

Abstract
Up to 85% of patients with pancreatic cancer have diabetes or hyperglycaemia, which frequently manifests as early as 2–3 years before a diagnosis of pancreatic cancer. Conversely, patients with new-onset diabetes have a 5–8-fold increased risk of being diagnosed with pancreatic cancer within 1–3 years of developing diabetes. Emerging evidence now indicates that pancreatic cancer causes diabetes. As in type 2 diabetes, β-cell dysfunction and peripheral insulin resistance are seen in pancreatic cancer-induced diabetes. However, unlike in patients with type 2 diabetes, glucose control worsens in patients with pancreatic cancer in the face of ongoing, often profound, weight loss. Diabetes and weight loss, which precede cachexia onset by several months, are paraneoplastic phenomena induced by pancreatic cancer. Although the pathogenesis of these pancreatic cancer-induced metabolic alterations is only beginning to be understood, these are likely mechanisms to promote the survival and growth of pancreatic cancer in a hostile and highly desmoplastic microenvironment. Interestingly, these metabolic changes could enable early diagnosis of pancreatic cancer, if they can be distinguished from the ones that occur in patients with type 2 diabetes. One such possible biomarker is adrenomedullin, which is a potential mediator of β-cell dysfunction in pancreatic cancer-induced diabetes.
Introduction
Pancreatic cancer is the fourth leading cause of cancer-related deaths in the USA.1 The incidence and mortality rates of pancreatic cancer are similar (~40,000 people per year in the USA),1 highlighting its dismal survival and prognosis, principally because the tumour frequently presents at an advanced stage (85% unresectable). 2,3 The relationship between diabetes and pancreatic cancer, a subject investigated for more than a century, has been complicated by the existence of a bidirectional association between the two entities (Figure 1a).4–6 Indeed, compelling epidemiological, clinical and experimental evidence now supports the concept that diabetes is induced by pancreatic cancer, and precedes the onset of cancer-specific symptoms by several months (Figure 1b). Pancreatic cancer-induced diabetes, which by definition is new-onset diabetes associated with pancreatic cancer, seems to be associated with paradoxical weight loss, which often manifests before the development of diabetes (Figure 1b). Understanding the mechanism of diabetes and weight loss in pancreatic cancer not only has broader implications for the field of obesity and diabetes, but also for early diagnosis of pancreatic cancer. In this Review, we summarize the evidence for paraneoplastic diabetes and associated weight loss in pancreatic cancer, and focus on the emerging concepts in the pathogenesis of these metabolic changes.
Figure 1.

Bidirectional association between pancreatic cancer and diabetes. a | Risk of diabetes after diabetes diagnosis. The risk of pancreatic cancer is high with new-onset diabetes (5–8-fold) whereas the risk levels out at about 1.5-fold 4 years after diabetes diagnosis.13 b | Timecourse of paraneoplastic diabetes and weight loss in relation to pancreatic cancer diagnosis, onset of symptoms and cachexia.45,61
Pancreatic cancer-induced diabetes
Epidemiological evidence
The association between pancreatic cancer and diabetes was noted as early as 1833,7 was clearly documented by the 1930s,8,9 and was described in a large cohort of patients with pancreatic cancer from Mayo Clinic (MN, USA) in 1958.10 Numerous epidemiological studies have explored the association between diabetes and cancer since the 1980s and three meta-analyses have been published (in 1995,11 200512 and 201113). The European Prospective Investigation in Cancer and Nutrition (EPIC) cohort correlated increased baseline haemoglobin A1C (HbA1C) levels with subsequent development of pancreatic cancer (OR 2.4 for HbA1C ≥6.5% compared with HbA1C <5.4%).14 The 1995 meta-analysis reported a twofold increased risk of pancreatic cancer in patients with long-standing (>5 years) diabetes and a greater risk in individuals with diabetes duration <5 years.11 In the 2005 meta-analysis, patients with diabetes had an overall relative risk of two for pancreatic cancer, but this risk increased to 4–7-fold in those with diabetes duration <3 years.12 The initial 3-year period after diabetes diagnosis was also found to be important for the development of pancreatic cancer when prospective pancreatographic screening was used.15 The 2011 meta-analysis confirmed a relative risk of 5.4 (95% CI 3.5–8.3) associated with diabetes duration <1 year with levelling of the risk at ~1.5-fold after 5 years.13 Thus, long-standing diabetes modestly increases the risk of pancreatic cancer. In fact, long-standing diabetes, insulin resistance and obesity have been shown to modestly increase the risk of several other cancers,16–21 and the risk might be further modulated by antidiabetic medications.22–27
However, the markedly higher risk of pancreatic cancer in patients with new-onset diabetes when compared with long-standing diabetes indicates that pancreatic cancer itself can cause diabetes. Support for this concept was provided by a population-based study by Chari et al.28 of 2,122 patients >50 years of age with new-onset diabetes in which 1 in 125 (0.85%) of the patients was diagnosed with pancreatic cancer within 3 years of diabetes onset (eightfold higher risk than expected for the population). Another population-based study among veterans in the San Francisco area from 2006 reported consistent results, although with a lower relative risk.29,30
Diabetes prevalence in pancreatic cancer
Increased prevalence of new-onset diabetes in patients with pancreatic cancer has been consistently seen in most case series, although the reported values have varied depending on the methodology of patient selection and criteria for diabetes diagnosis. Studies that screened for diabetes in patients with pancreatic cancer have reported considerably higher rates of diabetes31–35 than those relying on chart reviews for physician-diagnosed diabetes.10,30,36–41 This difference probably reflects the fact that one-third of new-onset diabetes in patients with pancreatic cancer remains unrecognized.42
Among studies that have screened for diabetes in pancreatic cancer, an even higher prevalence of diabetes is noted when an oral glucose tolerance test is performed, as opposed to analysis of fasting blood glucose levels.35 Preoperative oral glucose tolerance testing in 44 patients with resectable pancreatic cancer showed diabetes in 28 (64%) and impaired glucose tolerance in an additional four (11%) patients.43 Our group investigated the prevalence of diabetes in a prospectively recruited series of 512 patients with newly diagnosed pancreatic cancer by recording fasting glucose measurements within 30 days of diagnosis.31 Only 14% (56 patients) of these patients had normal fasting glucose values, whereas diabetes was present in 243 (47%) patients.31 Among patients with pancreatic cancer and diabetes, the duration of diabetes was <2 years in 74% (177 of 243). Pancreatic cancer-induced diabetes was therefore present in 34% of patients (177 of 512).31 Additionally, a large proportion of patients with pancreatic cancer (38%) had impaired fasting glucose levels but did not meet the diagnostic criteria for diabetes.31
Time course
The majority of diabetes in pancreatic cancer is of new onset.29,30,36,44,45 In a large retrospective study, fasting glucose measurements up to 5 years prior to the diagnosis of pancreatic cancer (n = 765) and in matched controls (n = 1,865) were reviewed and the prevalence of diabetes compared in the two groups.45 Although a trend towards a higher prevalence of diabetes was noted in patients as early as 36–48 months prior to diagnosis of pancreatic cancer, a significant increase (when compared with controls) was observed in months 24–36, 12–24 and 0–12.45 Thus, diabetes caused by pancreatic cancer starts up to 2–3 years before diagnosis of pancreatic cancer.
Examination of possible hypotheses
Unmasking of type 2 diabetes
Many forms of stress, such as pregnancy, weight gain and steroid therapy, can unmask type 2 diabetes. Arguments for this hypothesis as a cause of diabetes in pancreatic cancer include the fact that canonical risk factors for type 2 diabetes (such as older age, obesity and family history of diabetes) are also risk factors for pancreatic cancer-induced diabetes;31 pancreatic cancer-induced diabetes can also be resolved by successful treatment of the cancer.31,46 However, the high frequency of new-onset diabetes and hyperglycaemia in patients with pancreatic cancer point to a pancreatic cancer-specific stressor that profoundly, and consistently, decompensates glucose homeostasis.
Consequence of cachexia
A study in 2012 comparing diabetes prevalence among various cancers found diabetes in ~20% of patients with lung, prostate, breast and colon cancer, which was not significantly different from that in the matched control population.47 By contrast, the prevalence of diabetes was higher in patients with pancreatic cancer than in controls and was noted in ~66% of patients. Although it is well-recognized that cachexia in any cancer is a dysmetabolic state in which diabetes can occur,48,49 the much higher prevalence in pancreatic cancer compared with other cancers suggests a unique relationship. Moreover, pancreatic cancer has one of the highest incidences of cachexia in any type of cancer.48,50 However, the onset of diabetes in pancreatic cancer occurs 2–3 years prior to the diagnosis of cancer, whereas cachexia-associated symptoms in pancreatic cancer manifest on average 2 months prior to cancer diagnosis (Figure 1b);45 therefore, pancreatic cancer-induced diabetes cannot be attributed to cachexia.
