

Abstract
Objective
To investigate whether β-catenin signaling in chondrocytes regulates osteoclastogenesis, thereby contributing to postnatal bone growth and bone remodeling.
Methods
Mice with conditional knockout (cKO) or conditional activation (cAct) of chondrocyte-specific β-catenin were generated. Changes in bone mass, osteoclast numbers, and osteoblast activity were examined. The mechanisms by which β-catenin signaling in chondrocytes regulates osteoclast formation were determined.
Results
The β-catenin cKO mice developed localized bone loss, whereas cAct mice developed a high bone mass phenotype. Histologic findings suggested that these phenotypes were caused primarily by impaired osteoclast formation, rather than impaired bone formation. Further molecular signaling analyses revealed that β-catenin signaling controlled this process by regulating the expression of the RANKL and osteoprotegerin (OPG) genes in chondrocytes. Activation of β-catenin signaling in chondrocytes suppressed Rankl gene transcription through a glucocorticoid receptor–dependent mechanism. The severe bone loss phenotype observed in β-catenin cKO mice was largely restored by treatment with human recombinant OPG or transgenic overexpression of Opg in chondrocytes.
Conclusion
β-catenin signaling in chondrocytes plays a key role in postnatal bone growth and bone remodeling through its regulation of osteoclast formation.
Growth plate chondrocytes contribute to the longitudinal growth of skeletal development via a process called endochondral ossification, whereby an intermediate cartilaginous template is made in the growth plate and then replaced by trabecular bone in the adjacent metaphysis (1). This process is initiated when progenitor cells in the resting zone are stimulated to proliferate, and following proliferation, the cells proceed through stages of maturation and hypertrophy. Hypertrophic chondrocytes produce a matrix that undergoes calcification, forming a calcified cartilaginous template for new bone formation. Thereafter, the hypertrophic chondrocytes undergo apoptosis at the lower region of the hypertrophic zone as new blood vessels begin to invade the calcified cartilage and bring in osteoblasts and osteoclasts (2,3). The calcified cartilage is remodeled by these cells and converted into trabecular bone.
Although the principles of endochondral bone formation are well established, the mechanisms by which chondrocytes signal to adjacent osteoclasts (or chondroclasts) to trigger bone resorption and to remove calcified cartilage are poorly understood. Several growth factors and their signaling molecules play important roles in the regulation of proliferation and differentiation of chondrocytes in the growth plate (4–7). Recently, emerging evidence has shown that canonical Wnt/β-catenin signaling, through regulation of mesenchymal progenitor cell differentiation, contributes to endochondral bone formation and bone remodeling (8–10). It has been reported that Dkk1, a regulatory protein of the Wnt pathway, plays important roles in the bone destruction that occurs in rheumatoid arthritis and in osteophyte formation and vascularity in the synovium during development of osteoarthritis in adult animals (11,12). However the role of β-catenin signaling in postnatal bone growth and bone remodeling has not been fully characterized. Postnatal bone growth is a unique process. It continues the expansion of the skeletal developmental process. At the same time, the bone remodeling process becomes active at this stage.
Wnt proteins act on targeting cells by binding to the Frizzled and low-density lipoprotein receptor–related protein complex at the cell surface (10,13,14). In the canonical Wnt pathway, in the absence of appropriate Wnt ligands, β-catenin is phosphorylated and polyubiquitinated, followed by proteasome degradation by a destruction complex comprising glycogen synthase kinase 3β(GSK-3β), adenomatous polyposis coli, and axin (15–17). The appropriate Wnt ligand binding to the receptor complex leads to activation of the intracellular proteins and causes the inhibition of GSK-3β and the dissociation of the destruction complex. As a result, β-catenin cannot be targeted for degradation, and accumulated β-catenin molecules translocate to the nucleus and serve as a coactivator of lymphoid enhancer factor/T cell factor (TCF) transcription factors and activate downstream target genes (18,19).
To determine the molecular mechanisms by which β-catenin signaling regulates postnatal bone remodeling, we specifically deleted or overexpressed the β-catenin gene in mouse postnatal chondrocytes. Mice with conditional knockout (cKO) of the β-catenin gene developed severe bone destruction, whereas mice with conditional activation (cAct) of the gene had a localized high bone mass phenotype. Through comprehensive in vivo and in vitro studies, we found that these phenotypes were primarily caused by impaired bone resorption, rather than impaired bone formation. Our findings suggest that β-catenin is a key molecule through which chondrocytes regulate osteoclast formation and bone remodeling at the postnatal stage.
MATERIALS AND METHODS
Generation of β-catenin cKO and cAct mice
Mice with conditional deletion of the β-catenin gene in chondrocytes (cKO mice) were created by mating Col2-CreER–transgenic mice with β-cateninflox/flox mice (all obtained from The Jackson Laboratory) (20,21). β-cateninflox/flox is a mouse model in which the flanking loxP sites are cloned in introns 1 and 6 of the β-catenin gene. Tamoxifen (1 mg/10 gm body weight, intraperitoneally, once per day for 5 days) was administered to 2-week-old cKO mice; the mice were killed at various time points thereafter. Mice with conditional activation of the β-catenin gene in chondrocytes (cAct mice) were created by mating Col2-CreER–transgenic mice with β-cateninflox(Ex3)/flox(Ex3) mice (22). The peptide encoded by β-catenin exon 3 contains 4 phosphorylation sites, and deletion of this peptide will prevent phosphorylation and subsequent proteasome degradation of β-catenin protein. Tamoxifen was administered to 2-week-old cAct mice using the same dosing protocol as that used for cKO mice.
Micro–computed tomography (micro-CT) analysis
In 3-month-old mice, quantitative micro-CT analysis was performed on a VivaCT 40 Scanner (Scanco Medical) at a resolution of 10 μm. Briefly, scanning in the tibia began approximately at the lower growth plate, and then extended proximally for 350 slices (10-μm thickness for each slice). Morphometric analysis was performed on 100 slices extending proximally, beginning with the first slice in which the tibial condyles had fully merged. The trabecular bone was segmented from the cortical shell manually on key slices using a contouring tool, and the contours were morphed automatically to segment the trabecular bone on all slices. The 3-dimensional structure and morphometry were reconstructed and analyzed.
Histology
Histologic analyses were performed on the tibiae of β-catenin cKO and cAct mice and Cre-negative control mice. Tibial samples were dissected, fixed in 10% formalin, decalcified in 5% formic acid, embedded in paraffin, and stained with Alcian blue/hematoxylin and orange G. Histomorphometric analyses of the proximal tibial metaphyses were performed to determine the number of osteoclasts and the percentage of osteoclast surface (measured as osteoclast surface/bone surface), using the OsteoMeasure system (OsteoMetrics). The bone formation rate (BFR) was determined using calcein double-labeling of the tibial tissue.
Immunohistochemistry
Tibial tissue sections were deparaffinized by immersing the tissue in xylene, fixing it with 4% paraformaldehyde for 15 minutes, and treating it with 0.5% Triton for 15 minutes, followed by fixation with 4% paraformaldehyde for another 5 minutes. Sections were incubated with a rabbit anti–β-catenin polyclonal antibody (1:50 dilution; Cell Signaling) and anti–type X collagen polyclonal antibody (1:80 dilution; Chemicon International) overnight, and then incubated with a horseradish peroxidase (HRP)–conjugated secondary antibody for 30 minutes. Slides were mounted with Permount (Electron Microscopy Sciences) and visualized under a light microscope.
