

Summary
Intracellular pathogens directly alter host cells in order to replicate and survive. While infection-induced changes in host transcription can be readily assessed, post-transcriptional alterations are more difficult to catalog. We applied the global protein stability (GPS) platform, which assesses protein stability based on relative changes in an adjoining fluorescent tag, to identify changes in the host proteome following infection with the obligate intracellular bacteria Chlamydia trachomatis. Our results indicate that C. trachomatis profoundly remodels the host proteome independently of changes in transcription. Additionally, C. trachomatis replication depends on a subset of altered proteins, such as Pin1 and Men1, which regulate the host transcription factor AP-1 controlling host inflammation, stress, and cell survival. Furthermore, AP-1-dependent transcription is activated during infection, and required for efficient Chlamydia growth. In summary, this experimental approach revealed that C. trachomatis broadly alters host proteins and can be applied to examine host-pathogen interactions and develop host-based therapeutics.
Introduction
Pathogens manipulate host cells for survival, intracellular replication, and transmission (Ham et al., 2011; Rohmer et al., 2011). Many intracellular pathogens translocate virulence factors into the host cytosol (Bingle et al., 2008; Cornelis, 2010), which is important for survival and replication (Barry et al., 2011; Cossart and Roy, 2010). A complication to understanding how bacterial effectors function is redundancy in the effector repertoire and in host pathways targeted (Galan, 2009; O'Connor et al., 2011). Although transcriptional profiling has been an important tool for studying host pathogen interactions (Burrack and Higgins, 2007), the host response to infection and the function of effectors cannot be determined by microarray analysis alone because many bacterial effectors act post-transcriptionally to modify or alter the stability of host proteins directly (Ensminger and Isberg, 2010; Misaghi et al., 2006; Mukherjee et al., 2006).
The obligate intracellular bacterium Chlamydia trachomatis is a leading cause of bacterial sexually transmitted disease and preventable blindness worldwide (Holmes, 2008). The elementary body (EB) is the infectious form of C. trachomatis that develops into the replicative form called the reticulate body (RB) upon uptake into host cells (Moulder, 1991). Replication occurs within a specialized vacuole known as the “inclusion”. A type III secretion system (TTSS) is used by C. trachomatis to deliver bacterial effectors into the host cytosol during intracellular infection (Peters et al., 2007). Proteins delivered into the cytosol by C. trachomatis include two deubiquitinating enzymes and several proteases. This suggests that C. trachomatis directly manipulates host protein turnover during infection however, the targets and activities of most secreted effectors remain largely unknown (Chellas-Gery et al., 2007; Misaghi et al., 2006; Zhong, 2011).
Understanding manipulation of host proteins by C. trachomatis would benefit from an analysis of host cell protein turnover, however, the methods available to study global changes to host proteins following infection are limited. Thus, we adapted the global protein stability (GPS) platform to screen the human ORFeome following infection with C. trachomatis (Emanuele et al., 2011; Yen et al., 2008). Hundreds of host proteins that are altered in stability during infection were identified, which led to the elucidation of host pathways that are manipulated during infection. Thus, GPS screening is a powerful approach that can be applied to the study of bacterial effectors and for the identification of host pathways manipulated post-transcriptionally during pathogen infection.
Results
The global protein stability platform identifies hundreds of host proteins that are altered following infection with C. trachomatis
To search for host proteins whose stability is altered following infection with C. trachomatis, we adapted the Global Protein Stability (GPS) screening platform. GPS is a system originally designed as a fluorescent protein-based method to identify the substrates of mammalian ubiquitin ligases (Emanuele et al., 2011; Yen et al., 2008). The GPS library was assembled from an arrayed set (ORFeome v3.1) of 12,000 human ORFs, each engineered into a retroviral reporter construct whose general structure is shown in figure 1A (Emanuele et al., 2011). The reporter expression cassette contains a single pCMV promoter and an internal ribosome entry site (IRES), permitting the translation of two fluorescent proteins from one mRNA transcript. The first fluorescent protein is the internal control DsRed and the second is an enhanced GFP (EGFP) expressed as a fusion to one of 12,000 host cell proteins. When integrated into the genome of cells, DsRed and EGFP-X are produced at a constant ratio (independent of transcriptional regulation) because they are translated from the same mRNA and lack regulatory 5′ and 3′ UTRs. However, once translated, the amount of DsRed remains constant, but the level of EGFP changes based on the stability of the protein fused to its C-terminus. When measured by flow cytometery, this baseline EGFP:DsRed ratio reflects relative protein stability: the lower the ratio, the less-stable the protein fused to EGFP.
Figure 1. GPS profiling identifies host proteins altered during C. trachomatis infection.
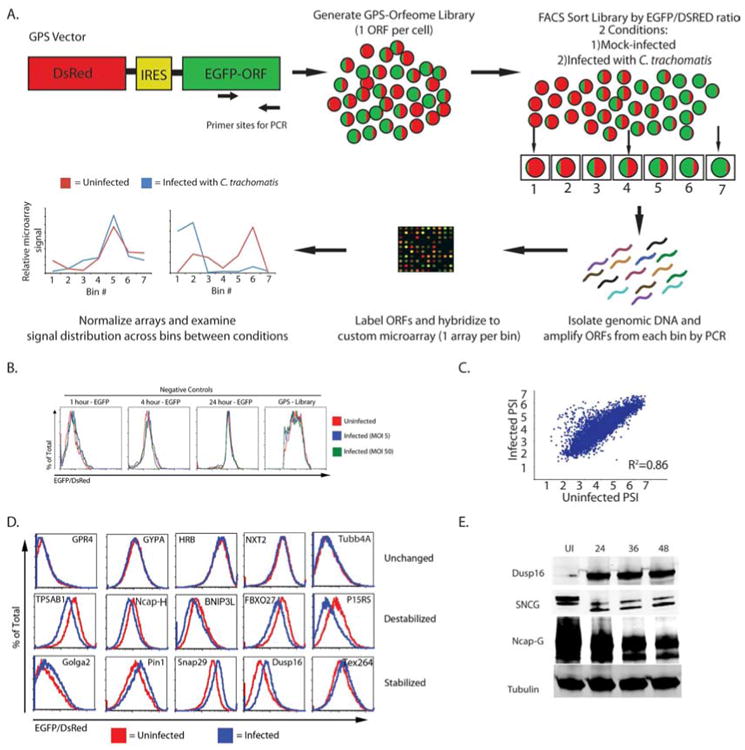
A) Schematic representation of the GPS screen.
B) Histograms show the EGFP/DsRed ratio for cells expressing GFP for varying half-life and the entire GPS library. The cell lines were mock-infected (red line) or infected with C. trachomatis at an MOI of 5 (blue line) or 50 (green line) for 24 hours and analyzed by flow cytometry.
C) Scatter plot for the PSI for each probe in the screen under the two conditions (mock-infected vs. infected).
D) A subset of candidates that were tested by expressing the host gene in HEK293T cells. Cells were mock-infected (red line) or infected with C. trachomatis (blue line) for 24 hours and evaluated using flow cytometry. Shown is a representative plot from three independent experiments.
E) HEK293T cells were mock-infected or infected with C. trachomatis for the indicated amount of time. Cells were analyzed by immunoblot for the expression of the experimental proteins (Dusp16, SNCG, and NCAP-G) as well as beta-tubulin (loading control). Blots are representative of at least 3 independent experiments. See also Figure S1, Table S1, S2, and S4.