Destruction of the pancreas
Although destruction and loss of normal pancreatic tissue owing to pancreatic cancer is possible in advanced stages, three pieces of evidence argue against such a mechanism for pancreatic cancer-induced diabetes. First, diabetes occurs even before the tumour is radiologically detectable.51,52 Second, insulin levels are relatively high in pancreatic cancer when compared with healthy controls, suggesting insulin resistance.33,53–56 Third, diabetes improves after resection of the cancerous parts of the pancreas.31,46
A paraneoplastic phenomenon
Compelling clinical and laboratory evidence supports the hypothesis that pancreatic cancer-induced diabetes is a paraneoplastic phenomenon caused by the cancer. Evidence for this hypothesis is presented in Box 1.
Box 1. Evidence for pancreatic cancer-induced diabetes.
Diabetes or impaired glucose tolerance occurs in the majority of patients with pancreatic cancer31 and precedes clinical presentation of cancer by several months to a few years45
Diabetes is prevalent in small pancreatic cancers142 and diabetes occurs before the tumour is radiologically detectable52
Worsening of diabetes occurs in patients with long-standing diabetes in the months preceding the diagnosis of pancreatic cancer59,60
Diabetes improves after surgical resection of pancreatic cancer31,46
Occurrence of diabetes preceding pancreatic cancer symptoms has been demonstrated in the hamster model of pancreatic cancer,78,143–145 which is consistent with clinical data
Insulin resistance and β-cell dysfunction has been reported in patients with pancreatic cancer by homeostasis model assessment66
Supernatants from pancreatic cancer cell lines have been shown to induce insulin resistance in cultured hepatocytes146,147 and myoblasts,148 as well as β-cell dysfunction in vivo149 and in vitro70,82–84,150
Skeletal muscle tissue obtained from patients with pancreatic cancer demonstrated insulin resistance in vitro when compared with tissue from healthy controls78,79
Paraneoplastic weight loss
Epidemiology and time course
Weight loss in pancreatic cancer occurring before the onset of cancer-related symptoms was recognized in reports from the 1980s and 1990s34,57–59 and in previously published cohorts from our centre.31,44 In the large retrospective cohort described earlier,45 serial BMI and fasting glucose values were trended up to 5 years prior to the diagnosis of pancreatic cancer.60 Surprisingly, a reduction in BMI began as early as 3 years before the diagnosis of cancer; despite this reduction, glycaemic control worsened over time in these patients, in contrast to what is observed in individuals with type 2 diabetes (in which glycaemia improves with weight loss).60 Interestingly, at the onset of diabetes, 59% of patients (17 of 29) with pancreatic cancer-induced diabetes had lost weight whereas weight gain was seen in 56% of patients (24 of 43) with new-onset noncancer-related type 2 diabetes matched for the prediabetes weight.61 Although most patients with type 2 diabetes continued to gain weight, progressive weight loss was seen in those with pancreatic cancer-induced diabetes, starting as early as 1 year prior to diabetes onset.61 The mean interval of diabetes onset in pancreatic cancer, and onset of symptoms, respectively, to the diagnosis of pancreatic cancer was 13 months61 and 2 months.45 These data suggest that weight loss is associated with occurrence of diabetes, and precedes onset of cancer-specific symptoms and onset of diabetes in pancreatic cancer by several months.
A paraneoplastic phenomenon
Loss of lean muscle mass is the signature feature of cachexia,48,49 which usually results in >10% weight loss and is seen in the advanced stages of cancer. However, cardinal symptoms, such as fatigue and anorexia, might start before the onset of muscle loss (pre-cachexia).49 In a report from 2010 examining cachexia in patients with lung and colorectal cancer, weight loss started only ~7 months prior to their death.62 Unfortunately, pancreatic cancer usually presents in the advanced stage with cachexia symptoms being invariably present at diagnosis (they can be its only clinical manifestation) (Figure 1). Therefore, weight loss in pancreatic cancer after the onset of symptoms is undoubtedly occurring in conjunction with cachexia (Figure 2).
Figure 2.

A model depicting the phases of weight loss in pancreatic cancer. Weight loss precedes any symptoms related to cancer or cachexia by several months. We propose that the weight loss, associated with diabetes and occurring prior to the onset of symptoms, is a paraneoplastic phenomenon induced by pancreatic cancer. Abbreviations: SAT, subcutaneous adipose tissue; VAT, visceral adipose tissue.
However, as discussed earlier, weight loss precedes the onset of symptoms in pancreatic cancer by several months. This initial period of weight loss cannot be attributed to cachexia. In fact, in our experience, patients deny feeling tired or eating less during this period, and are pleasantly surprised about having lost weight effortlessly. In our opinion, this weight loss, associated with diabetes and occurring prior to the onset of cachexia, is a paraneoplastic phenomenon induced by pancreatic cancer (Figure 2). In the absence of cachexia (and associated muscle loss), this paraneoplastic weight loss seems to result from loss of adipose tissue. We hypothesize that pancreatic cancer interacts with adipose tissue to induce this paraneoplastic weight loss that paradoxically occurs along with diabetes.
Mechanisms of paraneoplastic phenomena
Mechanisms analogous to type 2 diabetes
In individuals with obesity who are normoglycaemic, peripheral insulin resistance is present but compensated for by increased insulin secretion.63–65 Insulin resistance progressively worsens in the predisposed individuals along with progressive β-cell dysfunction and reduction of β-cell mass, eventually leading to type 2 diabetes. 63–65 Interestingly, a similar temporal relationship between insulin resistance, β-cell function and development of impaired glucose tolerance and diabetes was demonstrated in patients with pancreatic cancer.66
β-cell dysfunction
The existence of a diabetes-causing product of pancreatic cancer has been postulated for over two decades. Initial research led to isolation of amylin67 and S-100A8 N-terminal peptide,68,69 which were shown to cause insulin resistance in vitro, but their effects on β cells are unknown. A direct tumour-secreted mediator of β-cell dysfunction has been recognized in a collaborative study from Mayo Clinic (MN, USA) and MD Anderson Cancer Center (TX, USA) published in 2012.70 Gene profiling using microarray analysis of pancreatic cancer cell lines known to inhibit insulin secretion yielded 18 upregulated proteins70 among which adrenomedullin, a 52 amino acid peptide known to inhibit insulin secretion, 71,72 was identified. Adrenomedullin is a pluripotent hormone; in the pancreas, its receptors are found on β cells73 and its expression is seen specifically in the F cells of the islets,74 but the significance of these observations remain unclear.
Adrenomedullin was shown to mediate pancreatic cancer-induced inhibition of insulin secretion in β cells in various in vitro and in vivo orthotopic and subcutaneous tumour models.70 Interestingly, plasma adrenomedullin levels were higher in patients with pancreatic cancer than in patients with diabetes or healthy controls; the highest levels were seen in those with pancreatic cancer-induced diabetes.70 Moreover, overexpression of adrenomedullin was seen in surgically resected specimens of pancreatic cancer.70 Another group had previously shown that adrenomedullin is upregulated in pancreatic cancer in conditions of hypoxia75,76 and hypoglycaemia.76 Thus, adrenomedullin, secreted by the cancerous pancreas in its hostile microenvironment, is a mediator of β-cell dysfunction. However, it is possible that other (as yet unrecognized) adrenomedullin-independent mediators of β-cell dysfunction might exist.
Insulin resistance in β cells, hyperglycaemia and non-esterified fatty acids (NEFA) are known to indirectly lead to β-cell dysfunction and loss of β-cell mass in type 2 diabetes. 65 As discussed below, these indirect mechanisms also seem to be operational in pancreatic cancer-induced diabetes. The direct and indirect effects of pancreatic cancer on β cells are summarized in Figure 3.
Figure 3.

A model demonstrating pancreatic cancer and β-cell interactions resulting in the pathogenesis of paraneoplastic diabetes. Pancreatic cancer-derived products might directly affect β cells. Indirect effects resulting from the consequences of insulin resistance and adipose tissue interactions on β cells might also be important. Abbreviation: NEFA, non-esterified fatty acids.
Insulin resistance
Insulin resistance is consistently seen in patients with pancreatic cancer (even in those with normal fasting glucose levels66) and resolves after resection of the cancer.31 At the postreceptor level, insulin signalling is conveyed via insulin receptor substrate proteins through distinct downstream pathways for the control of metabolism and for regulation of cell proliferation in insulin-sensitive cells.65,77 In type 2 diabetes, selective resistance in the metabolic pathways but continued sensitivity in the proliferation pathways in observed,77 and the resistance occurs at the postreceptor level.65,77 Similar to type 2 diabetes, insulin resistance in pancreatic cancer is thought to occur at the postreceptor level. Evidence supporting this assertion was provided in a study78 that revealed differences in glycogen synthesis and glycogen breakdown in skeletal muscles obtained from patients with pancreatic cancer-induced diabetes compared with those with pancreatic cancer without diabetes and healthy controls. By contrast, insulin receptor binding, tyrosine kinase activity, insulin receptor substrate 1 and glucose transporter type (GLUT) 4 levels were similar.78 Furthermore, impaired action of phosphoinositide 3-kinase (a downstream effector in the insulin-regulated metabolic pathways) and impaired glucose uptake was observed in the skeletal muscle of patients with pancreatic cancer.79
The search for a putative mediator of insulin resistance in pancreatic cancer was boosted in the 1990s with the demonstration that insulin resistance induced by pancreatic cancer-conditioned media could be localized to a <10 kDa fraction.80 Subsequently, islet amyloid polypeptide (IAPP) was identified; levels of this putative mediator were higher in patients with pancreatic cancer than in patients with other cancers, diabetes or healthy controls67 and is known to cause insulin resistance in skeletal muscles.81 IAPP is normally secreted with insulin by β cells and pancreatic cancer was found to cause β cells to selectively secrete IAPP through direct stimulation82 and by altering responsiveness of β cells to other secretagogues.83,84 However, it was subsequently shown that IAPP does not have good diagnostic or discriminative potential in patients with pancreatic cancer.44 No subsubsequent studies have been conducted to explore the pathophysiological role of IAPP in pancreatic cancer. Another potential mediator identified in patients with pancreatic cancer-induced diabetes was S-100A8 N-terminal peptide,68,69 which induces insulin resistance in vitro, but further research is needed to explore its importance in pancreatic cancer. Therefore, at the moment, a biochemical mediator of insulin resistance secreted by pancreatic cancer remains a hypothesis.