Enzyme-linked immunosorbent assay (ELISA)
Blood samples were obtained just before the mice were killed, and the serum was prepared. Levels of mouse osteocalcin were measured using an ELISA kit (Biomedical Technologies), to evaluate the osteoblast activity in the mice. Assays were performed in accordance with the manufacturer’s recommendations.
Chondrocyte–spleen cell coculture assay
Primary sternal chondrocytes were isolated from the anterior rib cage and sternum of 3-day-old neonatal mice, as described previously (23). The cells were treated with 4-hydroxytamoxifen (1 μM) for 24 hours. Spleen cells were isolated from 2-month-old wild-type (WT) mice and were seeded in a 48-well culture plate at a density of 4 × 105/well.
Primary sternal chondrocytes were seeded at a density of 2,000 cells/well and cocultured with the spleen cells for 2 days in α-minimum essential medium, in the presence of 20 ng/ml macrophage colony-stimulating factor (M-CSF) (R&D Systems). Cells were then treated with M-CSF (20 ng/ml) and 1,25-dihydroxyvitamin D3 (1,25[OH]2D3) (10−8M; Sigma) for an additional 5 days. The adherent cells were fixed and stained for tartrate-resistant acid phosphatase (TRAP), and the numbers of cells that were TRAP-positive and with ≥3 nuclei were scored.
Real-time polymerase chain reaction (PCR) analysis
Total RNA was prepared using a PureLink RNA Mini kit (Invitrogen) according to the manufacturer’s protocol. One microgram of total RNA was used to synthesize complementary DNA (cDNA) using an iScripts cDNA Synthesis kit (Bio-Rad). Real-time PCR amplification was performed using gene-specific primers and a SYBR Green real-time PCR kit. Expression levels of the target gene were normalized to those of β-actin in the cDNA sample.
Western blot analysis
Cells were lysed on ice for 30 minutes in a buffer containing 50 mM Tris HCl (pH 7.4), 150 mM NaCl, 1% Nonidet P40, and 0.1% sodium dodecyl sulfate (SDS) supplemented with protease inhibitors (10 μg/ml leu-peptin, 10 μg/ml pepstatin A, and 10 μg/ml aprotinin) and phosphatase inhibitors (1 mM NaF and 1 mM Na3VO4). Proteins were fractionated by SDS–polyacrylamide gel electrophoresis, transferred to a nitrocellulose membrane, and detected using β-catenin antibodies (1:1,000 dilution; BD Biosciences) and β-actin antibodies (1:5,000 dilution; Sigma). The expression of β-catenin protein was then detected with HRP–conjugated secondary antibodies, followed by enhanced chemiluminescence–mediated visualization (Amersham).
Reporter constructs and luciferase assay
Deletion mutation of the GR binding sequences in the Rankl promoter (−705/+111-Luc) was made using a Quikchange II XL site-directed mutagenesis kit (Stratagene). The WT or mutant Rankl promoter construct (0.6 μg/well, 24-well plate) and control Renilla luciferase construct (0.01 μg/well) were cotransfected into ATDC5 cells using Lipofectamine 2000 reagent (Invitrogen). Eight hours after transfection, the cells were treated with dexamethasone (10−7M), Wnt-3a (50 μg/ml), LiCl (10 mM), or vehicle. Twenty-four hours after transfection, the cell extracts were harvested, and luciferase activity was measured using a Dual Luciferase assay system (Promega). The activity of firefly luciferase was normalized to that of Renilla luciferase.
Chromatin immunoprecipitation (ChIP) assay
For the ChIP assay, we used a ChIP assay kit from Millipore, in accordance with the manufacturer’s instructions. Briefly, ATDC5 cells were fixed with 1% formaldehyde for 10 minutes, washed, and harvested in SDS lysis buffer. After sonification, lysates containing soluble chromatin were incubated overnight with 4 μg of anti-GR antibody (Abcam) or rabbit IgG. DNA–protein immune complexes were precipitated with protein G magnetic beads, and then washed and eluted. Protein–DNA crosslinks were reversed by treatment with proteinase K. The DNA was subsequently purified and used as a template in the PCR reactions, to amplify mouse Rankl–specific sequences.
Osteoprotegerin (OPG) treatment in β-catenin cKO mice
Three-week-old β-catenin cKO mice and Cre-negative control mice were treated, via subcutaneous injection, with 5 mg/kg human recombinant OPG (rOPG) protein (obtained from Amgen), or with phosphate buffered saline as vehicle control, once weekly for 10 weeks. At the age of 3 months, the mice were killed, and long bone samples were harvested for micro-CT and histologic analyses.
RESULTS
Proximal metaphyseal bone loss in β-catenin cKO mice
We created β-catenin cKO mice by breeding Col2-CreER–transgenic mice with β-cateninflox/flox mice. As noted earlier, in β-cateninflox/flox mice, the loxP sites are inserted in introns 1 and 6 of the β-catenin gene (20,21). It has been reported that the CreERT2 fusion protein can be translocated into the nuclei of mouse chondrocytes in response to treatment with tamoxifen, an estrogen antagonist, or its metabolite, 4-hydroxytamoxifen (24). Our findings, as well as those in previous studies (25,26), demonstrated that there was efficient targeting of growth plate chondrocytes in Col2-CreER mice at the postnatal stage. The β-catenin cKO mice were viable and fertile, and had normal body size.
Three-month-old cKO mice showed significantly reduced radiodensity in the subchondral and proximal metaphyseal regions of the long bones, especially in the proximal region. In contrast, the radiodensity in the mid-shaft of the long bones was not changed (Figure 1A). Consistent with this, micro-CT imaging clearly revealed significant bone loss in the proximal metaphysis beneath the growth plate in 3-month-old β-catenin cKO mice. The trabecular bone structure was markedly destroyed in the cKO mice (Figure 1B).
Figure 1.

Reduced bone mass in mice with conditional knockout (cKO) of chondrocyte-specific β-catenin. A–C, Radiographic imaging (A) and micro–computed tomography imaging (B and C) display evidence of bone loss in the proximal metaphysis of the long bones of 3-month-old β-catenin cKO mice compared with Cre-negative control littermates. Red arrows indicate areas with bone destruction, and green arrows indicate areas with normal bone structure. D–I, Histomorphometric analyses show changes in bone parameters, such as bone volume (bone volume/total volume [BV/TV]) (D), bone mineral density (BMD) (bone mineral content/total volume [BC/TV]) (E), trabecular number (Tb.N.) (F), trabecular separation (Tb.Sp.) (G), structural model index (SMI) (H), and cortical thickness (Th.) (I), in cKO mice compared with controls (n = 8 per group). J, Histologic analysis shows reductions in the trabecular and cortical bone volumes of the proximal metaphysis of cKO mice compared with controls. Lower panels (a and b) are higher-magnification views of the boxed areas. K, Disorganization of growth plate chondrocytes is evident in the tibiae of 3-month-old cKO mice compared with controls. Lower panels (a and b) are higher-magnification views of the boxed areas. L, Immunohistochemical staining shows decreased β-catenin protein levels in the growth plate chondrocytes of cKO mice compared with controls. Arrows indicate β-catenin–positive cells. M, Real-time polymerase chain reaction analysis shows that chondrocyte marker gene expression is reduced in cKO mice compared with controls (n = 3 per group). N, Immunohistochemical staining shows that type X collagen (ColX) protein expression is reduced in the hypertrophic zone of the growth plates of cKO mice compared with controls. Arrows indicate ColX–positive hypertrophic chondrocytes. Results are the mean ± SD. * = P < 0.05 versus controls, by Student’s unpaired t-test.
