

Abstract
Purpose
To examine the role of survivin as a therapeutic target in preclinical models of human malignant peripheral nerve sheath tumors (MPNSTs)
Experimental Design
Survivin protein expression levels and sub-cellular localization were examined immunohistochemically in a MPNST tissue microarray. Human MPNST cells were studied in vitro and in vivo; RTPCR, WB, and immunocytochemical analyses were used to evaluate survivin expression and localization activation. Cell culture assays were used to evaluate the impact of anti-survivin-specific siRNA inhibition on cell growth, cell cycle progression and survival. The effect of the small molecule survivin inhibitor YM155 on local and metastatic MPNST growth was examined in vivo.
Results
Survivin was found to be highly expressed in human MPNSTs; enhanced cytoplasmic sub-cellular localization differentiated MPNSTs from their plexiform neurofibroma pre-malignant counterparts. Human MPNST cell lines exhibited survivin mRNA and protein over-expression; expression in both nuclear and cytoplasmic compartments was noted. Survivin knockdown abrogated MPNST cell growth, inducing G2 cell cycle arrest and marked apoptosis. YM155 inhibited human MPNST xenograft growth and metastasis in SCID mice. Anti-tumor effects were more pronounced in fast growing xenografts.
Conclusions
Our studies demonstrate an important role for survivin in human MPNST biology. MPNST patients should be considered for ongoing or future clinical trials that evaluate anti-survivin therapeutic strategies. Most importantly, future investigations should evaluate additional pathways that can be targeted in combination with survivin for maximal synergistic anti-MPNST effects.
Keywords: MPNST, Survivin, TMA, YM155, Targeted Therapy, preclinical models
Introduction
Characterized by aggressive local growth, propensity for systemic spread, and marked resistance to conventional chemo- and radiotherapy, malignant peripheral nerve sheath tumors (MPNSTs) cause remarkable morbidity and mortality in afflicted individuals (1, 2). Development of more efficacious therapeutic strategies is critically needed yet requires more comprehensive knowledge of molecular constituents driving MPNST. The tight association between MPNST and neurofibromatosis type 1 (NF1) is suggested by the reality that more than 50% of MPNST develop against the backdrop of this common genetic disorder (2) and that 8-13% of NF1 patients will develop MPNST (2), suggesting a fundamental role for neurofibromin loss of function underlying MPNST inception (3). Deactivating mutations in the NF1 tumor suppressor gene which encodes for the Ras-GTPase neurofibromin protein are the hallmark of NF1; plexiform neurofibromas developing in NF1 patients and MPNSTs arising within these typically deep seated lesions exhibit bi-allelic NF1 inactivation and consequential enhanced Ras pathway signaling. NF1 mutations also are observed in a portion of sporadic, non-NF1-associated MPNSTs as well (3). While neurofibromin loss and/or Ras pathway activation are early molecular events driving the development of MPNST pre-malignant stages, additional genetic and epigenetic alterations are most likely necessary for malignant transformation, disease progression, and metastasis (4). The disappointing results of MPNST clinical trials singularly targeting the Ras pathway or molecules within this signaling cascade (5) highlight the need to further identify additional MPNST-associated molecular aberrations, preferentially those that would be easily amenable to therapeutic targeting.
Originally identified in 1997 as a member of the Inhibitor of Apoptosis (IAP) family (6), survivin, encoded by the BIRC5 gene, has since been found to contribute to a multitude of critical biological functions including cellular division, survival, and adaptation to stress (6-7). While highly expressed during embryogenesis, survivin is largely undetectable in normal adult tissues and is restricted to the thymus, placenta, stem cell compartment, and basal epithelium of the colon (6, 8). However, survivin re-expression is commonly observed in transformed cells, and increased survivin levels have been found in multiple cancer types (9). Moreover, increased survivin expression levels have been found to correlate with adverse patient outcomes and resistance to therapy (10-13). Importantly, survivin has been shown to play a critical role in cancer, functioning as a convergence point for multiple signaling pathways controlling tumor maintenance and growth promotion (8). These attributes have rendered survivin a focus of intense investigation as a potentially worthy target for personalized molecular therapeutics (8). Anti-survivin treatment regimens have recently reached the clinic (http://clinicaltrials.gov/ct2/show/NCT00664677,NCT01186328,NCT01100931,NCT01250470) with encouraging results noted in early human cancer clinical trials (14-15).