Prior to running the GPS screen, we confirmed that infection with C. trachomatis does not alter the inherent stability of EGFP or DsRed. First we infected cells containing the GPS cassette with EGFP of varying half-lives at a multiplicity of infection (MOI) of 5 and 50 for 24 hours and then analyzed the samples by flow cytometry (Figure 1B). Overlays of the EGFP:DsRed ratio show no noticeable shifts under either infection condition, indicating that infection does not alter the fluorescent reporter proteins alone.
We next executed a full-scale GPS screen to identify host proteins that are altered in stability during infection with C. trachomatis. We first sorted the uninfected HEK293T cells by high-throughput Fluorescence Activated Cell Sorting (FACS) into 7 bins based on their individual EGFP:DsRed ratio, with increasing EGFP:DsRed ratios corresponding to bins 1-7 respectively (low to high). The ORFs in each bin were then amplified from genomic DNA using PCR, fluorescently labeled and hybridized to custom-designed microarrays (one microarray per bin). The intensities for each individual probe from all 7 arrays were measured and graphed across all seven bins. The presence of a cell bearing a particular ORF in higher number bins indicates that the protein is stable in the cells; the presence of a cell bearing a particular ORF in lower number bins indicates that the protein is unstable in the cells. Using these distributions we calculated a protein stability index (PSI) for each ORF. In a parallel experiment, we infected the HEK 293T cells with C. trachomatis EBs at an MOI of 3 for 24 hours. Using the PSI value for each protein in the library under uninfected and infected conditions, we calculated a ΔPSI resulting from infection (Yen et al., 2008). We used a priority rank system to identify high confidence hits using a combination of ΔPSI, probe intensity, agreement among probes for a single ORF, and visual inspection of ΔPSI graphs (described in the supplementary methods). We first compared the agreement of each probe for a given ORF between uninfected and infected conditions which showed a linear relationship and an R2 value > 0.85 (Figure 1C). When comparing probe distributions across the microarrays, the majority of ORFs examined were indistinguishable, increasing our confidence in shifts seen following infection (Supplementary Figure 1A). We next ordered every ORF by the ΔPSI and graphed them from most stabilized (negative ΔPSI) to most destabilized (positive ΔPSI) (Figure 1C) and identified over 600 proteins (out of 8000 that passed initial filtering from the microarrays) that are altered in stability (absolute value of ΔPSI > .25) following infection with C. trachomatis (Table S1 and described in detail in the supplementary methods). This was far more proteins than we anticipated based on previously reported transcription data and suggested that C. trachomatis profoundly remodels the host proteome upon infection (Xia et al., 2003).
In order to both estimate robustness of the GPS screen and validate the proteins identified, we individually cloned a random subset of 175 (out of 600) unique ORFs into the GPS vector and transduced HEK293T cells. Cell lines individually expressing a single EGFP-ORF fusion were then mock-infected or infected with C. trachomatis for 24 hours and analyzed by flow cytometry to examine changes in the EGFP/DsRed ratio. In these experiments, 109 cell lines (62%) had altered ratios following infection with C. trachomatis in at least two out of three replicates (Figure 1D, Supplementary Table 1). To ensure that these changes accurately reflected changes at the protein level, 20 random ORFs were cloned into a retroviral FLAG-tagged vector and used to create stable cells expressing tagged versions of proteins. We chose a short epitope tag instead of EGFP to limit concern about the tag interfering with function of the protein. We then mock-infected or infected these cell lines and probed for tagged proteins in uninfected and infected lysates. Fourteen of 20 (70%) showed quantifiable changes by immunoblot, indicating the reproducibility of our results (Supplementary Figure 1B). Since GPS is a screening platform that expresses each ORF independently of normal transcriptional regulation, we wanted to confirm that a subset of protein changes were occurring at endogenous levels of expression following infection with C. trachomatis. Using immunoblots, we examined the endogenous levels of 9 proteins and found that 7 were altered following infection (Figure 1E and Supplementary Figure 1C). We also confirmed several of these protein changes following infection of HeLa cells (data not shown).
Interestingly, we found that while the GFP fusions accurately informed us when a specific protein was altered, they did not always predict whether a protein was stabilized or destabilized. For example, we identified the host protein SNCG as stabilized following infection with C. trachomatis using the GPS reporter, yet immunoblot analysis showed that this protein is actually cleaved following infection (Figure 1D and 1E). These data suggest that cleavage of host proteins in the GPS platform can remove specific degrons, leading to the stabilization of the EGFP reporter even though cleavage has occurred.
To help integrate our GPS hit data into the overall biology of the host cell, we conducted bioinformatics analysis using DAVID clustering analysis (details in supplemental methods) (Huang da et al., 2009). Many proteins in enriched clusters recapitulated previous studies of Chlamydia infection (Supplementary Table S2). These included chromatin remodeling proteins, the Golgi apparatus, cell cycle control proteins, and host cytoskeleton networks, indicating that our GPS data can identify pathways important for Chlamydia growth (Supplementary Table S2) (Balsara et al., 2006; Chumduri et al., 2013; Heuer et al., 2009; Hybiske and Stephens, 2007; Rejman Lipinski et al., 2009). Top clusters that include protein families not previously associated with Chlamydia infection contained transcription factors and host-signaling cascades such as JNK (Supplementary Table S2). Additionally, a number of altered proteins were enriched in the nuclear membrane or mitochondria, suggesting that Chlamydia infection directly alters the makeup of these intracellular organelles (Table S2).
We next used the data from the GPS screen and our bioinformatics analysis to execute a small-scale proof-of-principle study to test the whether the subtler interactions we identified could reveal broader infection-induced alterations to host pathways. To do this we mined the GPS dataset for proteins that had robust shifts in EGFP:DsRed and were members of a larger family of proteins that were otherwise not identified in the screen. Two protein families we explored were the dual specificity phosphatases (Dusps) and nuclear pore proteins (Nups) that both had family members shift (Dusp15/16 and Nup50/160) (Supplementary table S1), but did not have broad coverage of the entire protein family. To test whether Chlamydia infection more broadly alters this protein family, we cloned a larger panel of Dusps (12 members) and Nups (8 members) into the GPS vector and created stable reporter cell lines for each member. We then infected these cells with Chlamydia and examined the shift of EGFP:DsRed 24 hours later by flow cytometry. We found that infection with Chlamydia broadly influenced the stability of 11 out of 12 Dusps and 6 out of 8 Nups, suggesting this family of proteins may be broadly altered during infection (Figure S2). Hence the GPS platform can drive additional studies that reveal mechanisms in play during Chlamydia interaction with the host.
Knockdown of a subset of altered host proteins inhibits the growth of C. trachomatis
To assess the functional consequences of C. trachomatis-induced altered protein stability, we employed RNAi-mediated depletion of host proteins that were stabilized during C. trachomatis infection. If stabilization of these proteins is important for the intracellular developmental process, then we would predict that depletion of these proteins would have a deleterious effect on bacterial growth or replication. We optimized this screen using siRNAs to PTEN, as it has been previously shown that PTEN depletion leads to decreased bacterial growth (Figure S2) (Gurumurthy et al., 2010). Cells were transfected with pools of 4 siRNAs for 72 hours, infected with C. trachomatis, and subsequently evaluated for bacterial growth by measuring the production of infectious progeny (inclusion forming units, IFU). We conducted a siRNA screen of 200 genes in triplicate, including 147 host genes stabilized during infection and 50 genes whose stability was not affected by infection. Of the 147 genes whose stability was altered by Chlamydia, depletion of 26 of them resulted in at least a 2-fold decrease in C. trachomatis growth as assessed by IFU analysis. In contrast, depletion of only 2 out of 50 of the control proteins affected C. trachomatis growth, suggesting an enrichment among stabilized proteins of host proteins essential for bacterial growth (Supplementary Table S3). To further validate these findings, we conducted an RNAi screen with four individual duplexes per gene, and we identified 13 host genes whose knockdown inhibited C. trachomatis growth with at least two independent duplexes (Supplementary Table S3). These data illustrate that a subset of host proteins that were identified as stabilized following infection are required for normal Chlamydia growth, and suggest that these are host proteins or pathways that could be targets for host-based therapeutics.