In comparison with Cre-negative control littermates, trabecular bone volume (measured as bone volume/total volume) was 69% lower in β-catenin cKO mice (P < 0.05; n = 8) (Figures 1C and D) and bone mineral density (BMD) (measured as bone mineral content/total volume) was 67% lower in β-catenin cKO mice (P < 0.05; n = 8) (Figure 1E). Consistent results were also obtained when other bone structure parameters were analyzed by micro-CT. The trabecular number was 57% lower in β-catenin cKO mice (P < 0.05 versus controls; n = 8) (Figure 1F). In contrast, trabecular separation was significantly increased, by 2.6-fold, in β-catenin cKO mice (P < 0.05 versus controls; n = 8) (Figure 1G). The structural model index was significantly increased, by 1.6-fold, in β-catenin cKO mice (P < 0.05 versus controls; n = 8) (Figure 1H), suggesting that the proximal trabeculae of cKO mice have a rod-like morphology, rather than the normal plate-like morphology. The cortical bone thickness was 54% lower in β-catenin cKO mice (P < 0.05 versus controls; n = 8) (Figures 1C and I).
We performed Alcian blue/hematoxylin and orange G staining on histologic sections of the tibiae of 3-month-old mice. Consistent with the micro-CT findings, histology studies demonstrated significant bone loss in the metaphyseal and diaphyseal regions of the tibia in β-catenin cKO mice (Figure 1J). Compared with the growth plates of Cre-negative control littermates, the growth plates of cKO mice were slightly disorganized, with abnormal alignment and abnormal columnar structure (Figure 1K).
To determine the expression of β-catenin in the growth plate, we performed immunohistochemical analyses of the histologic tissue sections, using an anti–β-catenin antibody. The β-catenin expression levels were significantly reduced in the growth plate chondrocytes from 3-month-old cKO mice (Figure 1L). These results demonstrate the efficient deletion of the β-catenin gene in cKO mice following tamoxifen induction.
Messenger RNA (mRNA) expression of chondrocyte marker genes was examined by real-time PCR, using primary sternal chondrocytes isolated from cKO mice and Cre-negative controls. Specifically, expression levels of the genes for type X collagen (ColX), alkaline phosphatase (Alp), matrix metalloproteinase 9 (Mmp9), matrix metalloproteinase 13 (Mmp13), and osteocalcin (Oc) were significantly reduced in β-catenin–deficient mouse chondrocytes, showing reductions of 52%, 78%, 68%, 53%, and 33%, respectively, compared with controls (Figure 1M). Furthermore, expression of the type X collagen protein was further confirmed by immunohistochemical staining of the chondrocytes, with the results showing that type X collagen protein levels were significantly reduced in β-catenin–deficient mouse chondrocytes compared with controls (Figure 1N). These results indicate that the maturation process of growth plate chondrocytes was impaired in β-catenin cKO mice.
Increase in proximal metaphyseal bone mass in β-catenin cAct mice
We also generated β-catenin cAct mice. As noted earlier, because amino acids encoded by exon 3 contain critical GSK-3β phosphorylation sites, deletion of exon 3 in the β-catenin gene results in the production of a stabilized and truncated β-catenin protein, which is resistant to phosphorylation by GSK-3β (22,27). The β-catenin cAct mice appeared viable and fertile, with normal body size at birth and in their post-natal life. After tamoxifen induction in β-catenin cAct mice at age 2 weeks, the mice displayed a delay in growth. At age 1 month, these mice were smaller in size when compared with their control littermates (Figure 2A).
Figure 2.

Increased bone mass in mice with conditional activation (cAct) of chondrocyte-specific β-catenin. A, The body size of a 1-month-old cAct mouse was smaller than that of a 1-month-old Cre-negative control littermate. B, Micro–computed tomography (micro-CT) imaging of the tibiae shows areas of high bone mass (arrows) under the growth plate of a 1-month-old cAct mouse compared with a control mouse. With increasing age in cAct mice (2 months and 3 months old), the high bone mass area was separated from the growth plate. C–H, Findings of micro-CT show that bone parameters, such as bone volume (C), bone mineral density (D), trabecular number (E), trabecular thickness (F), and connectivity density (H), were increased in 3-month-old cAct mice compared with controls. In contrast, trabecular separation (G) was decreased in cAct mice (n = 6 per group). I and J, Histologic staining of tibial sections (I) from 3-month-old mice reveals that the proximal metaphyseal bone volume (J) was increased in cAct mice compared with controls. In I, right panels (a and b) are higher-magnification views of the boxed areas. Arrows indicate high bone mass areas. K, Morphologic analysis reveals that the growth plate cartilage morphology (arrows) was disorganized in cAct mice compared with controls. L, Primary sternal chondrocytes were isolated from 3-day-old cAct mice and control littermates and cultured for 24 hours, followed by real-time polymerase chain reaction. The results show that expression of chondrocyte marker genes was increased in β-catenin–overexpressing mouse chondrocytes compared with controls (n = 3 per group). Results are the mean ± SD. * = P < 0.05 versus controls, by Student’s unpaired t-test. See Figure 1 for other definitions.
Micro-CT analysis of the cAct mice showed an increase in bone mass in the proximal metaphyseal regions of the long bones. In the 1-month-old mice, the high bone mass was observed beneath the growth plate. With increasing age, the high bone mass area began moving away from the growth plate, due to growth of the longitudinal bone (Figure 2B). When compared with Cre-negative control littermates, the bone volume and BMD were 96% and 93% higher, respectively, in the β-catenin cAct mice (P < 0.05; n = 6) (Figures 2C and D). The trabecular number and trabecular thickness were 40% and 45% higher, respectively, in β-catenin cAct mice (P < 0.05 versus controls; n = 6) (Figures 2E and F). In contrast, trabecular separation was significantly decreased, by 33%, in cAct mice (P < 0.05 versus controls; n = 6) (Figure 2G). The connectivity density was 44% higher in the cAct mice (P < 0.05 versus controls; n = 6) (Figure 2H).
Consistent with the micro-CT findings, histologic analyses of the tibia showed that bone mass was increased in the proximal metaphysis of cAct mice (Figure 2I). Moreover, there was an 8.2-fold increase in trabecular bone volume in the high bone mass area of the tibiae of 3-month-old cAct mice (P < 0.05 versus controls; n = 6) (Figure 2J). We also examined morphologic changes in the growth plate cartilage, and found disorganization of the growth plate cartilage in cAct mice (Figure 2K).
Analysis of mRNA expression demonstrated that the expression of ColX, Alp, Mmp9, Mmp13, and Oc was significantly increased in β-catenin cAct mouse chondrocytes, by 3.8-, 2.2-, 3.9-, 6.0-, and 4.8-fold, respectively, compared with controls (Figure 2L). Levels of osteocalcin protein were also increased in cAct mice (results available at http://www.rushu.rush.edu/biochem). In addition, we observed that the proteoglycan content was reduced in the articular cartilage of cAct mice, as demonstrated by a decrease in Alcian blue staining (results not shown). These findings demonstrate that, in addition to alterations in osteoclast formation, functions of the growth plate and articular cartilage chondrocytes were also altered in β-catenin cAct mice.