While extensively studied in a wide range of human malignancies, a role for survivin deregulation in MPNST biology has yet to be determined. Several previously published reports suggest that survivin is aberrantly expressed in MPNST (16-17). Specifically, Storlazzi et al identified the BIRC5 genetic region on chromosome 17 as commonly amplified in human MPNST samples; high level amplification correlated with poor prognosis (17). Overexpression of survivin mRNA in MPNST compared to neurofibroma has been observed by three independent groups (16, 18, 19). Finally, a recent immunohistochemistry-based study demonstrated survivin protein expression in 52 human MPNST samples (20). Building on these initial observations, the current study sought to further determine the potential role of survivin as a MPNST biomarker, to elucidate the functional consequences of survivin over-expression in these tumors, and, most importantly, to assess the efficacy of survivin blockade as an anti-MPNST therapeutic strategy.
Materials and Methods
Cell-lines and reagents
MPNST cell lines utilized for our studies included the NF1-associated: S462 (provided by Dr Lan Kluwe, University Hospital Eppendorf, Hamburg, Germany), ST88-14 (provided by Dr Jonathan Fletcher, Brigham and Women's Hospital, Boston, MA), the MPNST642 isolated in our laboratory (21), and the sporadic MPNST cell lines: STS26T (provided by Dr Steven Porcelli, Albert Einstein College of Medicine, Bronx, NY) and MPNST724 (provided by Dr Jonathan Fletcher); these were propagated and maintained as previously described (22). Primary human adult Schwann cell cultures established from human cauda equina nerves were provided by Dr Patrick Wood (Miami Project, University of Miami, Miami, FL) and maintained as previously described (23). DNA fingerprinting (short tandem repeat) was conducted as previously described (24) for all MPNST cell lines, confirming that no cross contamination has occurred.
The small molecule survivin inhibitor YM155 was purchased from Chemietek (Indianapolis, IN). For in vitro studies the drug was dissolved in dimethyl sulfoxide (DMSO) and stored in -20°C. For in vivo experiments YM155 (dosed at 6mg/kg/d) was dissolved and diluted in saline immediately prior to administration. Commercially available antibodies were used for immunoblot, immunohistochemical, or immunocytochemical detection of the full length wild type (WT) survivin (polyclonal; Abcam, Cambridge, MA), XIAP (polyclonal; Abcam, Cambridge, MA), cIAP1 (polyclonal; Abcam, Cambridge, MA), cIAP2 (polyclonal; Abcam, Cambridge, MA), the 85 kDa fragment of cleaved PARP (clone Y34; Abcam), total PARP (clone 46D11; Cell Signaling, Danvers, MA), Cleaved caspase- 3 (polyclonal; BioCare Medical, Concord, CA), α-Tubulin (Santa Cruz Biotechnology, Santa Cruz, CA), Ki67 (Thermo/Lab Vision, Fremont, CA), Lamin A/C (Santa Cruz Biotechnology) and β-actin (Santa Cruz Biotechnology). Hoechst (Invitrogen, Carlsbad, CA) was used as a nuclear stain for immuncytochemical analysis.
Immunohistochemistry and immunocytochemistry
A previously reported (25) tissue microarray (TMA) containing specimens retrieved from human MPNST and plexiform neurofibroma surgical resections was used to assess survivin expression. After excluding spots with insufficient material, 63 different patient MPNST samples, 21 plexiform neurofibromas, and two normal peripheral nerve samples were available for analysis. A comprehensive clinical database containing patient, tumor, treatment, and follow up information linked to the TMA has previously been constructed and updated to enable this current analysis. The median follow-up time was 2.5 years (ranging between 3 months to 15.25 years). TMA immunostaining, xenograft-derived specimen immunohistochemistry (IHC), and immunocytochemistry were conducted as previously described (25-27). For TMA analysis survivin expression was scored by two independent observers (AJL and MPHG). Intensity was graded as none (=0), weak/low (=1), moderate (=2), and high (=3); % of positively staining cells was also determined. Scoring was conducted separately for the nuclear and cytoplasmic intracellular compartments.
siRNA (small interfering RNA) transfection procedures
siRNAs (20nM pools targeting survivin and control non-targeting constructs; Thermo Scientific, Waltham, MA) were introduced into cells using X-treme Gene per manufacturer's instructions (Roche, Mannheim, Germany). Briefly, 2×105 cells were plated in each well of a six-well plate and incubated overnight. A mixture of siRNA (20nM) and X-treme Gene (6μl) diluted in 100μl Dulbecco's modified Eagle medium (DMEM) was added for 24 hr, followed by incubation in regular medium. Cells were harvested at indicated time points for specific experiments.