As an alternative knockdown method, we selected three genes, Pin1, Men1 and MAGEA11 that were stabilized in the GPS screen and created stable knockdown cell lines using two independent lentivirus-delivered shRNAs for each gene of interest or for control genes. Pin1 and Men1 are involved in regulating the magnitude host signaling networks including AP-1, while MAGEA11 regulates HIF1α activity (Aprelikova et al., 2009; Ikeo et al., 2004; Lee et al., 2009; Monje et al., 2005). Both cellular functions have been implicated as important during C. trachomatis infection, and we wanted to confirm their role. Positive transductants were selected with puromycin, and knockdown was confirmed using immunoblot (Figure S2E). We then examined the ability of C. trachomatis to replicate and produce infectious progeny in each knockdown cell line using two distinct assays: qPCR to quantify the number of Chlamydia genomes present, and titration of infected lysates to measure IFU production. This allowed us to simultaneously query the entire Chlamydia life cycle, including re-differentiation into EBs. When we examined the levels of C. trachomatis by qPCR, we found modest but reproducible 1.5-2-fold defects in replication in each knockdown (Figure 2B). However, these deficiencies were amplified when we examined the production of infectious progeny, with some knockdowns causing almost a ten-fold reduction in IFU (Figures 2C and 2D). We also examined the development of inclusions in knockdown cells by fluorescence microscopy. Thirty hours after infection there was no significant difference in the morphology of inclusions between control and experimental knockdown cells. When we quantified inclusion size, there was trend toward smaller inclusions yet not of statistical significance. This suggests that the pathways interacting with these host proteins may be required for late bacterial replication and/or the re-differentiation of RBs to EBs (Figure S2). Together these data confirm that a subset of proteins that are stabilized during infection with C. trachomatis are also required for intracellular growth of the pathogen.
Figure 2. Loss-of-function screen identifies host proteins that are necessary for C. trachomatis propagation.
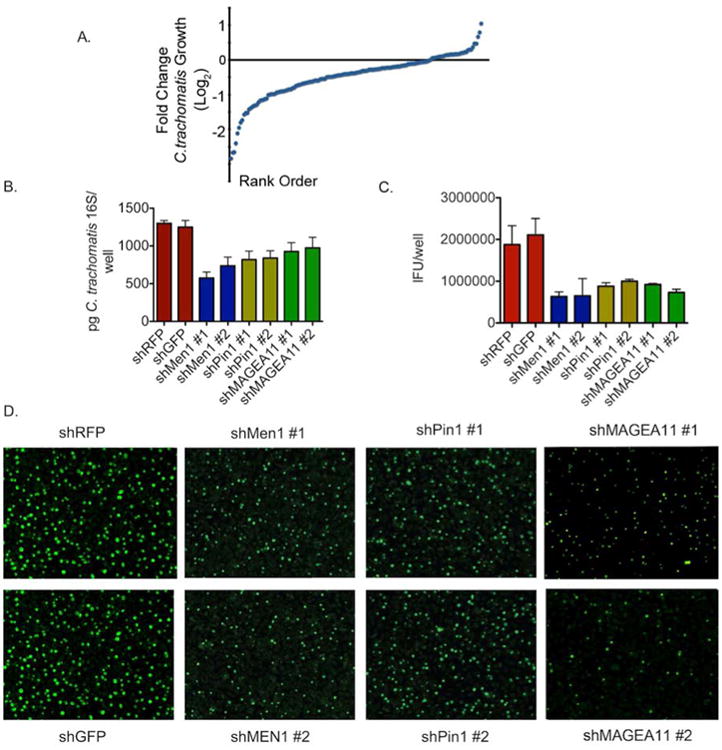
A) Distribution of the mean fold-change in C. trachomatis IFU production in experimental siRNA knockdown cell lines relative to negative controls.
B) A subset of stable shRNA knockdown cell lines was infected with C. trachomatis for 48 hours. Levels of C. trachomatis were determined by qPCR of the 16s gene and compared to non-targeting controls. The graphs show the mean levels of C. trachomatis from two independent hairpins +/- the standard. Shown is one of four independent experiments (n=4).
C) A subset of stable shRNA knockdowns was infected with C. trachomatis for 48 hours. Levels of C. trachomatis were determined by IFU production and compared to non-targeting control hairpins. The graphs show the mean IFU of C. trachomatis produced +/- the standard deviation for each individual hairpin. All experimental hairpins are p<.05 compared to control knockdown by one way ANOVA. Shown is one of four independent experiments (n=4).
D) Representative immunofluorescence micrographs from IFU analysis in (D) (Magnification 10×) from one of three independent experiments (n=4). See also Figure S2 and Table S3
AP-1 dependent transcription is activated following infection with C. trachomatis
The bioinformatics analysis of the GPS screen showed a strong enrichment in kinase signaling cascades and transcription factors. When we individually examined the role of the 13 genes identified in the loss-of-function screen using natural language processing we also found several genes, including Pin1 and Men1, that directly influence the activation and amplitude of the host transcription factor AP-1 (Chen et al., 2009; Chittenden et al., 2008; Ikeo et al., 2004; Park et al., 2012)(Figure S3). AP-1 is a heterodimeric transcription factor usually made up of a Jun and Fos protein that together regulate the expression of a large range of host genes related to inflammation, stress, and cell survival (Schonthaler et al., 2011). The AP-1 complex is activated following infection with a wide range of bacterial pathogens, but its role in C. trachomatis intracellular growth has not been described. Both Pin1 and Men1 have been shown to augment c-Jun signaling in human cells (Ikeo et al., 2004; Lee et al., 2009; Monje et al., 2005). Pin1, which is a prolyl isomerase, stabilizes phosphorylation-dependent signaling events through the isomerization of phosphorylated proteins (Lee et al., 2009; Monje et al., 2005). Men1 prevents transcription of the AP-1 component JunD, but augments the signaling cascades driven by c-Jun (Ikeo et al., 2004). Therefore, the stabilization of both Pin1 and Men1 protein during Chlamydia infection would enhance c-Jun/c-Fos dependent AP-1 signaling. We first examined whether changes in AP-1 dependent transcription occurred following infection by C. trachomatis by using a reporter cell line that expresses EGFP in an AP-1 dependent manner. We mock-treated these reporter cells, treated cells with PMA (a known activator of AP-1 driven transcription), or infected them with C. trachomatis. Thirty-six hours after infection, we analyzed cells by flow cytometry in order to determine the levels of EGFP. Relative to untreated controls, there was distinct up-regulation of EGFP in Ct-infected cells, which was even more pronounced than PMA-treated cells (Figure 3A). Quantification revealed more than a three-fold increase in the MFI of EGFP in Chlamydia-infected cells compared to control cells (Figure 3B). These data show that following infection with C. trachomatis, AP-1-directed transcription is activated.
Figure 3. C. trachomatis infection leads to the activation of the AP-1 complex.