Alterations in osteoclast formation in β-catenin–mutant mice
To determine changes in osteoclast formation, we performed TRAP staining of the tibial tissue from 5-week-old mice. The number of TRAP-positive multinucleated osteoclasts and the percentage of osteoclast surface were significantly increased in β-catenin cKO mice (increases of 55% and 65%, respectively) compared with Cre-negative control littermates (Figures 3A–C). In contrast, the number of TRAP-positive multi-nucleated osteoclasts and the percentage of osteoclast surface were significantly reduced in 5-week-old cAct mice (decreases of 48% and 57%, respectively) compared with Cre-negative controls (Figures 3D–F).
Figure 3.

Changes in osteoclast formation in β-catenin–mutant mice. A–F, Tartrate-resistant acid phosphatase (TRAP) staining was performed on tibial tissue of 5-week-old β-catenin cKO mice (A–C) or cAct mice (D–F) and Cre-negative control mice. In cKO mice, the numbers of TRAP-positive multinucleated osteoclasts (A and B) and percentage of osteoclast surface (C) in the proximal metaphysis were increased relative to controls. In cAct mice, osteoclast numbers (D and E) and percentage of osteoclast surface (F) in the proximal metaphysis were decreased relative to controls. Arrows indicate TRAP-positive osteoclasts. G–I, Primary sternal chondrocytes were isolated from β-catenin cKO mice or control mice and treated with or without 4-hydroxytamoxifen, and expression of β-catenin was analyzed by Western blotting (G). The chondrocytes were then cocultured with spleen cells from 2-month-old C57 mice in the presence of macrophage colony-stimulating factor (M-CSF) and 1,25-dihydroxyvitamin D3 (1,25[OH]2D3). The increase in osteoclast numbers observed in the spleen cell–cKO mouse chondrocyte cocultures was reversed by addition of human recombinant osteoprotegerin (rOPG) (H and I). J–L, Expression of Opg, Rankl, and T cell factor 4 (TCF-4) was examined by real-time polymerase chain reaction (PCR) (J and K) and Western blotting (L) in cKO mouse chondrocytes compared with controls. M and N, Primary sternal chondrocytes were isolated from β-cateninflox(Ex3)/flox(Ex3) cKO or control mice and treated with or without 4-hydroxytamoxifen. Expression of floxed (F) or truncated (tr) β-catenin protein (M) and levels of nuclear β-catenin (N) were examined by Western blotting. Lamin A2 was used as a positive control in the Western blots. O–R, Chondrocytes from β-catenin cAct mice or control mice were cocultured with spleen cells isolated from wild-type mice in the presence of M-CSF and 1,25(OH)2D3. Osteoclast numbers (O and P) and expression of Opg (Q) and Rankl (R) in the spleen cell–chondrocyte cocultures were examined by real-time PCR. Results are the mean ± SD of 5 mice per group in B, C, E, and F, and 4 mice per group in I, J, K, P, and Q. * = P < 0.05 versus controls, by Student’s unpaired t-test. See Figure 1 for other definitions.
We confirmed, by Western blotting, the deletion of the β-catenin gene in chondrocytes derived from β-catenin cKO mice, and also found that β-catenin protein levels were significantly decreased in β-catenin–deficient mouse chondrocytes (Figure 3G). To determine the effect of β-catenin signaling in chondrocytes on the regulation of osteoclast formation, we performed chondrocyte–spleen cell coculture experiments. When β-catenin–deficient mouse chondrocytes were cocultured with WT mouse spleen cells in the presence of 10−8M 1,25(OH)2D3, the formation of TRAP-positive osteoclasts was increased by 2-fold relative to controls, and the addition of rOPG to the coculture significantly reversed this increase in osteoclast formation, reducing the number of osteoclasts per well by 70% (Figures 3H and I).
We then analyzed mouse primary chondrocytes using a real-time PCR assay. The results showed that expression of Opg mRNA was decreased by 60% and expression of Rankl mRNA was increased by 1.9-fold in primary chondrocytes derived from β-catenin cKO mice relative to controls (Figures 3J and K).
β-catenin interacts with TCF-4 when it is translocated into the nucleus. In this study, we also examined whether the expression of TCF-4 was changed in β-catenin–deficient mouse chondrocytes. The results of Western blotting revealed that there were no significant changes in TCF-4 expression in the chondrocytes derived from β-catenin cKO mice compared with controls (Figure 3L).
We also performed a coculture assay using the chondrocytes derived from β-catenin cAct mice. We first analyzed the chondrocytes by Western blotting to determine the expression of β-catenin protein in these cells. Western blot analyses detected the presence of N-terminal–deleted β-catenin in the chondrocytes derived from β-catenin cAct mice, and also showed increased nuclear expression of β-catenin protein in the cAct mouse chondrocytes compared with controls (Figures 3M and N). In contrast to the findings in cocultures with β-catenin cKO mouse chondrocytes, coculture of β-catenin–overexpressing mouse chondrocytes with WT mouse spleen cells showed that the formation of TRAP-positive multinucleated osteoclasts was reduced by 50% in cAct mouse chondrocyte cocultures compared with controls (Figures 3O and P).
We also observed a significant increase in Opg expression and a significant decrease in Rankl expression in chondrocytes derived from β-catenin cAct mice (Figures 3Q and R). These findings further support the role of β-catenin signaling in the regulation of Opg and Rankl in chondrocytes.
In contrast to the findings mentioned above, we did not observe a significant change in osteoclast formation when bone marrow cells from β-catenin cKO mice and Cre-negative control mice were cultured in an osteoclast formation assay (results available at http://www.rushu.rush.edu/biochem). Consistently, no significant changes in the expression of Opg and Rankl mRNA and in the levels of β-catenin protein were found in β-catenin cKO mice (results available at http://www.rushu.rush.edu/biochem).
Lack of change in bone formation in β-catenin–mutant mice
We examined changes in the BFR in the tibiae of β-catenin–mutant mice. The results of calcein double-labeling of the tibial tissue showed that the BFR, an indicator of osteoblast activity, was not changed in β-catenin cKO mice compared with controls. Moreover, consistently, the serum osteocalcin level was not significantly changed in β-catenin cKO mice compared with controls, as measured by ELISA. Since bone marrow stromal cells, but not chondrocytes, are the major contributors to serum osteocalcin levels, these results suggest that deletion of β-catenin, as studied in the mutant mice herein, mainly affects chondrocytes, but not bone marrow stromal cells. Similarly, there was no significant change in the BFR in β-catenin cAct mice. (All of these results are available at http://www.rushu.rush.edu/biochem).
We also performed an alkaline phosphatase activity assay and alizarin red staining using the bone marrow cells isolated from Cre-negative control mice and β-catenin cKO mice. The results of these analyses indicated that there were no significant changes in alkaline phosphatase activity and no significant changes in mineralized bone matrix formation in the bone marrow cells derived from β-catenin cKO mice (results available at http://www.rushu.rush.edu/biochem).