The role of adipose tissue
Interactions between adipose tissue and pancreatic cancer might explain the occurrence of insulin resistance as well as paraneoplastic weight loss in pancreatic cancer. The role of adipose tissue in the development of the metabolic syndrome and type 2 diabetes is only starting to be elucidated.65,85,86 Here, insights from the field of type 2 diabetes and the metabolic syndrome are presented, with a discussion of how pancreatic cancer could induce similar pathogenic processes.
Adipose tissue inflammation
A key feature of the metabolic syndrome is inflammation of adipose tissue and alteration of adipokine secretion and sensitivity.87,88 Insulin resistance precedes and accompanies type 2 diabetes. Adiponectin, leptin, resistin and numerous other adipokines have been identified as possible mediators of insulin resistance within the past decade, although leptin and adiponectin are now believed to be the important ones in diabetes.65,77,89–91 Accumulation of visceral fat is associated with low-grade chronic inflammation in adipose tissue65,92 resulting from an interplay between inflammasome activation within adipocytes and sensitization of adipose tissue macrophages.87,93,94 Macrophages release inflammatory cytokines (which can comprise up to 90% of the hormonal output of adipose tissue) such as tumour necrosis factor (TNF), IL-6 and monocyte chemoattractant protein 1 that contribute to peripheral insulin resistance.65,77,92,94 Local inflammatory signals alter adipocyte secretion (drop in adiponectin, increase in leptin secretion)86,95 and responsiveness (resistance to leptin),65 which ultimately lead to the development of insulin resistance (Figure 4).77,92,94
Figure 4.

A model demonstrating pancreatic cancer and adipose tissue interactions resulting in the pathogenesis of paraneoplastic diabetes and associated weight loss. Interactions between pancreatic cancer and adipose tissue might lead to adipose tissue inflammation resulting in a systemic cytokine response, altered adipokine secretion and lipolysis. Eventually, these factors cause peripheral insulin resistance and β-cell dysfunction. Abbreviation: NEFA, non-esterified fatty acids.
Inflammation of adipose tissue has not been directly studied in pancreatic cancer. One small study reported an increased adiponectin:leptin ratio in patients with newly diagnosed pancreatic cancer, which is comparable to patients with and without diabetes.32 Prediagnostic inflammatory markers and subsequent development of pancreatic cancer was studied in the EPIC cohort revealing an association with soluble TNF receptor levels (sTNF-R1 in females; sTNF-R2 in individuals with obesity and diabetes of either sex).96 A review discussing the existence of pancreatic steatosis in the metabolic syndrome and type 2 diabetes argued for the possible relationship of pancreatic adipose tissue and pancreatic cancer,97 although further research is needed to confirm this relationship.
Lipolysis and NEFA toxicity
Increased lipolysis occurs with excessive fat accumulation in the metabolic syndrome and obesity, leading to the generation of NEFA.65 The release of NEFA has been regarded as a crucial factor in causing peripheral insulin resistance.65 Furthermore, direct NEFA toxicity to β cells,65 as well as insulin resistance in β cells,98 contribute to β-cell dysfunction and β-cell loss. As discussed earlier, the paraneoplastic phase of weight loss in pancreatic cancer seems to be predominantly mediated by adipose tissue lipolysis, which might be an essential mechanism in the development of insulin resistance, β-cell dysfunction and diabetes in pancreatic cancer (Figure 4). We hypothesize that inflammation of adipose tissue occurs in pancreatic cancer leading to adipokine, cytokine and NEFA-mediated insulin resistance (Figure 4). How pancreatic cancer interacts with adipose tissue remains an intriguing subject. Some possible mechanisms of how pancreatic cancer can cause inflammation of adipose tissue include: direct effects of factors released from a cancerous pancreas on adipose tissue; activation of pancreatic macrophages through inflammasomes, which might seed adipose tissue; and sensitization of naive circulating macrophages that might reach adipocytes. Furthermore, lipolysis of adipose tissue might be induced by pancreatic cancer to supply metabolic substrates for tumour growth and survival. A number of lipid-mobilizing factors, including zinc-α-2 glycoprotein,99,100 have been suggested in cachexia-related adipolysis (observed in the advanced stages of many cancers). We have proposed the presence of a lipid-mobilizing factor in pancreatic cancer that is secreted by adipose tissue, possibly in response to inflammation of adipose tissue (Figure 4).61
Exploring the paradox
Pancreatic cancer-induced diabetes is associated with weight loss, unlike type 2 diabetes, which is associated with weight gain and obesity. How could this paradox be explained?
Topographic differences
The visceral depot of adipose tissue, as compared to the subcutaneous depot, is accepted as being important in the development of insulin resistance and the metabolic syndrome.86,101–103 In fact, McLaughlin et al.104 showed that each standard deviation increase in subcutaneous adipose tissue reduced the risk of insulin resistance by 48%, whereas a standard deviation increase in visceral adipose tissue mass increased the risk of insulin resistance by 80%. Surgical removal of subcutaneous fat by liposuction did not affect insulin resistance,105 whereas removal of visceral fat led to improvement106 or an equivocal response.107 Multiple structural and functional differences exist between subcutaneous and visceral adipocytes (reviewed elsewhere108), which might account for their differential roles in health and disease. Differences in inflammatory gene expression profile between visceral and subcutaneous adipocytes taken from patients with type 2 diabetes (and healthy controls) have also been reported.109
Differential responses of adipose fat
Our group has started to focus on the differential responses of adipose tissue compartments in pancreatic cancer. Preliminary data indicate a differential response of visceral and subcutaneous fat compartments, with greater loss of subcutaneous compartment and relative preservation of visceral compartment in patients with pancreatic cancer (S. T. Chari, unpublished work). Progressive worsening of glycaemia and insulin resistance in pancreatic cancer is probably driven by the fairly preserved visceral compartment, whereas selective loss of the subcutaneous compartment might explain the paradoxical weight loss. Although cachexia-associated weight loss is defined by loss of lean muscle mass, marked loss of adipose tissue also occurs and similar loss occurs from both compartments.62 This paraneoplastic phase of weight loss in pancreatic cancer therefore seems to be unique. Could this feature be related to the difference in diabetes occurrence in pancreatic cancer versus other cancers? This provocative hypothesis needs further investigation.
Enhanced survival and tumour growth
Why do these paraneoplastic metabolic alterations occur in pancreatic cancer? Our understanding of the significance of these alterations is still in its early stages. Pancreatic cancer cells have a hostile microenvironment with poor vasculature, low nutrient supply and hypoxia. Yet these cells show the most aggressive behaviour— resistance to apoptosis, high proliferation rate and invasiveness. In our opinion, pancreatic cancer induces diabetes, lipolysis and weight loss for enhanced survival, proliferation and tumour growth, and possibly carcinogenicity (Figure 5).
Figure 5.
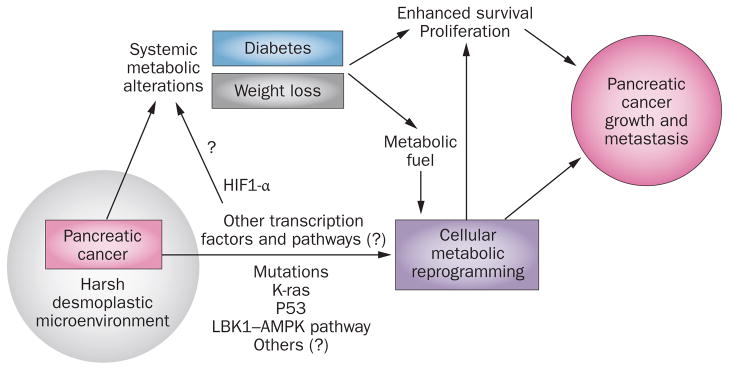
Significance of metabolic alterations in pancreatic cancer. Paraneoplastic diabetes and weight loss are induced by pancreatic cancer possibly to enhance survival, proliferation, tumour growth and carcinogenesis. Pancreatic cancer is highly desmoplastic with an extremely hostile microenvironment. The pancreatic cancer cell is metabolically reprogrammed for survival and proliferation in harsh conditions. Multiple cellular pathways have been identified that contribute to aerobic glycolysis and might lead to the release of tumour factors, which mediate systemic metabolic alterations. Diabetes and lipolysis might supply substrates for reprogrammed metabolic machinery resulting in enhanced survival of cancer cells and tumour growth whereas hyperglycaemia and hyperinsulinaemia might directly enhance proliferation and carcinogenicity.