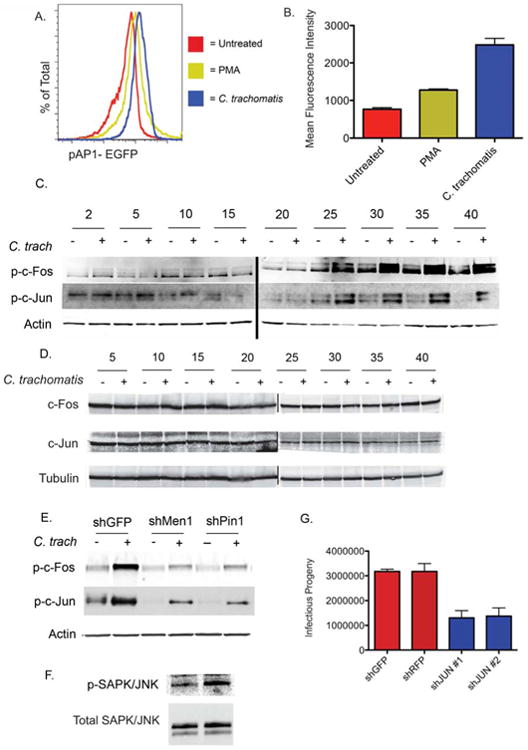
A) pAP-1 EGFP reporter cells were mock-infected, infected with C. trachomatis, or treated with PMA. Thirty hours later EGFP expression was determined by flow cytometry; A representative plot overlaying the three conditions is shown from three experiments (n=3).
B) Quantification of the mean-fluorescence intensity for each condition from (A) +/- the standard deviation (n=3). Both PMA and infection are p<.05 compared to Mock by two-tailed students t-test. Shown is a representative experiment from three completed.
C) Immunoblot analysis of and phosphorylated (top) c-Fos and (Bottom) c-Jun in mock-infected HeLa cells or HeLa cells infected with C. trachomatis every five hours for 40 hours. Loading was normalized to Actin. Shown is representative blot from one of three independent experiments.
D) Total c-Jun and c-Fos remain unchanged during infection with Chlamydia. HeLa cells were infected with Chlamydia or mock-infected. At the indicated timepoints cells were lysed and analyzed for total levels of c-Fos and c-Jun. All lysates were normalized using tubulin. Shown is a representative blot from three independent experiments.
E) Indicated shRNA knockdown cells were infected or mock-infected with Chlamydia for 40 hours. Cells were lysed and the levels of phosphorylated c-Fos and c-Jun were determined by immunoblot. Loading was normalized to host Actin. Shown is representative blot from one of three independent experiments.
F) Immunoblot analysis of HeLa cells infected mock-infected or infected with C. trachomatis for 30 hours to determine the levels of total SAPK/JNK (bottom) and phosphorylated SAPK/JNK (top). Shown is a representative blot from four independent experiments.
G) shJUN knockdowns were infected with C. trachomatis for 48 hours. Levels of C. trachomatis were determined by IFU production and compared to non-targeting control hairpins. The graphs show the mean IFU of C. trachomatis produced +/- the standard deviation for each individual hairpin (n=6). Shown is one representative experiment from two completed. All experimental hairpins have p<.05 compared to control knockdown by one way ANOVA. See also Figure S3.
The phosphorylation of the AP-1 components c-Jun and c-Fos is required for AP-1 dependent transcription, so we next examined these phosphorylation events in cells infected with C. trachomatis. We mock-infected or infected HeLa cells with C. trachomatis and examined the levels of total and phosphorylated c-Jun and c-Fos by immunoblot every five hours throughout infection (Figure 3C). Cells infected with C. trachomatis showed minimal phosphorylation of c-Fos or c-Jun for the first 20 hours of infection. However, 20-25 hours after infection, there was a significant increase the phosphorylation of both c-Fos and c-Jun that increased in magnitude through the remainder of the timecourse (Figure 3C). When we examined the levels of total c-Jun and c-Fos, we found minimal changes in the basal levels of both proteins throughout infection with C. trachomatis (Figure 3D). These experiments validated our findings using the AP-1 reporter and showed that AP-1 is activated late in host cells infected with C. trachomatis.
We next confirmed the timing of AP-1 activation by examining the transcriptional activation of two known AP-1 targets using quantitative RT-PCR. HeLa cells were mock-infected or infected with C. trachomatis for 20 or 30 hours. Cells were then lysed and RNA was purified for expression analysis. Using pre-mixed primer pairs, we amplified individual genes by RT-PCR in technical duplicates to analyze changes in transcription. Experimental genes were then normalized to host beta-actin expression, and the fold-change in infected cells was determined. In line with our immunoblot analysis, we saw no changes in mRNA levels of two AP-1 target genes, c-Fos or JunB, twenty hours post-infection or at any earlier timepoints examined (Figure S3 and data not shown). However, 30 hours after infection we saw a strong induction of the expression of both c-Fos and JunB mRNA. These data support our findings that AP-1 dependent transcription is activated between 20 and 30 hours following C. trachomatis infection.
Because our loss-of-function screen identified genes that altered C. trachomatis growth and also influence AP-1 signaling (Fig S3), we were curious whether Chlamydia restriction in these knockdown cells might be in part due to alterations in the activation of AP-1. To test this, we mock infected or infected stable control knockdown cells or cells containing shRNA against Men1 or Pin1 with C. trachomatis for 40 hours. Cells were then lysed and probed for the activation of AP-1 components by immunoblot. Both shPin1 and shMen1 knockdown cells showed a significant decrease in the phosphorylation of c-Fos and c-Jun (Fig 3E) compared to shGFP controls. These data show a direct link between the loss-of-function data and the activation of the AP-1 signaling cascade and suggests one potential mechanism preventing efficient Chlamydia growth in these cells.
If AP-1 dependent transcription is required for C. trachomatis growth, we hypothesized that upstream signaling components, such as the MAPK pathways p38, ERK, and JNK, would also be activated during infection. These pathways have previously been shown to mediate c-Jun phosphorylation, and both p38 and ERK are activated throughout infection with C. trachomatis (Buchholz and Stephens, 2007; Chen et al., 2010). One previous study showed that inhibition of ERK and p38 leads to continued c-Jun phosphorylation (Chen et al., 2010), but we hypothesized that the JNK pathway may also play a role in this process. Two previous studies examined JNK activation early following infection with C. trachomatis and did not observe activation. However these reports may have overlooked subtle activation of this cascade that occurs later during infection (Buchholz and Stephens, 2007; Chen et al., 2010). Therefore, we examined JNK activation throughout infection with C. trachomatis. We infected HeLa cells with C. trachomatis and then lysed the cells at various timepoints. These lysates were then used to perform a sandwich ELISA to detect total levels of SAPK/JNK as well as phosphorylated levels of SAPK/JNK. We found that following infection with Chlamydia there was a negligible change in the levels of total SAPK/JNK (Figure S3). However, there was a general accumulation in the levels of phosphorylated SAPK/JNK that increased as infection progressed similar to what was observed with c-Fos and c-Jun (Figure S3). We confirmed these findings by immunoblot and determined that infection with C. trachomatis leads to the phosphorylation of SAPK/JNK compared to mock-treated cells (Figure 3F). Taken together these data show that infection with C. trachomatis induces AP-1 dependent transcription late in the developmental cycle and may be influenced by the JNK signaling cascade.