We then examined changes in osteoblast numbers by performing Runx2 immunostaining, and also evaluated changes in osteocyte numbers and morphologic features of the tibiae, in β-catenin–mutant mice. We found that the numbers of Runx2-positive osteoblasts in the trabecular bone of the primary spongiosa area, the numbers of osteocytes, and the morphologic features of the cortical bone below the growth plate were not changed in β-catenin–mutant mice compared with controls (results available at http://www.rushu.rush.edu/biochem). These results suggest that it is the osteoclast, but not the osteoblast or osteocyte, that contributes to the altered bone mass in β-catenin–mutant mice.
Regulation of Rankl expression in a GR-dependent manner by β-catenin in chondrocytes
We examined changes in the expression of Rankl and Opg mRNA in chondrogenic ATDC5 cells in the presence of Wnt-3a or the GSK-3β inhibitor LiCl. Wnt-3a inhibited Rankl expression by 51% and 48% in ATDC5 cells after 24 hours and 48 hours of culture, respectively, and stimulated Opg expression by 6.1-fold and 9.3-fold at 24 hours and 48 hours, respectively, compared with vehicle-treated cells (Figures 4A and B). Similarly, LiCl inhibited Rankl expression by 52% and 88% after 24 hours and 48 hours of culture, respectively, and stimulated Opg expression by 2.5-fold and 2.3-fold at 24 hours and 48 hours, respectively, compared with vehicle-treated cells (Figures 4A and B). Interestingly, neither Wnt-3a nor LiCl inhibited Rankl expression after 12 hours of culture, and only slightly suppressed Opg expression at the 12-hour time point (Figures 4A and B). Consistently, transfection of ATDC5 cells with β-catenin small interfering RNA (siRNA) led to an increase in Rankl expression at 24 hours and 48 hours of β-catenin siRNA transfection (Figure 4C).
Figure 4.

The β-catenin signaling pathway down-regulates Rankl expression in a GR-dependent manner. A–D, ATDC5 cells were cultured with Wnt-3a (50 μg/ml), LiCl (10 mM), or vehicle (A and B), transfected with β-catenin or control small interfering RNA (siRNA) (C), or treated with dexamethasone (Dex) (10−7M) or vehicle (D) for 12, 24, and 48 hours. The fold change in Rankl mRNA expression (A and C), Opg mRNA expression (B), and Rankl promoter activity (D) was significantly different between the groups after 24 or 48 hours of culture. E, Treatment of ATCD5 cells with dexamethasone enhanced Rankl mRNA expression at all time points. F, Transfection of ATDC5 cells with a mutant Rankl promoter construct (mutation of the GR binding site in the Rankl promoter) inhibited basal and dexamethasone-induced Rankl promoter activity, as compared to transfection with a wild-type (WT) reporter construct. G and H, Wnt-3a and LiCl inhibited GR mRNA expression in WT reporter–transfected cells (G) and inhibited Rankl promoter activity in WT reporter–transfected cells, but not in mutant (Mut) reporter–transfected cells (H). I and J, Transfection of cells with a Rankl-luc reporter construct and β-catenin siRNA up-regulated Rankl promoter activity (I), whereas treatment of these cells with Wnt-3a inhibited Rankl promoter activity (J). No effects were observed on empty vector–transfected cells. K, Interaction of β-catenin with GR was detected by chromatin immunoprecipitation (ChIP) assay. L, ChIP assay was performed using sonicated chromatin extracted from ATDC5 cells treated with either anti-GR antibody or normal rabbit IgG. Purified DNA was analyzed by standard polymerase chain reaction, using mouse Rankl–specific primer set 1 (−682/−562), which amplifies the fragment spanning the GR binding sequence (−647/−633). Primer set 2 was used as a negative control, amplifying a fragment downstream of the transcription start site (+2463/+2592). GR specifically binds to the proximal promoter region of the Rankl promoter, and treatment of cells with Wnt-3a inhibited GR binding to the Rankl promoter. Results are the mean ± SD of 3 mice per group. * = P < 0.05 versus controls, by Student’s unpaired t-test. WB = Western blotting.
Recent studies have shown that in osteoblasts, β-catenin signaling induces Opg expression by targeting the TCF binding sites on the promoter of the OPG gene (28). However, it remains unknown how β-catenin signaling affects Rankl expression. Although several putative TCF binding sites were identified in the Rankl promoter, we found that TCF-4 expression was not changed in β-catenin cKO mice relative to controls (Figure 3L). There is a GR binding sequence at the −647/−633 site of the Rankl promoter. Based on the fact that glucocorticoids are a well-known Rankl regulator, we hypothesized that β-catenin may interfere with the GR–DNA interaction to achieve its inhibitory effect on Rankl expression. To test this hypothesis, we examined the effects of dexamethasone on Rankl expression, and found that dexamethasone promoted Rankl promoter activity and significantly increased Rankl mRNA expression at 12 hours of treatment, compared with vehicle-treated cells (Figures 4D and E). Deletion of the GR binding sequence reduced Rankl promoter activity by 30% and abolished the effect of dexamethasone on Rankl promoter activity (Figure 4F).
We then sought to investigate whether β-catenin signaling affects Rankl promoter activity, and if so, whether the inhibition of the Rankl promoter involves inactivation of GR signaling. We first examined whether activation of β-catenin affects the expression of GR. At 12 hours of treatment with either Wnt-3a or LiCl, GR mRNA expression in ATDC5 cells was repressed by 69% and 88%, respectively, compared with vehicle-treated controls (Figure 4G). Consistent with these findings, Wnt-3a and LiCl inhibited Rankl promoter activity by 32% and 44%, respectively, at 24 hours of treatment (Figure 4H). In contrast, the inhibitory activity of Wnt-3a and LiCl was completely abolished when the GR binding site in the Rankl promoter was mutated in chondrogenic ATDC5 cells (Figure 4H).
Furthermore, we found that transfection of the ATDC5 cells with β-catenin siRNA up-regulated Rankl promoter activity, and addition of Wnt-3a in the ATDC5 cell cultures inhibited Rankl promoter activity (Figures 4I and J). Both β-catenin siRNA and Wnt-3a had no effect on the activity of the cells transfected with empty vector (Figures 4I and J), and Wnt-3a had no effect on the luciferase activity in cells transfected with the pGL3-SV40 basal reporter (results not shown). These results suggest that β-catenin and Wnt-3a specifically regulate Rankl promoter activity.
To further determine whether β-catenin directly interacts with GR in ATDC5 cells, we performed a ChIP assay. Results of the ChIP assay showed that β-catenin indeed interacts with GR in chondrocytes (Figure 4K), suggesting that β-catenin may affect GR activity by directly binding to GR in the nucleus. The findings demonstrated that GR directly bound its response element at the Rankl promoter, and addition of Wnt-3a inhibited GR binding (Figure 4L). These findings suggest that inhibition of glucocorticoid receptor signaling may be the major mechanism by which β-catenin regulates Rankl expression.