Supply of metabolic substrates
In addition to metabolic changes in the patient, the intracellular metabolic machinery of pancreatic cancer is reprogrammed towards a metabolism more suitable for proliferation in the midst of hostile conditions. Preferential aerobic glycolysis (Warburg effect110) is one such intracellular reprogramming of glucose metabolism in pancreatic cancer cells, which is accompanied by aberrant expression and activity of metabolic enzymes and of cellular receptors for glucose uptake.111–113 Despite high uptake of glucose in pancreatic cancer cells, it is metabolized inefficiently by aerobic glycolysis.111 Biosynthetic intermediates are generated in exchange for this inefficient energy production, which sustains proliferation and confers a survival advantage.111 Hyperglycaemia and lipolysis might therefore provide a supply of glucose and substrates for the metabolic and growth demands of the reprogrammed ‘hungry’ pancreatic cancer cells (Figure 5).
The mechanisms of such metabolic reprogramming is only starting to be unravelled.111 Oncogenic K-ras protein mutations strongly associated with pancreatic cancer seem to be important in reprogramming cells to anabolic glucose metabolism and maintaining tumorigenicity in a harsh environment.114 Other important pathways recognized to link cellular metabolism to proliferation and carcinogenicity are: p53;111 c-Myc;111 liver kinase B1 (LKB1, encoded by STK11)–AMPK;111 mammalian target of rapamycin (mTOR; activated by insulin signalling); and hypoxia-inducible factor 1-α (HIF1-α). An involvement of microRNAs in fine regulation of these pathways has also been suggested to occur in pancreatic cancer.115
Proliferation and tumorigenesis
Emerging evidence indicates that glucose116 and advanced glycation end products (AGE)117,118 are mitogenic. In fact, hyperglycaemia has been shown to enhance proliferation,119,120 local invasiveness and metastatic potential in pancreatic cancer.121,122 The expression of receptor for AGE (RAGE) was shown to promote pancreatic cancer tumorigenesis.123,124 However, a case–control study failed to show any association between AGE and pancreatic cancer risk, but found an inverse association with RAGE levels at onset, in patients diagnosed with pancreatic cancer within 2 years of follow up.125 Another study from Finland reported a similar inverse association.126
Hyperinsulinaemia also stimulates proliferation of pancreatic cancer,24,122 which is known to overexpress insulin-like growth factor receptor and G-protein coupled receptors.127 These proliferative effects of hyperglycaemia and hyperinsulinaemia might also explain the increased risk of pancreatic cancer in long-standing diabetes, which is even higher among insulin users.122 Insulin and insulin-like growth factors are implicated in proliferation and neoplastic transformations in other cancers.128 Interestingly, metformin has been shown to reduce the risk of pancreatic cancer,26,129 as well as several other cancers. This phenomenon has provided interesting insights into the links between energy supply, metabolism and proliferation.129 Metformin is known to activate the LKB1–AMPK pathway, a cellular energy stress-sensing mechanism, and blocks proliferation through inhibition of mTOR (which drives the proliferation effects of the insulin signalling pathway).129 In fact, STK11 is a tumour suppressor gene and its germline mutations are associated with Peutz–Jeghers syndrome (patients with this syndrome have an increased risk of pancreatic cancer);130 mutations in this gene have also been reported in sporadic pancreatic cancers.131
Furthermore, pathway analysis of genome-wide association study data in patients with pancreatic cancer suggested an association between susceptibility for pancreatic cancer and the pancreatic development genes HNF1A, HNF1B and PDX1 and also another development gene NR5A2 that is involved in lipid and glucose metabolism.132 HNF1A, HNF1B and PDX1 are known to be associated with maturity-onset diabetes of the young type 3, type 5 and type 4 respectively; HNF1A and HNF1B are also associated with type 2 diabetes.132 These data highlight the complex relationship between diabetes and pancreatic cancer, and provide another mechanism of increased risk of pancreatic cancer with long-term diabetes and conversely, might partly explain the metabolic alterations in pancreatic cancer.
Desmoplasia and hypoxia
Pancreatic cancer is a highly desmoplastic tumour with a stressful microenvironment. In response to hypoxia and other cellular stressors, HIFs are induced that modulate a range of cellular responses conferring survival advantage in the hostile microenvironment. HIF1-α is thought to be particularly important in pancreatic cancer.133–135 Constitutive expression of HIF1-α was seen in the majority of pancreatic cancer cell lines, but not in most cell lines of other cancers.134
HIF1-α conferred resistance to apoptosis, upregulated glycolytic proteins and selectively upregulated GLUT subtypes that favour glycolysis.134 Dominant negative HIF1-α expression was shown to inhibit tumorigenicity of pancreatic cancer and led to suppression of glycolytic metabolism.135 A hypoxia-independent mechanism might also regulate HIF1-α in pancreatic cancer.133 Mucin-1, which is known to be overexpressed in pancreatic cancer, activates and stabilizes HIF1-α, and its expression enhances glycolytic activity both in vitro and in vivo.133 Thus, HIF1-α might be important in metabolic reprogramming of pancreatic cells suitable for its hostile microenvironment, as well as providing resistance to apoptosis (Figure 5). In addition, HIF1-α is also a transcription factor for adrenomedullin76 and might mediate the overexpression of adrenomedullin in pancreatic cancer. Adrenomedullin, shown to be induced in hypoxic conditions,75,76 mediates β-cell dysfunction70 and enhances cancer invasion,75 and therefore might be a survival mechanism (Figure 5).
Predicting pancreatic cancer
Prognosis of pancreatic cancer is extremely poor despite surgical resection and chemotherapy. Early diagnosis might be the best hope of increasing survival in pancreatic cancer.3 Individuals with new-onset diabetes are at high risk of developing pancreatic cancer with ~1% of patients developing pancreatic cancer within 3 years.28 However, type 2 diabetes is at least 100 times more common than pancreatic cancer-induced diabetes. To utilize its potential in screening for pancreatic cancer, one has to be able to distinguish between pancreatic cancer-induced diabetes and type 2 diabetes.136
New-onset type 2 diabetes is classically associated with the metabolic syndrome, weight gain and family history of diabetes. By contrast, pancreatic cancer-induced diabetes is seen even in individuals without associated family history and other manifestations of the metabolic syndrome.31,45 However, these epidemiological features alone are insufficient to distinguish between these two types of diabetes. Presence of weight loss in the preceding months prior to onset of diabetes might be an important predictor of the development of pancreatic cancer-induced diabetes,61 although this feature will probably have limited diagnostic utility in clinical practice. Adrenomedullin levels are higher in pancreatic cancer-induced diabetes than in new-onset type 2 diabetes,70 but its diagnostic utility remains to be explored in a large cohort. A second biochemical marker for pancreatic cancer-induced diabetes is essential to distinguish those at high risk of pancreatic cancer from all patients with new-onset diabetes.3 The search for a good biomarker remains in progress (discussed extensively elsewhere3,136).
Type 3 diabetes (or pancreatogenous diabetes) is defined as diabetes resulting from diseases of the exocrine pancreas137–139 and is probably an underdiagnosed entity.137,138 By this anatomical definition, pancreatic cancer-induced diabetes has been classified as type 3c diabetes.4,137,139 Type 3 diabetes is characterized by low insulin and pancreatic polypeptide levels, increased peripheral insulin sensitivity, but reduced hepatic insulin sensitivity,4,137,138 which is in contrast to type 2 diabetes.140,141 However, as discussed previously, patients with pancreatic cancer-induced diabetes have high insulin levels and peripheral insulin resistance similar to type 2 diabetes rather than type 3 diabetes.33,53–56 Rightfully, the American Diabetes Association139 acknowledges that pancreatic cancer-induced diabetes differs from the other diseases listed in the type 3 category (such as diabetes related to chronic pancreatitis), although further studies are needed to better characterize these differences.
Conclusions
Pancreatic cancer is associated with paraneoplastic diabetes. Furthermore, paradoxical weight loss occurs in the face of worsening diabetes in pancreatic cancer. The mechanism and significance of these metabolic alterations is only starting to be uncovered but such understanding might yield biomarkers for the early diagnosis of pancreatic cancer.
Key points.
Compelling evidence now indicates that pancreatic cancer causes paraneoplastic diabetes
As in type 2 diabetes, β-cell dysfunction and peripheral insulin resistance occur in pancreatic cancer-induced diabetes; however, unlike type 2 diabetes, weight loss occurs alongside worsening diabetes in pancreatic cancer
Paraneoplastic diabetes and weight loss manifest many months prior to the onset of cachexia or clinical presentation of pancreatic cancer
Differential responses of visceral and subcutaneous adipose tissue compartments in pancreatic cancer might underlie the development of insulin resistance and paradoxical weight loss
These metabolic alterations might be induced by pancreatic cancer for enhanced survival and tumour growth in an otherwise hostile microenvironment
Review criteria.
We searched PubMed and Ovid databases using the following key words alone or in various combinations: “pancreatic cancer”, “diabetes”, “diabetes mellitus”, “pancreatic cancer associated diabetes” and “weight loss”, and retrieved articles from 1985 to 2012. We reviewed original studies and reviews for relevance and included all pertinent studies in the preparation of the manuscript. We also reviewed the bibliographies of the selected articles for other relevant articles.