Since AP-1 complex regulators and robust AP-1 activation were found to be required for normal Chlamydia growth (Figure 2), we next directly tested the necessity of AP-1 components for Chlamydia replication. We created shRNA knockdowns of c-Jun using two independent hairpins (Figure S3) and compared the production of infective C. trachomatis progeny in these cells compared to control shRNA knockdowns (Figure 3G). Consistent with the knockdowns of AP-1 regulating components (Figure 2D and 2E) depletion of c-Jun significantly reduced the production of IFU. These data confirm that AP-1 is activated late during infection and required for C. trachomatis to efficiently complete its developmental cycle.
AP-1 activation is required for C. trachomatis growth in vitro and in vivo
One goal of the GPS screen was to identify host proteins or pathways that are possible targets of small molecules, to develop host-based therapeutics that inhibit bacterial growth. As shown above, the AP-1 complex is activated during infection with C. trachomatis, and through loss-of-functions screens we identified that AP-1 activation is required for growth. To test whether pharmacological inhibitors of this pathway inhibit the growth and replication of C. trachomatis, we used two distinct inhibitors to prevent AP-1 dependent transcription: SP600125 and Juglone (Figure S4). SP600125 is a JNK inhibitor that blocks upstream activation of the AP-1 complex, while Juglone is a specific inhibitor of Pin1 and prevents prolonged amplification of AP-1 dependent transcription (Lee et al., 2009). We first tested the specificity of these inhibitors in infected cells by using phosphoarray analysis. Infected cells treated with 1uM SP600125 or Juglone did not alter the activation of a wide range of host-pathways during infection. We also examined the phosphorylation of c-Jun by immunoblot 40 hours post-infection for all conditions. In the presence of either SP600125 or Juglone, there was a significant decrease in the phosphorylation of c-Jun compared to the vehicle control, indicating the inhibitors directly alter AP-1 signaling. While these findings do not eliminate the possibility of potential off target effects not included in our analysis, they suggest that the inhibition of c-Jun phosphorylation is partially dependent on JNK signaling.
We next infected HeLa cells with C. trachomatis for one hour then added media containing various concentrations of SP600125, Juglone or the vehicle in which they were prepared (DMSO or EtOH alone). We examined bacterial replication using qPCR 48 hours following infection. The presence of either SP600125 or Juglone at concentrations of 1 uM or higher significantly decreased the levels of C. trachomatis by qPCR at 48 hours post-infection (Figure 4A), yet low concentrations of either inhibitor (100 nM) did not alter C. trachomatis growth compared to control treatments. When we examined inclusions using immunofluorescence microscopy, we noted a significant decrease in the size of inclusions in the presence of inhibitors (Figure 4B). These data suggest that inhibiting AP-1 activation can directly alter C. trachomatis growth.
Figure 4. Inhibition of AP-1 signaling prevents C. trachomatis growth and the production of elementary bodies.
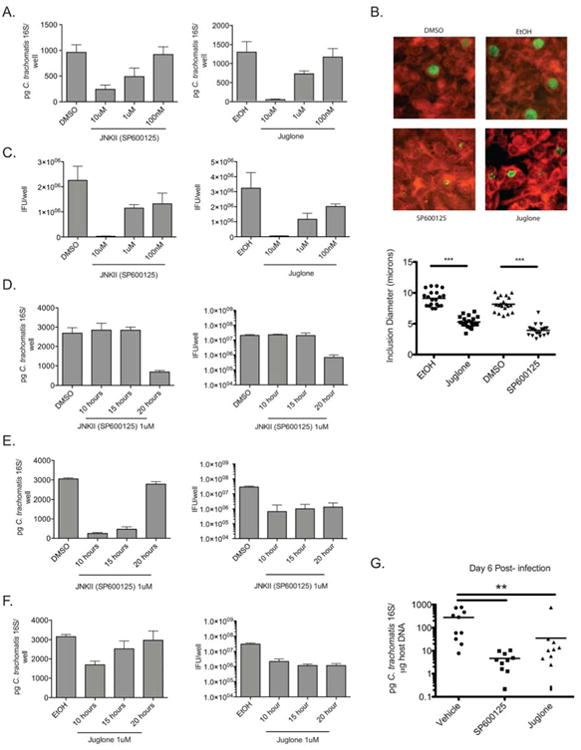
A) HeLa cells were infected with C. trachomatis and then treated with the indicated concentration of JNK inhibitor (SP600125), Pin1 inhibitor (Juglone), or vehicle alone (DMSO or EtOH). The number of Chlamydia genomes was quantified using qPCR at 48 hours post-infection. Shown is a representative experiment of 4 completed (n=4). All experimental inhibitor treatments except 100nM are p<.05 compared to vehicle control by one way ANOVA.
B) Infected HeLa cells were treated with vehicle alone or indicated inhibitors and fixed 30 hours post-infection. Cells were stained for C. trachomatis major outer membrane protein (MOMP). (Top) Representative images of C. trachomatis inclusions treated with DMSO, EtOH, SP600125 or Juglone (10 uM each at Magnification 20×) from one of two independent experiments. (Bottom) Quantification of average inclusion diameter following inhibitor treatment. p<.001 by students t-test (n=20).
C) Production of IFU from cells infected with C. trachomatis then treated with the indicated inhibitors for 48 hours (n=4). All experimental inhibitor treatments are p<.05 compared to vehicle control by one way ANOVA. Shown is a representative experiment of four independent experiments.
D) HeLa cells were infected with C. trachomatis and then treated with SP600125. At the indicated times post-infection, the inhibitor was removed and replaced with fresh media. C. trachomatis levels were determined by qPCR (Left) and IFU analysis (Right) 48 hours post-infection (n=4). Only washout at 20 hours following infection had p<.05 compared to vehicle control by one way ANOVA. Shown is a representative experiment of four independent experiments.
E) HeLa cells were infected with C. trachomatis and at the indicated timepoints were treated with SP600125. 48 hours post infection cells were lysed and the levels of C. trachomatis were determined by qPCR (Left) and IFU analysis (Right) (n=4). All experimental inhibitor treatments in the IFU experiment are p<.05 compared to vehicle control by one way ANOVA. Shown is a representative experiment of four independent experiments.
F) HeLa cells were infected with C. trachomatis, and then at the indicated timepoints the cells were treated with Juglone. 48 hours post infection cells were lysed and the levels of C. trachomatis were determined by qPCR (Left) and IFU production (Right).
In all experiments the graphs represent the mean level of C. trachomatis +/- the standard deviation (n=4). All experimental inhibitor treatments in the IFU experiment are p<.05 compared to vehicle control by one way ANOVA. Shown is a representative experiment of four independent experiments.
G) Mice were infected transcervically with 106 IFU of C. trachomatis. Twenty-four, 48 and 72 hours post-infection mice were treated with indicated inhibitors or vehicle alone. Six days after infection the levels of C. trachomatis were determined using qPCR. Graphs show bacterial levels in individual mice and mean distribution for each treatment group p< 0.01 (n=10). Shown are the data from one of two independent experiments. See also Figure S4.