Reversal of the bone loss phenotype by rOPG in β-catenin cKO mice
We treated β-catenin cKO mice with rOPG, starting when the mice were 3 weeks old, to determine whether treatment with rOPG could reverse the bone destruction phenotype observed in β-catenin cKO mice. We found that treatment with rOPG resulted in a severe osteopetrosis phenotype in both Cre-negative control mice and β-catenin cKO mice (Figures 5A and E). Specifically, treatment of control mice with rOPG caused a 3.8-fold and 4.7-fold increase in the trabecular bone volume and BMD, respectively (P < 0.05 versus vehicle-treated control mice; n = 6) (Figures 5B and C), as well as a 1.7-fold increase in cortical bone thickness (P < 0.05 versus vehicle-treated control mice; n = 6) (Figure 5D). Treatment of cKO mice with rOPG caused a 16-fold and 20-fold increase in the trabecular bone volume and BMD, respectively (P < 0.05 versus vehicle-treated cKO mice; n = 6) (Figures 5B and C), as well as a 3.2-fold increase in cortical bone thickness (P < 0.05 versus vehicle-treated cKO mice; n = 6) (Figure 5D). Interestingly, although vehicle-treated cKO mice had much lower trabecular and cortical bone volumes when compared with vehicle-treated control mice, no significant difference in bone volumes was observed between the cKO mice and their Cre-negative controls after rOPG treatment (Figure 5E).
Figure 5.

Treatment with human recombinant osteoprotegerin (rOPG) reverses the bone loss phenotype observed in β-catenin cKO mice. Two-week-old β-catenin cKO mice or Cre-negative control mice were treated with rOPG or phosphate buffered saline as vehicle control, after tamoxifen induction. A–D, Micro–computed tomography was used to analyze the effects of rOPG treatment on trabecular and cortical bone mass (A) and on the parameters of bone volume (B), bone mineral density (C), and cortical bone thickness (D) in β-catenin cKO and control mice. E, Histologic staining of tibial tissue also revealed the effects of rOPG on bone mass in β-catenin cKO and control mice. Results are the mean ± SD of 6 mice per group. * = P < 0.05 versus vehicle-treated mice in the same group; # = P < 0.05 versus Cre-negative control mice, by Student’s unpaired t-test. See Figure 1 for other definitions.
To further determine the role of chondrocyte-derived OPG in the regulation of osteoclast formation, we generated Col2-Opg–transgenic mice. Overexpression of Opg in these mice was confirmed by detection of increased serum levels of OPG. There was an ~2-fold increase in the OPG levels in Col2-Opg–transgenic mice (Figure 6A).
Figure 6.
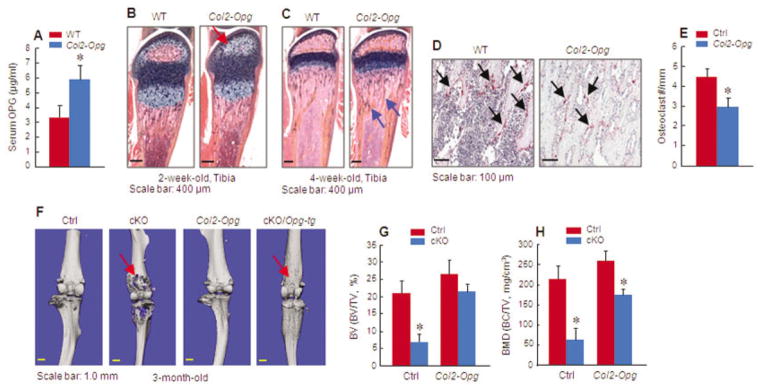
Overexpression of Opg reverses the bone loss phenotype observed in β-catenin cKO mice. A, Serum osteoprotegerin (OPG) production in Col2-Opg–transgenic mice and wild-type (WT) mice (n = 7 per group) was examined by enzyme-linked immunosorbent assay. B–E, Histologic staining of the tibiae of 2-week-old Col2-Opg–transgenic or WT mice (n = 5 per group) was used to assess formation of the secondary ossification center (B) and changes in bone mass (arrows) underneath the growth plate (C). In addition, osteoclast formation (arrows) was assessed by tartrate-resistant acid phosphatase (TRAP) staining in 4-week-old Col2-Opg–transgenic and WT mice (D and E). F–H, Col2a1-Opg–transgenic mice were bred with β-catenin cKO mice, and the tibiae of 3-month-old mice in each group (n = 6 per group) were assessed by micro–computed tomography for bone destruction (arrows) (F) and changes in bone volume (G) and bone mineral density (H). Results are the mean ± SD. * = P < 0.05 versus controls, by Student’s unpaired t-test. See Figure 1 for other definitions.
Histologic analysis of the tibiae showed that formation of the secondary ossification center was delayed in 2-week-old Col2-Opg–transgenic mice (Figure 6B). In 4-week-old transgenic mice, the trabecular bone volume was slightly increased and osteoclast formation was reduced (Figures 6C–E).
Breeding the Col2-Opg–transgenic mice with β-catenin cKO mice significantly reversed the bone destruction phenotype observed in β-catenin cKO mice (Figure 6F). Bone volume and BMD were significantly increased in β-catenin cKO;Col2-Opg double-mutant mice when compared with β-catenin cKO mice (Figures 6G and H). These findings provide additional evidence to confirm that chondrocyte OPG production does play an important role in the regulation of osteoclast formation.
DISCUSSION
The contribution of Wnt/β-catenin signaling to early embryonic skeletal development has been well established. In contrast, the role of β-catenin signaling in chondrocytes during late embryonic skeletal development and postnatal bone growth is not fully understood. Several Wnt genes are expressed in the postnatal growth plate (29), suggesting that Wnt/β-catenin signaling may play important roles in the regulation of postnatal cartilage and bone growth. In previous studies by our group, we found that activation of cartilage-specific β-catenin signaling promotes chondrocyte maturation and facilitates formation of the primary ossification center in day 18.5 mouse embryos (30). We also found that adult β-catenin cAct mice exhibited the cartilage degradation phenotype (27). In a study by Yasuhara et al, in which β-catenin conditional transgenic mice were generated using the Col11 promoter, the thickness of the superficial zone of articular cartilage was increased in 1-month-old β-catenin conditional transgenic mice (31). In the present study, no significant changes in articular cartilage thickness were evident in β-catenin–mutant mice, whereas proteoglycan content was reduced in 3-month-old β-catenin cAct mice.
We also observed significant changes in chondrocyte morphologic features (disorganized chondrocyte columns and irregular shapes of hypertrophic chondrocytes) and changes in the expression levels of chondrocyte marker genes (increased chondrocyte marker gene expression in cAct mice and decreased chondrocyte marker gene expression in cKO mice). Moreover, the length of the growth plate cartilage was not significantly changed in 3-month-old β-catenin cKO mice, but was slightly reduced in β-catenin cAct mice. However, the most prominent and obvious feature observed in the chondrocyte-specific β-catenin cKO mice was the severe bone destruction that was localized to the proximal metaphyseal area beneath the growth plate, while the bone mass in the mid-shaft area remained normal. Since we used chondrocyte-specific Cre–expressing transgenic mice to target the floxed β-catenin gene, the reasonable interpretation of this bone loss phenotype is that it can be attributed to impairment of β-catenin function in chondrocytes, which subsequently causes abnormal regulation of adjacent osteoclasts in the proximal metaphyseal bone area.
Since chondrocytes are the major cells targeted in Col2-CreER–transgenic mice, only a small proportion of osteoclast precursor cells, which have direct contact with hypertrophic chondrocytes, are affected in β-catenin–mutant mice. In contrast, the majority of bone marrow cells are not affected. This may explain why the regions far away from the growth plate chondrocytes escape destruction. In contrast, the β-catenin cAct mice displayed an obvious increase in bone mass, as observed in 1-, 2-, and 3-month-old β-catenin cAct mice. Because of chondrocyte turnover, chondrocytes may be targeted in a transient manner in Col2-CreER–transgenic mice. When β-catenin–activated cells were replaced by non-targeting cells at the later time points, the growth plate chondrocytes could not inhibit osteoclast (or chondroclast) formation, and therefore isolated and enhanced bone mass was observed. Concurrent with the growth of the long bone, this area of high bone mass was observed to be moving away from the growth plate.