Acknowledgments
D. Mukhopadhyay was supported by funding from NIH (R01 CA150190) and the Mayo Clinic Pancreas Cancer SPORE (P50 CA 102701). S. T. Chari was supported by grants from the NIH (R01 CA 100685) and the Mayo Clinic Pancreas Cancer SPORE (P50 CA 102701).
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
All authors researched data for the article and reviewed and/or edited the manuscript before submission. R. P. Sah, D. Mukhopadhyay and S. T. Chari substantially contributed to the discussion of content. R. P. Sah, S. J. S. Nagpal and S. T. Chari wrote the Review.
References
- 1.Jemal A, et al. Cancer statistics, 2005. CA Cancer J Clin. 2005;55:10–30. doi: 10.3322/canjclin.55.1.10. [DOI] [PubMed] [Google Scholar]
- 2.Conlon KC, Klimstra DS, Brennan MF. Long-term survival after curative resection for pancreatic ductal adenocarcinoma. Clinicopathologic analysis of 5-year survivors. Ann Surg. 1996;223:273–279. doi: 10.1097/00000658-199603000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chari ST. Detecting early pancreatic cancer: problems and prospects. Semin Oncol. 2007;34:284–294. doi: 10.1053/j.seminoncol.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Magruder JT, Elahi D, Andersen DK. Diabetes and pancreatic cancer: chicken or egg? Pancreas. 2011;40:339–351. doi: 10.1097/MPA.0b013e318209e05d. [DOI] [PubMed] [Google Scholar]
- 5.Li D. Diabetes and pancreatic cancer. Mol Carcinog. 2012;51:64–74. doi: 10.1002/mc.20771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cui Y, Andersen DK. Diabetes and pancreatic cancer. Endocr Relat Cancer. 2012;19:F9–F26. doi: 10.1530/ERC-12-0105. [DOI] [PubMed] [Google Scholar]
- 7.Bright R. Cases and observations connected with disease of the pancreas and duodenum. Med Chir Trans. 1833;18:1–56. doi: 10.1177/09595287330180p102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marble A. Diabetes and pancreatic cancer. N Engl J Med. 1934;211:339–349. [Google Scholar]
- 9.Grauer FW. Pancreatic carcinoma: a review of thirty-four autopsies. Arch Intern Med. 1939;63:884–889. [Google Scholar]
- 10.Green RC, Jr, Baggenstoss AH, Sprague RG. Diabetes mellitus in association with primary carcinoma of the pancreas. Diabetes. 1958;7:308–311. doi: 10.2337/diab.7.4.308. [DOI] [PubMed] [Google Scholar]
- 11.Everhart J, Wright D. Diabetes mellitus as a risk factor for pancreatic cancer. A meta-analysis. JAMA. 1995;273:1605–1609. [PubMed] [Google Scholar]
- 12.Huxley R, Ansary-Moghaddam A, Berrington de Gonzalez A, Barzi F, Woodward M. Type-II diabetes and pancreatic cancer: a meta-analysis of 36 studies. Br J Cancer. 2005;92:2076–2083. doi: 10.1038/sj.bjc.6602619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ben Q, et al. Diabetes mellitus and risk of pancreatic cancer: A meta-analysis of cohort studies. Eur J Cancer. 2011;47:1928–1937. doi: 10.1016/j.ejca.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 14.Grote VA, et al. Diabetes mellitus, glycated haemoglobin and C-peptide levels in relation to pancreatic cancer risk: a study within the European Prospective Investigation into Cancer and Nutrition (EPIC) cohort. Diabetologia. 2011;54:3037–3046. doi: 10.1007/s00125-011-2316-0. [DOI] [PubMed] [Google Scholar]
- 15.Ogawa Y, et al. A prospective pancreatographic study of the prevalence of pancreatic carcinoma in patients with diabetes mellitus. Cancer. 2002;94:2344–2349. doi: 10.1002/cncr.10493. [DOI] [PubMed] [Google Scholar]
- 16.De Nunzio C, Tubaro A. Prostate cancer: diabetes and prostate cancer—an open debate. Nat Rev Urol. 2012;10:12–14. doi: 10.1038/nrurol.2012.239. [DOI] [PubMed] [Google Scholar]
- 17.Djiogue S, et al. Insulin resistance and cancer: the role of insulin and insulin-like growth factors. Endocr Relat Cancer. 2013;20:R1–R17. doi: 10.1530/ERC-12-0324. [DOI] [PubMed] [Google Scholar]
- 18.Yuhara H, et al. Is diabetes mellitus an independent risk factor for colon cancer and rectal cancer? Am J Gastroenterol. 2011;106:1911–1921. doi: 10.1038/ajg.2011.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang C, et al. Increased risk of hepatocellular carcinoma in patients with diabetes mellitus: a systematic review and meta-analysis of cohort studies. Int J Cancer. 2012;130:1639–1648. doi: 10.1002/ijc.26165. [DOI] [PubMed] [Google Scholar]
- 20.Vongsuvanh R, George J, Qiao L, Poorten DV. Visceral adiposity in gastrointestinal and hepatic carcinogenesis. Cancer Lett. 2013;330:1–10. doi: 10.1016/j.canlet.2012.11.038. [DOI] [PubMed] [Google Scholar]
- 21.McTiernan A. Obesity and cancer: the risks, science, and potential management strategies. Oncology. 2005;19:871–881. [PubMed] [Google Scholar]
- 22.Mannucci E. Insulin therapy and cancer in type 2 diabetes. ISRN Endocrinol. 2012;2012:240634. doi: 10.5402/2012/240634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bonelli L, et al. Exocrine pancreatic cancer, cigarette smoking, and diabetes mellitus: a case-control study in northern Italy. Pancreas. 2003;27:143–149. doi: 10.1097/00006676-200308000-00007. [DOI] [PubMed] [Google Scholar]
- 24.Ding XZ, Fehsenfeld DM, Murphy LO, Permert J, Adrian TE. Physiological concentrations of insulin augment pancreatic cancer cell proliferation and glucose utilization by activating MAP kinase, PI3 kinase and enhancing GLUT-1 expression. Pancreas. 2000;21:310–320. doi: 10.1097/00006676-200010000-00014. [DOI] [PubMed] [Google Scholar]
- 25.Maisonneuve P, et al. Past medical history and pancreatic cancer risk: results from a multicenter case-control study. Ann Epidemiol. 2010;20:92–98. doi: 10.1016/j.annepidem.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 26.Li D, Yeung SC, Hassan MM, Konopleva M, Abbruzzese JL. Antidiabetic therapies affect risk of pancreatic cancer. Gastroenterology. 2009;137:482–428. doi: 10.1053/j.gastro.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330:1304–1305. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chari ST, et al. Probability of pancreatic cancer following diabetes: a population-based study. Gastroenterology. 2005;129:504–511. doi: 10.1053/j.gastro.2005.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gupta S, et al. New-onset diabetes and pancreatic cancer. Clin Gastroenterol Hepatol. 2006;4:1366–1372. doi: 10.1016/j.cgh.2006.06.024. [DOI] [PubMed] [Google Scholar]
- 30.Wang F, Gupta S, Holly EA. Diabetes mellitus and pancreatic cancer in a population-based case-control study in the San Francisco Bay Area, California. Cancer Epidemiol Biomarkers Prev. 2006;15:1458–1463. doi: 10.1158/1055-9965.EPI-06-0188. [DOI] [PubMed] [Google Scholar]
- 31.Pannala R, et al. Prevalence and clinical profile of pancreatic cancer-associated diabetes mellitus. Gastroenterology. 2008;134:981–987. doi: 10.1053/j.gastro.2008.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krechler T, et al. Leptin and adiponectin in pancreatic cancer: connection with diabetes mellitus. Neoplasma. 2011;58:58–64. doi: 10.4149/neo_2011_01_58. [DOI] [PubMed] [Google Scholar]
- 33.Cersosimo E, et al. Insulin secretion and action in patients with pancreatic cancer. Cancer. 1991;67:486–493. doi: 10.1002/1097-0142(19910115)67:2<486::aid-cncr2820670228>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 34.Permert J, et al. Pancreatic cancer is associated with impaired glucose metabolism. Eur J Surg. 1993;159:101–107. [PubMed] [Google Scholar]
- 35.Saruc M, Pour PM. Diabetes and its relationship to pancreatic carcinoma. Pancreas. 2003;26:381–387. doi: 10.1097/00006676-200305000-00012. [DOI] [PubMed] [Google Scholar]
- 36.Kim TD, et al. Clinical characteristics of pancreatic cancer according to the presence of diabetes mellitus [Korean] Korean J Gastroenterol. 2004;43:35–40. [PubMed] [Google Scholar]
- 37.Wu JM, et al. Resolution of diabetes after pancreaticoduodenectomy in patients with and without pancreatic ductal cell adenocarcinoma. Ann Surg Oncol. 2013;20:242–249. doi: 10.1245/s10434-012-2577-y. [DOI] [PubMed] [Google Scholar]
- 38.Trna J, Dite P, Adamcova A, Crawford BJ, Hermanova M. Diabetes mellitus in pancreatic cancer patients in the Czech Republic: sex differences. Exp Diabetes Res. 2012;2012:414893. doi: 10.1155/2012/414893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cuzick J, Babiker AG. Pancreatic cancer, alcohol, diabetes mellitus and gall-bladder disease. Int J Cancer. 1989;43:415–421. doi: 10.1002/ijc.2910430312. [DOI] [PubMed] [Google Scholar]
