar]
- 27.Theobald DL, Wuttke DS. THESEUS: maximum likelihood superpositioning and analysis of macromolecular structures. Bioinformatics. 2006;22:2171–2172. doi: 10.1093/bioinformatics/btl332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Theobald DL, Wuttke DS. Empirical Bayes hierarchical models for regularizing maximum likelihood estimation in the matrix Gaussian Procrustes problem. Proc Natl Acad Sci U S A. 2006;103:18521–18527. doi: 10.1073/pnas.0508445103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kelley LA, Gardner SP, Sutcliffe MJ. An automated approach for defining core atoms and domains in an ensemble of NMR-derived protein structures. Protein Eng. 1997;10:737–741. doi: 10.1093/protein/10.6.737. [DOI] [PubMed] [Google Scholar]
- 30.Mao B, Guan R, Montelione GT. Improved technologies now routinely provide protein NMR structures useful for molecular replacement. Structure. 2011;19:757–766. doi: 10.1016/j.str.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu D, Zhang Y. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. PROTEINS: Struct. Funct. Bioinf. 2012;80:1715–1735. doi: 10.1002/prot.24065. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.