To address the question as to whether the bone destruction observed in β-catenin cKO mice was due to activation of osteoclast activity, we examined the formation of osteoclasts in 5-week-old mice. The number of osteoclasts and percentage of osteoclast surface were significantly increased in β-catenin cKO mice, whereas both were significantly decreased in β-catenin cAct mice. We also analyzed the BFR by calcein double-labeling in β-catenin cKO and cAct mice, and found no significant differences in the BFR between these mutant mice. Taken together, these findings suggest that impairment of both osteoclast formation and bone resorption is the major reason for the changes in bone mass observed in β-catenin–mutant mice.
The major molecules of osteoclast regulation under both physiologic and pathologic conditions are RANKL and OPG, and both factors are believed to be produced by osteoblasts and stromal cells under the control of several cytokines and hormones, including 1,25(OH)2D3. Results of immunohistochemical analyses and in situ hybridization studies by other groups have shown that RANKL and OPG are expressed by hypertrophic chondrocytes in murine and rat growth plates (32,33). Further investigations have demonstrated a novel role for chondrocytes in the regulation of osteoclast formation through the expression of RANKL, an effect mediated by 1,25(OH)2D3 and bone morphogenetic protein 2 (33,34). OPG acts as a key inhibitor of osteoclast differentiation through its interaction with RANKL, by antagonizing the function of RANKL. The role of OPG in chondrocytes in the regulation of osteoclast formation has not been demonstrated in previous studies, although OPG has been reported to be a direct downstream target of β-catenin in mature osteoblasts (28).
In the present study, we present novel evidence to demonstrate that the expression of both Rankl and Opg is regulated by β-catenin signaling in chondrocytes, and this plays a critical role in the regulation of cartilage and bone remodeling in vitro and in vivo. First, Rankl mRNA expression was increased and Opg mRNA expression was decreased in β-catenin–deficient mouse chondrocytes. Second, the increased osteoclast formation observed in cocultures of spleen cells and β-catenin–deficient mouse chondrocytes was significantly blocked by the addition of rOPG. Third, treatment of β-catenin cKO mice with rOPG completely blocked the bone destruction, resulting in a severe osteopetrosis phenotype, at a level of severity comparable to that in rOPG-treated Cre-negative control mice. In addition to inhibiting RANKL expression in chondrocytes, OPG could also inhibit RANKL expression in bone marrow cells from β-catenin cKO mice. However, since treatment with OPG could bring the bone volume and BMD to the same levels as those in Cre-negative control mice, this finding suggests that OPG reversed the bone destruction phenotype caused by enhanced RANKL expression in β-catenin–deficient mouse chondrocytes.
In this study, we did not observe bone destruction at the mid-shaft region of the long bone in β-catenin cKO mice, further suggesting that RANKL signaling in bone marrow cells was not affected by changes in chondrocyte β-catenin signaling. In addition, breeding Col2-Opg–transgenic mice with β-catenin cKO mice significantly reversed the bone destruction phenotype observed in β-catenin cKO mice. These findings thus suggest that specific overexpression of OPG in chondrocytes can block RANKL signaling and subsequently prevent high osteoclast activity. These findings genetically place RANKL/OPG downstream of the β-catenin signaling pathway in chondrocytes.
Our findings also demonstrated that β-catenin signaling in chondrocytes plays an indispensible role in the differentiation of osteoclasts. Depletion of β-catenin in mouse chondrocytes led to the bone loss phenotype through changes in OPG/RANKL expression, and chondrocyte-regulated osteoclast formation could not be compensated by the formation of osteoblasts. It is possible that the function of chondrocyte-regulated osteoclasts may play a more important role in bone growth during the postnatal period. Recent evidence has also demonstrated that osteocytes can express high levels of RANKL and have the capacity to regulate formation of osteoclasts (35,36). Thus, further studies are needed to address whether this regulatory mechanism is of prime importance during the processes of bone remodeling in adult mice.
Supplementary Material
Acknowledgments
Supported by the NIH (grants AR-055915 and AR-054465 to Dr. Chen), the New York State Department of Health, and the Empire State Stem Cell Board (contract C024320 to Dr. Chen). Dr. Wang’s work was supported in part by the Natural Science Foundation of China (grants 81071436 and 11JCZDJC16500) and the Tianjin Municipal Natural Science Foundation, China. Dr. Zhu’s work was supported in part by the Natural Science Foundation of China (grant 30973040).
We gratefully acknowledge the technical expertise of Ryan Tierney and Sarah Mack within the Center for Musculoskeletal Research Histology Core at the University of Rochester. We also would like to acknowledge Drs. Paul Kostenuik and David Ke at Amgen for generously providing us with the rOPG protein.
Footnotes
Dr. Boyce has received consulting fees, speaking fees, and/or honoraria from Merck (less than $10,000).
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Chen had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. Zhang, Xiao, Xing, Chen.
Acquisition of data. Wang, Jin, Zhu, Li, Zhao, Tang, Chen.
Analysis and interpretation of data. Zhang, Xiao, Xing, Boyce, Chen.