- 40.Hiatt RA, Klatsky AL, Armstrong MA. Pancreatic cancer, blood glucose and beverage consumption. Int J Cancer. 1988;41:794–797. doi: 10.1002/ijc.2910410603. [DOI] [PubMed] [Google Scholar]
- 41.Bell ET. Carcinoma of the pancreas. I. A clinical and pathologic study of 609 necropsied cases. II. The relation of carcinoma of the pancreas to diabetes mellitus. Am J Pathol. 1957;33:499–523. [PMC free article] [PubMed] [Google Scholar]
- 42.Aggarwal G, Rabe KG, Petersen GM, Chari ST. New-onset diabetes in pancreatic cancer: a study in the primary care setting. Pancreatology. 2012;12:156–161. doi: 10.1016/j.pan.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Permert J, et al. Pancreatic cancer is associated with impaired glucose metabolism. Eur J Surg. 1993;159:101–107. [PubMed] [Google Scholar]
- 44.Chari ST, Klee GG, Miller LJ, Raimondo M, DiMagno EP. Islet amyloid polypeptide is not a satisfactory marker for detecting pancreatic cancer. Gastroenterology. 2001;121:640–645. doi: 10.1053/gast.2001.27210. [DOI] [PubMed] [Google Scholar]
- 45.Chari ST, et al. Pancreatic cancer-associated diabetes mellitus: prevalence and temporal association with diagnosis of cancer. Gastroenterology. 2008;134:95–101. doi: 10.1053/j.gastro.2007.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Permert J, et al. Improved glucose metabolism after subtotal pancreatectomy for pancreatic cancer. Br J Surg. 1993;80:1047–1050. doi: 10.1002/bjs.1800800841. [DOI] [PubMed] [Google Scholar]
- 47.Aggarwal G, Kamada P, Chari ST. Prevalence of diabetes mellitus in pancreatic cancer compared to common cancers. Pancreas. 2013;42:198–201. doi: 10.1097/MPA.0b013e3182592c96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tsoli M, Robertson G. Cancer cachexia: malignant inflammation, tumorkines, and metabolic mayhem. Trends Endocrinol Metab. doi: 10.1016/j.tem.2012.10.006. http://dx.doi.org/10.1016/j.tem.2012.10.006. [DOI] [PubMed]
- 49.Fearon K, Arends J, Baracos V. Understanding the mechanisms and treatment options in cancer cachexia. Nat Rev Clin Oncol. 2013;10:90–99. doi: 10.1038/nrclinonc.2012.209. [DOI] [PubMed] [Google Scholar]
- 50.Fearon KC, et al. Pancreatic cancer as a model: inflammatory mediators, acute-phase response, and cancer cachexia. World J Surg. 1999;23:584–588. doi: 10.1007/pl00012351. [DOI] [PubMed] [Google Scholar]
- 51.Gangi S, et al. Time interval between abnormalities seen on CT and the clinical diagnosis of pancreatic cancer: retrospective review of CT scans obtained before diagnosis. AJR Am J Roentgenol. 2004;182:897–903. doi: 10.2214/ajr.182.4.1820897. [DOI] [PubMed] [Google Scholar]
- 52.Pelaez-Luna M, Takahashi N, Fletcher JG, Chari ST. Resectability of presymptomatic pancreatic cancer and its relationship to onset of diabetes: a retrospective review of CT scans and fasting glucose values prior to diagnosis. Am J Gastroenterol. 2007;102:2157–2163. doi: 10.1111/j.1572-0241.2007.01480.x. [DOI] [PubMed] [Google Scholar]
- 53.Permert J, et al. Islet hormone secretion in pancreatic cancer patients with diabetes. Pancreas. 1997;15:60–68. doi: 10.1097/00006676-199707000-00009. [DOI] [PubMed] [Google Scholar]
- 54.Basso D, et al. β-cell function in pancreatic adenocarcinoma. Pancreas. 1994;9:332–335. doi: 10.1097/00006676-199405000-00008. [DOI] [PubMed] [Google Scholar]
- 55.Schwarts SS, Zeidler A, Moossa AR, Kuku SF, Rubenstein AH. A prospective study of glucose tolerance, insulin, C-peptide, and glucagon responses in patients with pancreatic carcinoma. Am J Dig Dis. 1978;23:1107–1114. doi: 10.1007/BF01072886. [DOI] [PubMed] [Google Scholar]
- 56.Permert J, et al. Islet amyloid polypeptide in patients with pancreatic cancer and diabetes. N Engl J Med. 1994;330:313–318. doi: 10.1056/NEJM199402033300503. [DOI] [PubMed] [Google Scholar]
- 57.Rosa JA, Van Linda BM, Abourizk NN. New-onset diabetes mellitus as a harbinger of pancreatic carcinoma. A case report and literature review. J Clin Gastroenterol. 1989;11:211–215. doi: 10.1097/00004836-198904000-00020. [DOI] [PubMed] [Google Scholar]
- 58.Silverstein MD, Richter JM, Podolsky DK, Warshaw AL. Suspected pancreatic cancer presenting as pain or weight loss: analysis of diagnostic strategies. World J Surg. 1984;8:839–845. doi: 10.1007/BF01656023. [DOI] [PubMed] [Google Scholar]
- 59.Girelli CM, Reguzzoni G, Limido E, Savastano A, Rocca F. Pancreatic carcinoma: differences between patients with or without diabetes mellitus. Recenti Prog Med. 1995;86:143–146. [PubMed] [Google Scholar]
- 60.Pannala R, et al. Temporal association of changes in fasting blood glucose and body mass index with diagnosis of pancreatic cancer. Am J Gastroenterol. 2009;104:2318–2325. doi: 10.1038/ajg.2009.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hart PA, et al. Weight loss precedes cancer-specific symptoms in pancreatic cancer-associated diabetes mellitus. Pancreas. 2011;40:768–772. doi: 10.1097/MPA.0b013e318220816a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Murphy RA, et al. Loss of adipose tissue and plasma phospholipids: relationship to survival in advanced cancer patients. Clin Nutr. 2010;29:482–487. doi: 10.1016/j.clnu.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 63.Hermans MP, Levy JC, Morris RJ, Turner RC. Comparison of insulin sensitivity tests across a range of glucose tolerance from normal to diabetes. Diabetologia. 1999;42:678–687. doi: 10.1007/s001250051215. [DOI] [PubMed] [Google Scholar]
- 64.Hermans MP, Levy JC, Morris RJ, Turner RC. Comparison of tests of β-cell function across a range of glucose tolerance from normal to diabetes. Diabetes. 1999;48:1779–1786. doi: 10.2337/diabetes.48.9.1779. [DOI] [PubMed] [Google Scholar]
- 65.Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840–846. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- 66.Chari ST, Zapiach M, Yadav D, Rizza RA. β-cell function and insulin resistance evaluated by HOMA in pancreatic cancer subjects with varying degrees of glucose intolerance. Pancreatology. 2005;5:229–233. doi: 10.1159/000085276. [DOI] [PubMed] [Google Scholar]
- 67.Permert J, et al. Islet amyloid polypeptide in patients with pancreatic cancer and diabetes. N Engl J Med. 1994;330:313–318. doi: 10.1056/NEJM199402033300503. [DOI] [PubMed] [Google Scholar]
- 68.Basso D, et al. Pancreatic cancer-derived S-100A8 N-terminal peptide: a diabetes cause? Clin Chim Acta. 2006;372:120–128. doi: 10.1016/j.cca.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 69.Basso D, et al. Pancreatic cancer-associated diabetes mellitus: an open field for proteomic applications. Clin Chim Acta. 2005;357:184–189. doi: 10.1016/j.cccn.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 70.Aggarwal G, et al. Adrenomedullin is upregulated in patients with pancreatic cancer and causes insulin resistance in β cells and mice. Gastroenterology. 2012;143:1510–1517. doi: 10.1053/j.gastro.2012.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Martinez A, et al. Regulation of insulin secretion and blood glucose metabolism by adrenomedullin. Endocrinology. 1996;137:2626–2632. doi: 10.1210/endo.137.6.8641217. [DOI] [PubMed] [Google Scholar]
- 72.Sekine N, Takano K, Kimata-Hayashi N, Kadowaki T, Fujita T. Adrenomedullin inhibits insulin exocytosis via pertussis toxin-sensitive G protein-coupled mechanism. Am J Physiol Endocrinol Metab. 2006;291:E9–E14. doi: 10.1152/ajpendo.00213.2005. [DOI] [PubMed] [Google Scholar]
- 73.Zudaire E, Cuttitta F, Martinez A. Regulation of pancreatic physiology by adrenomedullin and its binding protein. Regu Pept. 2003;112:121–130. doi: 10.1016/s0167-0115(03)00030-2. [DOI] [PubMed] [Google Scholar]
- 74.Hong Y, Hay DL, Quirion R, Poyner DR. The pharmacology of adrenomedullin 2/intermedin. Br J Pharmacol. 2012;166:110–120. doi: 10.1111/j.1476-5381.2011.01530.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Keleg S, et al. Adrenomedullin is induced by hypoxia and enhances pancreatic cancer cell invasion. Int J Cancer. 2007;121:21–32. doi: 10.1002/ijc.22596. [DOI] [PubMed] [Google Scholar]