We next examined IFU production following treatment with these compounds and found that either inhibitor significantly decreased the production of infectious progeny at all concentrations tested (Figure 4C). This was surprising based on our data showing no growth defect as measured by qPCR when using 100 nM concentrations of either inhibitor, yet these data were consistent with those obtained using shRNA knockdowns where the defects in replication were also more pronounced when measured as IFU (Figure 2C and 2D). We further inhibited infectious progeny production by combining both inhibitors even at low concentrations (Figure S4). Since our Chlamydia infection timecourse revealed late activation of AP-1, we conducted a washout experiment to remove the inhibitor at certain timepoints after infection. Because Juglone is a non-reversible inhibitor, we chose to run these experiments only with the reversible JNK inhibitor. Cells were infected with C. trachomatis in the presence of 1 uM SP600125 or DMSO. Ten, 15 or 20 hours after infection the media was removed, cells were washed, and fresh media lacking the inhibitor was added. After allowing the infection to continue to 48 hours, the cells were lysed and the levels and infectivity of C. trachomatis were determined by qPCR and IFU titration (Figure 4D). When we removed the inhibitor as late as 15 hours post-infection we found no effect on C. trachomatis growth or infectivity. However, if we removed the inhibitor 20 hours following infection, we saw a profound defect in Chlamydia levels and IFU production. These data suggest that AP-1 dependent transcription is not required early in C. trachomatis infection, yet it is required by 20 hours post-infection.
As a complement to these experiments we next added inhibitors at certain time points after infection. Cells were infected with C. trachomatis and SP600125, Juglone, or vehicle alone were added 10, 15 or 20 hours post-infection. Forty-eight hours after infection, the cells were lysed and we determined the levels and infectivity of C. trachomatis by qPCR and IFU titration (Figure 4E and 4F). Similar to the washout experiment, addition of either SP600125 or Juglone to infected cells at 10 or 15 hours post-infection reduced C. trachomatis levels and IFU production. Interestingly, addition of either inhibitor 20 hours post-infection had a negligible effect on the growth of C. trachomatis by qPCR yet caused a significant decrease in the production of IFU, similar to the defect seen following inhibitor addition at earlier timepoints. These data show that AP-1 signaling is required for efficient C. trachomatis growth in vitro.
It remained possible that the activation of AP-1 components, not AP-1 mediated transcription, was required for Chlamydia growth. To examine whether AP-1 mediated transcription was also required for intracellular survival we employed a third inhibitor, Tanshinone IIa, which allows AP-1 phosphorylation but prevents DNA binding and subsequent transcriptional activity (Lee et al., 2008). Cells were infected with C. trachomatis for 20 hours then treated with various concentrations of Tanshione IIa to limit the time of inhibitor treatment. Cells were then lysed at 48 hours post-infection and bacterial levels and the production of infectious progeny were determined by qPCR and IFU analysis respectively. We saw minimal defects in bacterial growth by qPCR but observed a significant inhibition in the production of infectious progeny in a dose-dependent manner (Figure S4). These data show that preventing AP-1 mediated transcription directly inhibits the ability of Chlamydia to complete the developmental cycle.
We next wanted to determine whether activation of this pathway was required for C. trachomatis growth in vivo. We have recently described a murine model of C. trachomatis infection that allows the use of human Chlamydia strains in mice and recapitulates many aspects of human disease in the upper genital tract (Gondek et al., 2012). Using this model, mice were infected transcervically with C. trachomatis and 24, 48 and 72 hours post-infection were treated with vehicle alone, SP600125, or Juglone by both IP injection and transcervical application. Six days post-infection, mice were sacrificed, the upper genital tract was isolated, and the burden of C. trachomatis in the tissue was determined using qPCR. Treatment of mice with either SP600125 or Juglone led to a 10-100 fold decrease in the levels of C. trachomatis in the upper genital mucosa compared to vehicle alone (Figure 4G). This suggests that C. trachomatis requires AP-1 dependent signaling in order to survive in the murine upper genital mucosa. Together these results show that C. trachomatis manipulates host signaling cascades that are required for bacterial growth both in vitro and in vivo.
Discussion
In this report we applied GPS, a system that probes over 12,000 proteins simultaneously to evaluate how infection with the intracellular bacterial pathogen C. trachomatis alters host protein stability. Some of the proteins identified in the GPS screen including the Mcl1 protein, and proteins involved in histone modifications, and Golgi functions were known to be altered during infection with Chlamydia validating the screen (Chumduri et al., 2013; Heuer et al., 2009; Rajalingam et al., 2008). These findings suggest Chlamydia broadly remodels the host proteome during infection, most likely through a variety of mechanisms.
This technology could have potential to uncover important host therapeutic targets. Therefore we initially targeted the AP-1 transcription complex that both bioinformatics analysis of the GPS screen and natural language processing analysis of the loss-of-function screen indicated that the AP-1 complex is activated late during Chlamydia infection and required for robust intracellular growth. One of the most intriguing findings was that late blockade of AP-1 activation had much more dramatic effects on IFU production than early AP-1 blockade. This suggests that AP-1 activity may be required for RB replication and/or re-differentiation into the EB form. Perhaps the intracellular growth of C. trachomatis triggers host stress responses, and C. trachomatis senses these host stress pathways to initiate differentiation and exit from the failing host cell.
The AP-1 complex is a critical transcription factor that regulates the expression of a large range of host genes in response to diverse stimuli (Schonthaler et al., 2011). The AP-1 complex is targeted by several bacterial pathogens to alter cellular survival or prevent inflammation and cytokine production, with some pathogens preventing activation and some inducing activation of upstream host signaling cascades (Alto and Orth, 2012). Interestingly, a recent report using C. pneumoniae suggested that AP-1 activation is responsible for inflammation seen in vivo (Wang et al., 2012). While the effect of AP-1 on inflammation during infection in vivo remains to be confirmed for C. trachomatis, these data together suggest that finely tuned AP-1 signaling is required for robust intracellular growth. While our data suggest AP-1 activity influences C. trachomatis infection in vivo, we do not yet know whether this is due to restriction in the epithelial cell or due to alterations in the subsequent immune response.
Proteomic approaches are a robust tool to characterize unknown host-pathogen interactions. These techniques should be of particular interest to probe the interactions of the variety of other pathogens which deliver ubiquitin modifying proteins to the host cell cytoplasm during infection (Collins and Brown, 2010). In addition, investigators can compare changes to the proteome that occur during infection between wild-type bacteria and strains with loss or gain of function for ubiquitin modifying enzymes. Therefore, the experimental approaches described here provide a blueprint for cataloging host proteins and pathways that are altered in stability by intracellular pathogens. These pathways may be conserved among multiple pathogens and could drive the development of therapeutics for these difficult to treat infections.
Methods
Bacterial Strains, Tissue Culture, Reagents, and Procedures
Full description of Experimental Procedures and reagents used are available in supplementary material. C. trachomatis serovar L2 434/Bu was propagated as done previously. To examine C. trachomatis levels both IFU and qPCR procedures were used as done previously (Gondek et al., 2012). For IFU, infected cells were lysed, titered, stained, and quantified by fluorescence microscopy as done previously. For quantitative PCR we measured the level of genomic DNA in samples as done previously. Automated microscopy was conducted using a Cellomics ArrayScan VTI automated microscope as previously done.
For immunoblot analysis, cells were lysed in buffer containing 8M urea unless otherwise indicated, and separated via SDS-PAGE. For all westerns unless indicated the Odyssey two-color fluorescence imaging system (LiCor) was used. Changes in protein content were quantified using Odyssey software (LiCor) or ImageJ. All statistics were calculated using Prism Software (Graphpad) and unless indicated, one-way ANOVA with a Tukey post-test were used with p values under .05 considered significant.
GPS Screening and Scoring
GPS screens were performed essentially as described (Emanuele et al., 2011). Cells were infected with C. trachomatis at an MOI of 3 while control cells were mock-infected. 24 hours following infection, cells were detached and prepared for cell sorting. Cells were sorted into 7 bins per condition with at least one million cells per bin. Following hybridization, microarray data was normalized and filtered based on signal to noise ratio and a ΔPSI value was calculated. Probe distribution across bins for treated and untreated samples were graphed using the included macro.