References
- 1.Villemure I, Stokes IA. Growth plate mechanics and mechanobiology: a survey of present understanding. J Biomech. 2009;42:1793–803. doi: 10.1016/j.jbiomech.2009.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adams CS, Shapiro IM. The fate of the terminally differentiated chondrocyte: evidence for microenvironmental regulation of chondrocyte apoptosis. Crit Rev Oral Biol Med. 2002;13:465–73. doi: 10.1177/154411130201300604. [DOI] [PubMed] [Google Scholar]
- 3.Gibson G. Active role of chondrocyte apoptosis in endochondral ossification. Microsc Res Tech. 1998;43:191–204. doi: 10.1002/(SICI)1097-0029(19981015)43:2<191::AID-JEMT10>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 4.Chen M, Zhu M, Awad H, Li TF, Sheu TJ, Boyce BF, et al. Inhibition of β-catenin signaling causes defects in postnatal cartilage development. J Cell Sci. 2008;121:1455–65. doi: 10.1242/jcs.020362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maeda Y, Nakamura E, Nguyen MT, Suva LJ, Swain FL, Razzaque MS, et al. Indian Hedgehog produced by postnatal chondrocytes is essential for maintaining a growth plate and trabecular bone. Proc Natl Acad Sci U S A. 2007;104:6382–7. doi: 10.1073/pnas.0608449104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wuelling M, Vortkamp A. Chondrocyte proliferation and differentiation. Endocr Dev. 2011;21:1–11. doi: 10.1159/000328081. [DOI] [PubMed] [Google Scholar]
- 7.Xian CJ. Roles of epidermal growth factor family in the regulation of postnatal somatic growth. Endocr Rev. 2007;28:284–96. doi: 10.1210/er.2006-0049. [DOI] [PubMed] [Google Scholar]
- 8.Day TF, Guo X, Garrett-Beal L, Yang Y. Wnt/β-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell. 2005;8:739–50. doi: 10.1016/j.devcel.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 9.Hill TP, Spater D, Taketo MM, Birchmeier W, Hartmann C. Canonical Wnt/β-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev Cell. 2005;8:727–38. doi: 10.1016/j.devcel.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 10.Tamamura Y, Otani T, Kanatani N, Koyama E, Kitagaki J, Komori T, et al. Developmental regulation of Wnt/β-catenin signals is required for growth plate assembly, cartilage integrity, and endochondral ossification. J Biol Chem. 2005;280:19185–95. doi: 10.1074/jbc.M414275200. [DOI] [PubMed] [Google Scholar]
- 11.Diarra D, Stolina M, Polzer K, Zwerina J, Ominsky MS, Dwyer D, et al. Dickkopf-1 is a master regulator of joint remodeling. Nat Med. 2007;13:156–63. doi: 10.1038/nm1538. [DOI] [PubMed] [Google Scholar]
- 12.Weng LH, Ko JY, Wang CJ, Sun YC, Wang FS. Dkk-1 promotes angiogenic responses and cartilage matrix proteinase secretion in synovial fibroblasts from osteoarthritic joints. Arthritis Rheum. 2012;64:3267–77. doi: 10.1002/art.34602. [DOI] [PubMed] [Google Scholar]
- 13.Balemans W, Van Hul W. The genetics of low-density lipoprotein receptor-related protein 5 in bone: a story of extremes. Endocrinology. 2007;148:2622–9. doi: 10.1210/en.2006-1352. [DOI] [PubMed] [Google Scholar]
- 14.Kubota T, Michigami T, Ozono K. Wnt signaling in bone metabolism. J Bone Miner Metab. 2009;27:265–71. doi: 10.1007/s00774-009-0064-8. [DOI] [PubMed] [Google Scholar]
- 15.Behrens J. Cross-regulation of the Wnt signalling pathway: a role of MAP kinases. J Cell Sci. 2000;113:911–9. doi: 10.1242/jcs.113.6.911. [DOI] [PubMed] [Google Scholar]
- 16.Eastman Q, Grosschedl R. Regulation of LEF-1/TCF transcription factors by Wnt and other signals. Curr Opin Cell Biol. 1999;11:233–40. doi: 10.1016/s0955-0674(99)80031-3. [DOI] [PubMed] [Google Scholar]
- 17.Ikeda S, Kishida S, Yamamoto H, Murai H, Koyama S, Kikuchi A. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3β and β-catenin and promotes GSK-3β-dependent phosphorylation of β-catenin. EMBO J. 1998;17:1371–84. doi: 10.1093/emboj/17.5.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moon RT, Bowerman B, Boutros M, Perrimon N. The promise and perils of Wnt signaling through β-catenin. Science. 2002;296:1644–6. doi: 10.1126/science.1071549. [DOI] [PubMed] [Google Scholar]
- 19.Nelson WJ, Nusse R. Convergence of Wnt, β-catenin, and cadherin pathways. Science. 2004;303:1483–7. doi: 10.1126/science.1094291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, et al. Inactivation of the β-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–64. doi: 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- 21.Cattelino A, Liebner S, Gallini R, Zanetti A, Balconi G, Corsi A, et al. The conditional inactivation of the β-catenin gene in endothelial cells causes a defective vascular pattern and increased vascular fragility. J Cell Biol. 2003;162:1111–22. doi: 10.1083/jcb.200212157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harada N, Tamai Y, Ishikawa T, Sauer B, Takaku K, Oshima M, et al. Intestinal polyposis in mice with a dominant stable mutation of the β-catenin gene. EMBO J. 1999;18:5931–42. doi: 10.1093/emboj/18.21.5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li TF, Darowish M, Zuscik MJ, Chen D, Schwarz EM, Rosier RN, et al. Smad3-deficient chondrocytes have enhanced BMP signaling and accelerated differentiation. J Bone Miner Res. 2006;21:4–16. doi: 10.1359/JBMR.050911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Indra AK, Warot X, Brocard J, Bornert JM, Xiao JH, Chambon P, et al. Temporally-controlled site-specific mutagenesis in the basal layer of the epidermis: comparison of the recombinase activity of the tamoxifen-inducible Cre-ERT and Cre-ERT2 recombinases. Nucleic Acids Res. 1999;27:4324–7. doi: 10.1093/nar/27.22.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen M, Lichtler AC, Sheu TJ, Xie C, Zhang X, O’Keefe RJ, et al. Generation of a transgenic mouse model with chondrocyte-specific and tamoxifen-inducible expression of Cre recombinase. Genesis. 2007;45:44–50. doi: 10.1002/dvg.20261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen J, Li J, Wang B, Jin H, Wang M, Zhang Y, et al. Deletion of the transforming growth factor β receptor type II gene in articular chondrocytes leads to a progressive osteoarthritis-like phenotype in mice. Arthritis Rheum. 2013;65:3107–19. doi: 10.1002/art.38122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu M, Tang D, Wu Q, Hao S, Chen M, Xie C, et al. Activation of β-catenin signaling in articular chondrocytes leads to osteoarthritis-like phenotype in adult β-catenin conditional activation mice. J Bone Miner Res. 2009;24:12–21. doi: 10.1359/JBMR.080901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glass DA, II, Bialek P, Ahn JD, Starbuck M, Patel MS, Clevers H, et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell. 2005;8:751–64. doi: 10.1016/j.devcel.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 29.Andrade AC, Nilsson O, Barnes KM, Baron J. Wnt gene expression in the post-natal growth plate: regulation with chondrocyte differentiation. Bone. 2007;40:1361–9. doi: 10.1016/j.bone.2007.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dao DY, Jonason J, Zhang Y, Hsu W, Chen D, Hilton MJ, et al. Cartilage-specific β-catenin signaling regulates chondrocyte maturation, generation of ossification centers, and perichondrial bone formation during skeletal development. J Bone Miner Res. 2012;27:1680–94. doi: 10.1002/jbmr.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yasuhara R, Ohta Y, Yuasa T, Kondo N, Hoang T, Addya S, et al. Roles of β-catenin signaling in phenotypic expression and proliferation of articular cartilage superficial zone cells. Lab Invest. 2011;91:1739–52. doi: 10.1038/labinvest.2011.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Silvestrini G, Ballanti P, Patacchioli F, Leopizzi M, Gualtieri N, Monnazzi P, et al. Detection of osteoprotegerin (OPG) and its ligand (RANKL) mRNA and protein in femur and tibia of the rat. J Mol Histol. 2005;36:59–67. doi: 10.1007/s10735-004-3839-1. [DOI] [PubMed] [Google Scholar]
- 33.Usui M, Xing L, Drissi H, Zuscik M, O’Keefe R, Chen D, et al. Murine and chicken chondrocytes regulate osteoclastogenesis by producing RANKL in response to BMP2. J Bone Miner Res. 2008;23:314–25. doi: 10.1359/JBMR.071025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Masuyama R, Stockmans I, Torrekens S, Van Looveren R, Maes C, Carmeliet P, et al. Vitamin D receptor in chondrocytes promotes osteoclastogenesis and regulates FGF23 production in osteoblasts. J Clin Invest. 2006;116:3150–9. doi: 10.1172/JCI29463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med. 2011;17:1231–4. doi: 10.1038/nm.2452. [DOI] [PubMed] [Google Scholar]
- 36.Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O’Brien CA. Matrix-embedded cells control osteoclast formation. Nat Med. 2011;17:1235–41. doi: 10.1038/nm.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.