- 76.Natsuizaka M, et al. Synergistic up-regulation of Hexokinase-2, glucose transporters and angiogenic factors in pancreatic cancer cells by glucose deprivation and hypoxia. Exp Cell Res. 2007;313:3337–3348. doi: 10.1016/j.yexcr.2007.06.013. [DOI] [PubMed] [Google Scholar]
- 77.Konner AC, Bruning JC. Selective insulin and leptin resistance in metabolic disorders. Cell Metab. 2012;16:144–152. doi: 10.1016/j.cmet.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 78.Liu J, et al. The intracellular mechanism of insulin resistance in pancreatic cancer patients. J Clin Endocrinol Metab. 2000;85:1232–1238. doi: 10.1210/jcem.85.3.6400. [DOI] [PubMed] [Google Scholar]
- 79.Isaksson B, et al. Impaired insulin action on phosphatidylinositol 3-kinase activity and glucose transport in skeletal muscle of pancreatic cancer patients. Pancreas. 2003;26:173–177. doi: 10.1097/00006676-200303000-00014. [DOI] [PubMed] [Google Scholar]
- 80.Basso D, et al. Putative pancreatic cancer-associated diabetogenic factor: 2030 MW peptide. Pancreas. 2002;24:8–14. doi: 10.1097/00006676-200201000-00002. [DOI] [PubMed] [Google Scholar]
- 81.Tabata H, et al. Islet amyloid polypeptide (IAPP/amylin) causes insulin resistance in perfused rat hindlimb muscle. Diabetes Res Clin Pract. 1992;15:57–61. doi: 10.1016/0168-8227(92)90068-3. [DOI] [PubMed] [Google Scholar]
- 82.Ding X, Flatt PR, Permert J, Adrian TE. Pancreatic cancer cells selectively stimulate islet β cells to secrete amylin. Gastroenterology. 1998;114:130–138. doi: 10.1016/s0016-5085(98)70641-9. [DOI] [PubMed] [Google Scholar]
- 83.Wang F, Adrian TE, Westermark G, Gasslander T, Permert J. Dissociated insulin and islet amyloid polypeptide secretion from isolated rat pancreatic islets cocultured with human pancreatic adenocarcinoma cells. Pancreas. 1999;18:403–409. doi: 10.1097/00006676-199905000-00012. [DOI] [PubMed] [Google Scholar]
- 84.Wang F, et al. Dissociated secretion of islet amyloid polypeptide and insulin in serum-free culture media conditioned by human pancreatic adenocarcinoma cell lines. Int J Pancreatol. 1997;21:157–164. doi: 10.1007/BF02822387. [DOI] [PubMed] [Google Scholar]
- 85.Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148:852–871. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hardy OT, Czech MP, Corvera S. What causes the insulin resistance underlying obesity? Curr Opin Endocrinol Diabetes Obes. 2012;19:81–87. doi: 10.1097/MED.0b013e3283514e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Ann Rev Physiol. 2010;72:219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- 88.De Boer MP, et al. Microvascular dysfunction: a potential mechanism in the pathogenesis of obesity-associated insulin resistance and hypertension. Microcirculation. 2012;19:5–18. doi: 10.1111/j.1549-8719.2011.00130.x. [DOI] [PubMed] [Google Scholar]
- 89.Koerner A, Kratzsch J, Kiess W. Adipocytokines: leptin--the classical, resistin--the controversical, adiponectin--the promising, and more to come. Best Pract Res Clin Endocrinol Metab. 2005;19:525–546. doi: 10.1016/j.beem.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 90.Matsuzawa Y. Adiponectin: a key player in obesity related disorders. Curr Pharm Des. 2010;16:1896–1901. doi: 10.2174/138161210791208893. [DOI] [PubMed] [Google Scholar]
- 91.Oda N, et al. The ratio of leptin to adiponectin can be used as an index of insulin resistance. Metabolism. 2008;57:268–273. doi: 10.1016/j.metabol.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 92.Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Ann Rev Immunol. 2011;29:415–445. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- 93.Vandanmagsar B, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17:179–188. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Stienstra R, Tack CJ, Kanneganti TD, Joosten LA, Netea MG. The inflammasome puts obesity in the danger zone. Cell Metab. 2012;15:10–18. doi: 10.1016/j.cmet.2011.10.011. [DOI] [PubMed] [Google Scholar]
- 95.Matsuzawa Y. The metabolic syndrome and adipocytokines. FEBS Lett. 2006;580:2917–2921. doi: 10.1016/j.febslet.2006.04.028. [DOI] [PubMed] [Google Scholar]
- 96.Grote VA, et al. Inflammation marker and risk of pancreatic cancer: a nested case-control study within the EPIC cohort. Br J Cancer. 2012;106:1866–1874. doi: 10.1038/bjc.2012.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Smits MM, van Geenen EJ. The clinical significance of pancreatic steatosis. Nat Rev Gastroenterol Hepatol. 2011;8:169–177. doi: 10.1038/nrgastro.2011.4. [DOI] [PubMed] [Google Scholar]
- 98.Gupta D, Krueger CB, Lastra G. Over-nutrition, obesity and insulin resistance in the development of β-cell dysfunction. Curr Diabetes Rev. 2012;8:76–83. doi: 10.2174/157339912799424564. [DOI] [PubMed] [Google Scholar]
- 99.Tisdale MJ. Zinc- α-2-glycoprotein in cachexia and obesity. Curr Opin Support Palliat Care. 2009;3:288–293. doi: 10.1097/SPC.0b013e328331c897. [DOI] [PubMed] [Google Scholar]
- 100.Bing C, Mracek T, Gao D, Trayhurn P. Zinc- α-2-glycoprotein: an adipokine modulator of body fat mass? Int J Obes (Lond) 2010;34:1559–1565. doi: 10.1038/ijo.2010.105. [DOI] [PubMed] [Google Scholar]
- 101.Matsuzawa Y. The role of fat topology in the risk of disease. Int J Obes (Lond) 2008;32 (Suppl 7):S83–S92. doi: 10.1038/ijo.2008.243. [DOI] [PubMed] [Google Scholar]
- 102.Matsuzawa Y, Funahashi T, Nakamura T. The concept of metabolic syndrome: contribution of visceral fat accumulation and its molecular mechanism. J Atheroscler Thromb. 2011;18:629–639. doi: 10.5551/jat.7922. [DOI] [PubMed] [Google Scholar]
- 103.Matsuzawa Y. Establishment of a concept of visceral fat syndrome and discovery of adiponectin. Proc Jpn Acad Ser B Phys Biol Sci. 2010;86:131–141. doi: 10.2183/pjab.86.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.McLaughlin T, Lamendola C, Liu A, Abbasi F. Preferential fat deposition in subcutaneous versus visceral depots is associated with insulin sensitivity. J Clin Endocrinol Metab. 2011;96:E1756–E1760. doi: 10.1210/jc.2011-0615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Klein S, et al. Absence of an effect of liposuction on insulin action and risk factors for coronary heart disease. N Engl J Med. 2004;350:2549–2557. doi: 10.1056/NEJMoa033179. [DOI] [PubMed] [Google Scholar]
- 106.Gabriely I, et al. Removal of visceral fat prevents insulin resistance and glucose intolerance of aging: an adipokine-mediated process? Diabetes. 2002;51:2951–8. doi: 10.2337/diabetes.51.10.2951. [DOI] [PubMed] [Google Scholar]
- 107.Thorne A, Lonnqvist F, Apelman J, Hellers G, Arner P. A pilot study of long-term effects of a novel obesity treatment: omentectomy in connection with adjustable gastric banding. Int J Obes (Lond) 2002;26:193–199. doi: 10.1038/sj.ijo.0801871. [DOI] [PubMed] [Google Scholar]
- 108.Ibrahim MM. Subcutaneous and visceral adipose tissue: structural and functional differences. Int J Obes (Lond) 2010;11:11–18. doi: 10.1111/j.1467-789X.2009.00623.x. [DOI] [PubMed] [Google Scholar]
- 109.Samaras K, Botelho NK, Chisholm DJ, Lord RV. Subcutaneous and visceral adipose tissue gene expression of serum adipokines that predict type 2 diabetes. Obesity. 2010;18:884–889. doi: 10.1038/oby.2009.443. [DOI] [PubMed] [Google Scholar]
- 110.Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–270. [PubMed] [Google Scholar]
- 111.Regel I, et al. Energy metabolism and proliferation in pancreatic carcinogenesis. Langenbecks Arch Surg. 2012;397:507–512. doi: 10.1007/s00423-012-0933-9. [DOI] [PubMed] [Google Scholar]
- 112.Chaika NV, et al. Differential expression of metabolic genes in tumor and stromal components of primary and metastatic loci in pancreatic adenocarcinoma. PLoS ONE. 2012;7:e32996. doi: 10.1371/journal.pone.0032996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dong X, et al. Glucose metabolism gene variants modulate the risk of pancreatic cancer. Cancer Prev Res (Phila) 2011;4:758–766. doi: 10.1158/1940-6207.CAPR-10-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ying H, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–670. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Singh PK, Brand RE, Mehla K. MicroRNAs in pancreatic cancer metabolism. Nat Rev Gastroenterol Hepatol. 2012;9:334–344. doi: 10.1038/nrgastro.2012.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ferrer J. Glucose as a mitogenic hormone. Cell Metab. 2011;13:357–358. doi: 10.1016/j.cmet.2011.03.014. [DOI] [PubMed] [Google Scholar]