Bioinformatic Analysis
We used DAVID bioinformatics resources to identify enriched annotation clusters within the GPS dataset as done previously (Huang da et al., 2009). We uploaded our stabilized and destabilized protein lists independently into DAVID and used the human proteome as the background list. We used cluster analysis to identify redundant enriched terms for each group through several categories including KEGG pathway and Gene Ontology terms.
siRNA Screen
siRNA smart pools were picked into 96 well plates from the Dharmacon siRNA genome set at the Harvard Institute of Chemistry and Cell Biology. HeLa cells were seeded at a density of 2.5×103 into 96-well plates and transfected in triplicate for 72 hours when cell viability was evaluated. Cells were then infected with 104 IFU of C. trachomatis for 48 hours lysed diluted 1:1000 in fresh media and used to infect fresh HeLa cells. 24 hours later, cells were fixed with MeOH stained using DAPI and a FITC-antibody to C. trachomatis MOMP (Bio-Rad). Cells were quantified by automated microscopy as done previously. The mean number of IFU produced by non-targeting controls for each plate was calculated. A fold-change in C. trachomatis growth was calculated against non-targeting mean for each plate. A mean fold-change was then calculated for all triplicate samples.
Inhibitor Treatment
For in vitro experiments Juglone was resuspended in EtOH, for in vivo experiments Juglone was resuspended in DMSO. SP600125 and Tanshione IIa were dissolved in DMSO for all experiments. Vehicle controls contain the appropriate solvent for each experiment. Inhibitors were used at the indicated concentrations for each experiment.
Mice and in vivo inhibitor treatment
C57BL/6 mice were purchased from The Jackson Laboratory and were maintained and cared for within the Harvard Medical School Center for Animal Resources and Comparative Medicine (Boston, MA). Mice were treated with medroxyprogesterone subcutaneously seven days prior to infection in order to normalize the estrous cycle. Mice were infected transcervically with 106 IFU of C. trachomatis. For inhibitor treatments mice were injected IP and transcervically with vehicle alone, 10 mg/kg of SP600125 per injection route (Wang et al., 2004), and 1 mg/kg of Juglone per injection route (Kim et al., 2010) similar to previous studies. All experiments were approved by Institutional Animal Care and Use Committee. In all experiments ten mice per group were used.
Supplementary Material
Highlights.
C. trachomatis broadly alters the host proteome throughout infection
Several altered host proteins are necessary for productive C. trachomatis growth
Activation of the AP-1 transcription complex is required for C. trachomatis growth
Acknowledgments
We thank Jörn Coers and members of the Starnbach lab for valuable discussions. Craig Roy and Samuel Miller had helpful comments on the manuscript. This work was supported by National Institute of Health Grants R01 AI062827 and R01 AI039558 (to M.N.S.) as well as by a grant from Vertex Pharmaceuticals (to M.N.S.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alto NM, Orth K. Subversion of cell signaling by pathogens. Cold Spring Harbor perspectives in biology. 2012;4:a006114. doi: 10.1101/cshperspect.a006114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aprelikova O, Pandolfi S, Tackett S, Ferreira M, Salnikow K, Ward Y, Risinger JI, Barrett JC, Niederhuber J. Melanoma antigen-11 inhibits the hypoxia-inducible factor prolyl hydroxylase 2 and activates hypoxic response. Cancer research. 2009;69:616–624. doi: 10.1158/0008-5472.CAN-08-0811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balsara ZR, Misaghi S, Lafave JN, Starnbach MN. Chlamydia trachomatis infection induces cleavage of the mitotic cyclin B1. Infection and immunity. 2006;74:5602–5608. doi: 10.1128/IAI.00266-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry AO, Mege JL, Ghigo E. Hijacked phagosomes and leukocyte activation: an intimate relationship. Journal of leukocyte biology. 2011;89:373–382. doi: 10.1189/jlb.0510270. [DOI] [PubMed] [Google Scholar]
- Bingle LE, Bailey CM, Pallen MJ. Type VI secretion: a beginner's guide. Current opinion in microbiology. 2008;11:3–8. doi: 10.1016/j.mib.2008.01.006. [DOI] [PubMed] [Google Scholar]
- Buchholz KR, Stephens RS. The extracellular signal-regulated kinase/mitogen-activated protein kinase pathway induces the inflammatory factor interleukin-8 following Chlamydia trachomatis infection. Infection and immunity. 2007;75:5924–5929. doi: 10.1128/IAI.01029-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrack LS, Higgins DE. Genomic approaches to understanding bacterial virulence. Current opinion in microbiology. 2007;10:4–9. doi: 10.1016/j.mib.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Chellas-Gery B, Linton CN, Fields KA. Human GCIP interacts with CT847, a novel Chlamydia trachomatis type III secretion substrate, and is degraded in a tissue-culture infection model. Cellular microbiology. 2007;9:2417–2430. doi: 10.1111/j.1462-5822.2007.00970.x. [DOI] [PubMed] [Google Scholar]
- Chen CB, Ng JK, Choo PH, Wu W, Porter AG. Mammalian sterile 20-like kinase 3 (MST3) mediates oxidative-stress-induced cell death by modulating JNK activation. Bioscience reports. 2009;29:405–415. doi: 10.1042/BSR20090096. [DOI] [PubMed] [Google Scholar]
- Chen F, Cheng W, Zhang S, Zhong G, Yu P. Induction of IL-8 by Chlamydia trachomatis through MAPK pathway rather than NF-kappaB pathway. Zhong nan da xue xue bao Yi xue ban = Journal of Central South University Medical sciences. 2010;35:307–313. doi: 10.3969/j.issn.1672-7347.2010.04.005. [DOI] [PubMed] [Google Scholar]
- Chittenden TW, Howe EA, Culhane AC, Sultana R, Taylor JM, Holmes C, Quackenbush J. Functional classification analysis of somatically mutated genes in human breast and colorectal cancers. Genomics. 2008;91:508–511. doi: 10.1016/j.ygeno.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chumduri C, Gurumurthy RK, Zadora PK, Mi Y, Meyer TF. Chlamydia infection promotes host DNA damage and proliferation but impairs the DNA damage response. Cell host & microbe. 2013;13:746–758. doi: 10.1016/j.chom.2013.05.010. [DOI] [PubMed] [Google Scholar]
- Collins CA, Brown EJ. Cytosol as battleground: ubiquitin as a weapon for both host and pathogen. Trends in cell biology. 2010;20:205–213. doi: 10.1016/j.tcb.2010.01.002. [DOI] [PubMed] [Google Scholar]
- Cornelis GR. The type III secretion injectisome, a complex nanomachine for intracellular ‘toxin’ delivery. Biological chemistry. 2010;391:745–751. doi: 10.1515/BC.2010.079. [DOI] [PubMed] [Google Scholar]
- Cossart P, Roy CR. Manipulation of host membrane machinery by bacterial pathogens. Current opinion in cell biology. 2010;22:547–554. doi: 10.1016/j.ceb.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuele MJ, Elia AE, Xu Q, Thoma CR, Izhar L, Leng Y, Guo A, Chen YN, Rush J, Hsu PW, et al. Global identification of modular cullin-RING ligase substrates. Cell. 2011;147:459–474. doi: 10.1016/j.cell.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ensminger AW, Isberg RR. E3 ubiquitin ligase activity and targeting of BAT3 by multiple Legionella pneumophila translocated substrates. Infection and immunity. 2010;78:3905–3919. doi: 10.1128/IAI.00344-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan JE. Common themes in the design and function of bacterial effectors. Cell host & microbe. 2009;5:571–579. doi: 10.1016/j.chom.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gondek DC, Olive AJ, Stary G, Starnbach MN. CD4+ T cells are necessary and sufficient to confer protection against Chlamydia trachomatis infection in the murine upper genital tract. J Immunol. 2012;189:2441–2449. doi: 10.4049/jimmunol.1103032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurumurthy RK, Maurer AP, Machuy N, Hess S, Pleissner KP, Schuchhardt J, Rudel T, Meyer TF. A loss-of-function screen reveals Ras- and Raf-independent MEK-ERK signaling during Chlamydia trachomatis infection. Science signaling. 2010;3:ra21. doi: 10.1126/scisignal.2000651. [DOI] [PubMed] [Google Scholar]
- Ham H, Sreelatha A, Orth K. Manipulation of host membranes by bacterial effectors. Nature reviews Microbiology. 2011;9:635–646. doi: 10.1038/nrmicro2602. [DOI] [PubMed] [Google Scholar]
- Heuer D, Rejman Lipinski A, Machuy N, Karlas A, Wehrens A, Siedler F, Brinkmann V, Meyer TF. Chlamydia causes fragmentation of the Golgi compartment to ensure reproduction. Nature. 2009;457:731–735. doi: 10.1038/nature07578. [DOI] [PubMed] [Google Scholar]
- Holmes KK. Sexually transmitted diseases. 4th. New York: McGraw-Hill Medical; 2008. [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Hybiske K, Stephens RS. Mechanisms of Chlamydia trachomatis entry into nonphagocytic cells. Infection and immunity. 2007;75:3925–3934. doi: 10.1128/IAI.00106-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeo Y, Yumita W, Sakurai A, Hashizume K. JunD-menin interaction regulates c-Jun-mediated AP-1 transactivation. Endocrine journal. 2004;51:333–342. doi: 10.1507/endocrj.51.333. [DOI] [PubMed] [Google Scholar]
- Kim SE, Lee MY, Lim SC, Hien TT, Kim JW, Ahn SG, Yoon JH, Kim SK, Choi HS, Kang KW. Role of Pin1 in neointima formation: down-regulation of Nrf2-dependent heme oxygenase-1 expression by Pin1. Free radical biology & medicine. 2010;48:1644–1653. doi: 10.1016/j.freeradbiomed.2010.03.013. [DOI] [PubMed] [Google Scholar]
- Lee CY, Sher HF, Chen HW, Liu CC, Chen CH, Lin CS, Yang PC, Tsay HS, Chen JJ. Anticancer effects of tanshinone I in human non-small cell lung cancer. Molecular cancer therapeutics. 2008;7:3527–3538. doi: 10.1158/1535-7163.MCT-07-2288. [DOI] [PubMed] [Google Scholar]
- Lee NY, Choi HK, Shim JH, Kang KW, Dong Z, Choi HS. The prolyl isomerase Pin1 interacts with a ribosomal protein S6 kinase to enhance insulin-induced AP-1 activity and cellular transformation. Carcinogenesis. 2009;30:671–681. doi: 10.1093/carcin/bgp027. [DOI] [PubMed] [Google Scholar]
- Misaghi S, Balsara ZR, Catic A, Spooner E, Ploegh HL, Starnbach MN. Chlamydia trachomatis-derived deubiquitinating enzymes in mammalian cells during infection. Molecular microbiology. 2006;61:142–150. doi: 10.1111/j.1365-2958.2006.05199.x. [DOI] [PubMed] [Google Scholar]
- Monje P, Hernandez-Losa J, Lyons RJ, Castellone MD, Gutkind JS. Regulation of the transcriptional activity of c-Fos by ERK. A novel role for the prolyl isomerase PIN1. The Journal of biological chemistry. 2005;280:35081–35084. doi: 10.1074/jbc.C500353200. [DOI] [PubMed] [Google Scholar]
- Moulder JW. Interaction of chlamydiae and host cells in vitro. Microbiological reviews. 1991;55:143–190. doi: 10.1128/mr.55.1.143-190.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Keitany G, Li Y, Wang Y, Ball HL, Goldsmith EJ, Orth K. Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation. Science. 2006;312:1211–1214. doi: 10.1126/science.1126867. [DOI] [PubMed] [Google Scholar]
- O'Connor TJ, Adepoju Y, Boyd D, Isberg RR. Minimization of the Legionella pneumophila genome reveals chromosomal regions involved in host range expansion. Proc Natl Acad Sci U S A. 2011;108:14733–14740. doi: 10.1073/pnas.1111678108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JE, Lee JA, Park SG, Lee DH, Kim SJ, Kim HJ, Uchida C, Uchida T, Park BC, Cho S. A critical step for JNK activation: isomerization by the prolyl isomerase Pin1. Cell death and differentiation. 2012;19:153–161. doi: 10.1038/cdd.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J, Wilson DP, Myers G, Timms P, Bavoil PM. Type III secretion a la Chlamydia. Trends in microbiology. 2007;15:241–251. doi: 10.1016/j.tim.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Rajalingam K, Sharma M, Lohmann C, Oswald M, Thieck O, Froelich CJ, Rudel T. Mcl-1 is a key regulator of apoptosis resistance in Chlamydia trachomatis-infected cells. PloS one. 2008;3:e3102. doi: 10.1371/journal.pone.0003102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rejman Lipinski A, Heymann J, Meissner C, Karlas A, Brinkmann V, Meyer TF, Heuer D. Rab6 and Rab11 regulate Chlamydia trachomatis development and golgin-84-dependent Golgi fragmentation. PLoS pathogens. 2009;5:e1000615. doi: 10.1371/journal.ppat.1000615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohmer L, Hocquet D, Miller SI. Are pathogenic bacteria just looking for food? Metabolism and microbial pathogenesis. Trends in microbiology. 2011;19:341–348. doi: 10.1016/j.tim.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonthaler HB, Guinea-Viniegra J, Wagner EF. Targeting inflammation by modulating the Jun/AP-1 pathway. Annals of the rheumatic diseases. 2011;70(1):i109–112. doi: 10.1136/ard.2010.140533. [DOI] [PubMed] [Google Scholar]
- Wang A, Al-Kuhlani M, Johnston SC, Ojcius DM, Chou J, Dean D. Transcription factor complex AP-1 mediates inflammation initiated by Chlamydia pneumoniae infection. Cellular microbiology. 2012 doi: 10.1111/cmi.12071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Shi L, Xie Y, Ma C, Li W, Su X, Huang S, Chen R, Zhu Z, Mao Z, et al. SP600125, a new JNK inhibitor, protects dopaminergic neurons in the MPTP model of Parkinson's disease. Neuroscience research. 2004;48:195–202. doi: 10.1016/j.neures.2003.10.012. [DOI] [PubMed] [Google Scholar]
- Xia M, Bumgarner RE, Lampe MF, Stamm WE. Chlamydia trachomatis infection alters host cell transcription in diverse cellular pathways. The Journal of infectious diseases. 2003;187:424–434. doi: 10.1086/367962. [DOI] [PubMed] [Google Scholar]
- Yen HC, Xu Q, Chou DM, Zhao Z, Elledge SJ. Global protein stability profiling in mammalian cells. Science. 2008;322:918–923. doi: 10.1126/science.1160489. [DOI] [PubMed] [Google Scholar]
- Zhong G. Chlamydia trachomatis secretion of proteases for manipulating host signaling pathways. Frontiers in microbiology. 2011;2:14. doi: 10.3389/fmicb.2011.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.