- 117.Jiao L, et al. Advanced glycation end products, soluble receptor for advanced glycation end products, and risk of colorectal cancer. Cancer Epidemiol Biomarkers Prev. 2011;20:1430–1438. doi: 10.1158/1055-9965.EPI-11-0066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jiao L, et al. Evidence that serum levels of the soluble receptor for advanced glycation end products are inversely associated with pancreatic cancer risk: a prospective study. Cancer Res. 2011;71:3582–3589. doi: 10.1158/0008-5472.CAN-10-2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Han L, et al. High glucose promotes pancreatic cancer cell proliferation via the induction of EGF expression and transactivation of EGFR. PLoS ONE. 2011;6:e27074. doi: 10.1371/journal.pone.0027074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Butler AE, et al. Pancreatic duct replication is increased with obesity and type 2 diabetes in humans. Diabetologia. 2010;53:21–26. doi: 10.1007/s00125-009-1556-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Li J, et al. Relationship between neural alteration and perineural invasion in pancreatic cancer patients with hyperglycemia. PLoS ONE. 2011;6:e17385. doi: 10.1371/journal.pone.0017385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Stolzenberg-Solomon RZ, et al. Insulin, glucose, insulin resistance, and pancreatic cancer in male smokers. JAMA. 2005;294:2872–2878. doi: 10.1001/jama.294.22.2872. [DOI] [PubMed] [Google Scholar]
- 123.Kang R, et al. The expression of the receptor for advanced glycation endproducts (RAGE) is permissive for early pancreatic neoplasia. Proc Natl Acad Sci USA. 2012;109:7031–7036. doi: 10.1073/pnas.1113865109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Arumugam T, Ramachandran V, Gomez SB, Schmidt AM, Logsdon CD. S100P-derived RAGE antagonistic peptide reduces tumor growth and metastasis. Clin Cancer Res. 2012;18:4356–4364. doi: 10.1158/1078-0432.CCR-12-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Grote VA, et al. The associations of advanced glycation end products and its soluble receptor with pancreatic cancer risk: a case-control study within the prospective EPIC Cohort. Cancer Epidemiol Biomarkers Prev. 2012;21:619–628. doi: 10.1158/1055-9965.EPI-11-1139. [DOI] [PubMed] [Google Scholar]
- 126.Jiao L, et al. Evidence that serum levels of the soluble receptor for advanced glycation end products are inversely associated with pancreatic cancer risk: a prospective study. Cancer Res. 2011;71:3582–3589. doi: 10.1158/0008-5472.CAN-10-2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Muders MH, et al. Expression and regulatory role of GAIP-interacting protein GIPC in pancreatic adenocarcinoma. Cancer Res. 2006;66:10264–10268. doi: 10.1158/0008-5472.CAN-06-2321. [DOI] [PubMed] [Google Scholar]
- 128.Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. 2008;8:915–928. doi: 10.1038/nrc2536. [DOI] [PubMed] [Google Scholar]
- 129.Rozengurt E, Sinnett-Smith J, Kisfalvi K. Crosstalk between insulin/insulin-like growth factor-1 receptors and G protein-coupled receptor signaling systems: a novel target for the antidiabetic drug metformin in pancreatic cancer. Clin Cancer Res. 2010;16:2505–2511. doi: 10.1158/1078-0432.CCR-09-2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Mehenni H, et al. Cancer risks in LKB1 germline mutation carriers. Gut. 2006;55:984–990. doi: 10.1136/gut.2005.082990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Birnbaum DJ, et al. Genome profiling of pancreatic adenocarcinoma. Genes Chromosomes Cancer. 2011;50:456–465. doi: 10.1002/gcc.20870. [DOI] [PubMed] [Google Scholar]
- 132.Li D, et al. Pathway analysis of genome-wide association study data highlights pancreatic development genes as susceptibility factors for pancreatic cancer. Carcinogenesis. 2012;33:1384–1390. doi: 10.1093/carcin/bgs151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chaika NV, et al. MUC1 mucin stabilizes and activates hypoxia-inducible factor 1 α to regulate metabolism in pancreatic cancer. Proc Natl Acad Sci USA. 2012;109:13787–13792. doi: 10.1073/pnas.1203339109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Akakura N, et al. Constitutive expression of hypoxia-inducible factor-1 α renders pancreatic cancer cells resistant to apoptosis induced by hypoxia and nutrient deprivation. Cancer Res. 2001;61:6548–6554. [PubMed] [Google Scholar]
- 135.Chen J, et al. Dominant-negative hypoxia-inducible factor-1 α reduces tumorigenicity of pancreatic cancer cells through the suppression of glucose metabolism. Am J Pathol. 2003;162:1283–12891. doi: 10.1016/s0002-9440(10)63924-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Pannala R, Basu A, Petersen GM, Chari ST. New-onset diabetes: a potential clue to the early diagnosis of pancreatic cancer. Lancet Oncol. 2009;10:88–95. doi: 10.1016/S1470-2045(08)70337-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hardt PD, Brendel MD, Kloer HU, Bretzel RG. Is pancreatic diabetes (type 3c diabetes) underdiagnosed and misdiagnosed? Diabetes Care. 2008;31 (Suppl 2):S165–S169. doi: 10.2337/dc08-s244. [DOI] [PubMed] [Google Scholar]
- 138.Chen N, Unnikrishnan IR, Anjana RM, Mohan V, Pitchumoni CS. The complex exocrine-endocrine relationship and secondary diabetes in exocrine pancreatic disorders. J Clin Gastroenterol. 2011;45:850–861. doi: 10.1097/MCG.0b013e31822a2ae5. [DOI] [PubMed] [Google Scholar]
- 139.Diagnosis and classification of diabetes mellitus. Diabetes Care. 2010;33 (Suppl 1):S62–S69. doi: 10.2337/dc10-S062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cui Y, Andersen DK. Pancreatogenic diabetes: special considerations for management. Pancreatology. 2011;11:279–294. doi: 10.1159/000329188. [DOI] [PubMed] [Google Scholar]
- 141.Raue G, Keim V. Secondary diabetes in chronic pancreatitis [German] Z Gastroenterol. 1999;(Suppl 1):4–9. [PubMed] [Google Scholar]
- 142.Tsuchiya R, et al. Collective review of small carcinomas of the pancreas. Ann Surg. 1986;203:77–81. doi: 10.1097/00000658-198601000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Permert J, Herrington M, Kazakoff K, Pour PM, Adrian TE. Early changes in islet hormone secretion in the hamster pancreatic cancer model. Teratog Carcinog Mutagen. 2001;21:59–67. [PubMed] [Google Scholar]
- 144.Ahren B, Andren-Sandberg A. Glucose tolerance and insulin secretion in experimental pancreatic cancer in the Syrian hamster. Res Exp Med (Berl) 1993;193:21–26. doi: 10.1007/BF02576207. [DOI] [PubMed] [Google Scholar]
- 145.Pour PM, Bell RH. Alteration of pancreatic endocrine cell patterns and their secretion during pancreatic carcinogenesis in the hamster model. Cancer Res. 1989;49:6396–6400. [PubMed] [Google Scholar]
- 146.Basso D, et al. An unidentified pancreatic cancer cell product alters some intracellular pathways of glucose metabolism in isolated rat hepatocytes. Pancreas. 1997;15:132–138. doi: 10.1097/00006676-199708000-00004. [DOI] [PubMed] [Google Scholar]
- 147.Valerio A, et al. Glucose metabolic alterations in isolated and perfused rat hepatocytes induced by pancreatic cancer conditioned medium: a low molecular weight factor possibly involved. Biochem Biophys Res Commun. 1999;257:622–628. doi: 10.1006/bbrc.1999.0521. [DOI] [PubMed] [Google Scholar]
- 148.Basso D, et al. Altered glucose metabolism and proteolysis in pancreatic cancer cell conditioned myoblasts: searching for a gene expression pattern with a microarray analysis of 5000 skeletal muscle genes. Gut. 2004;53:1159–1166. doi: 10.1136/gut.2003.024471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Basso D, et al. The pancreatic cancer cell line MIA PaCa2 produces one or more factors able to induce hyperglycemia in SCID mice. Anticancer Res. 1995;15:2585–2588. [PubMed] [Google Scholar]
- 150.Wang F, Larsson J, Adrian TE, Gasslander T, Permert J. In vitro influences between pancreatic adenocarcinoma cells and pancreatic islets. J Surg Res. 1998;79:13–19. doi: 10.1006/jsre.1998.5393. [DOI] [PubMed] [Google Scholar]