In vitro growth assays
MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H tetrazolium) assays: these were conducted using CellTiter96 Aqueous Non-Radioactive Cell Proliferation Assay kit (Promega Corp, Madison, WI), per manufacturer's instructions. Absorbance was measured at a wavelength of 490nm, and the absorbance values of treated (or of survivin knocked down) cells are presented as a percentage of the absorbance of untreated cells. Colony formation assay: for pretreatment analysis, MPNST cells were treated in culture dishes for 24h with DMSO (control) or YM155. One hundred viable cells per well were re-plated and allowed to grow in normal medium (no drug) for 10 days and then stained for 30 min at room temperature with a 6% glutaraldehyde, 0.5% crystal violet solution. For continuous treatment studies one hundred viable cells per well were plated and treated with YM155 or DMSO alone for 10 consecutive days. Pictures were captured digitally and surface area covered by cells (as surrogate to colony number) was determined using image J software. Anchorage independent growth: MPNST cells were treated with DMSO (control) or YM155 for 24h in a 6-well plate. 1×103 viable cells were plated in a 24-well plate in culture medium containing 0.35% agarose overlying a 0.7% agarose layer. Cells were incubated for 3 weeks at 37°C. Cells were stained with p-iodonitrotetrazolium violet (1mg/ml) for 24 h at 37°.
Cellular assays
Western blot (WB) analyses were performed by standard methods (28). Subcellular (nuclear and cytoplasmic) protein fractionation and isolation was conducted using the Nuclear and Cytoplasmic Extraction Reagents Kit (NE-PER Thermo Scientific, Rockford, IL) per manufacturer's instructions. Cell cycle progression was measured via Propidium Iodide (PI) staining/fluorescence activated cell sorting (FACS) analysis (28) and apoptosis was measured via annexin V/PI staining FACS analysis using the Apoptosis Detection kit I (BD Biosciences) per manufacturers' recommendations. Real time PCR (RTPCR) and quantitative RTPCR (qRTPCR) were conducted as we have previously described (28); primers (survivin forward: 5′-GGACCACCGCATCTCTACAT-3′ and reverse: 5′-GTTCCTCTATGGGGTCGTCA-3′ were obtained from Sigma (Woodlands, TX).
In vivo animal models
All animal procedures and care were approved by the MD Anderson Cancer Center Institutional Animal Care and Usage Committee. Animals received humane care as per the Animal Welfare Act and the NIH “Guide for the Care and Use of Laboratory Animals.” For experiments evaluating effect of treatment on local tumor growth trypan blue staining confirmed viable MPNST cells (STS26T and MPNST724; 1-2 × 106/0.1 mL HBSS/mouse) were used. Cell suspensions were injected subcutaneously (s.c.) into the flank of six week old female hairless SCID mice (n = 7-8/treatment group) and growth was measured twice weekly; after establishment of palpable lesions (average diameter ∼4-5mm) mice were assigned to treatment groups as described below. An experimental lung metastasis MPNST model was used to evaluate growth of metastases. STS26T cells (1 × 106/0.1 mL HBSS/mouse) were injected into the tail vein of female SCID mice. Two weeks after injection mice were allocated to treatment groups as per below. Therapeutic regimen and dose followed previous reports (29, 30): YM155 (6mg/kg/d; several preclinical studies [30, 31] have demonstrated anti-tumor efficacy using doses of 1-10mg/kg/d, with the latter being the maximal tolerated dose) or saline were delivered via a micro-osmotic pump (Alzet® model 1003D, Durect Corporation, Cupertino, CA) that was implanted subcutaneously on day-1 of treatment into each mouse; these pumps enable continuous drug delivery for three consecutive days. Previous studies have demonstrated that this regimen results in steady state levels of YM155 in plasma and enhanced tumor bioavailability (31); YM155 continuous infusion was found superior to intravenous daily bolus or intermittent schedules (31). The pumps were replaced on day 8 for a total of six treatment days. Mice were followed for tumor size, well being, and body weight and sacrificed when control group tumors reached an average of 1.5 cm in their largest dimension. Tumors were resected, weighed, and fixed in formalin and paraffin-embedded for immunohistochemical studies. For lung metastasis studies, mice were followed for body weight and well being and sacrificed after three weeks after start of treatment. Lungs were resected, evaluated macroscopically for tumor load, and weighed and fixed in formalin and paraffin-embedded for H+E staining.
Statistics
Several alternative statistical tests were used to determine the correlation between survivin expression and clinical factors such as histology, NF1-status, and disease-status including Spearman's correlation coefficient, Chi-Square, and Fisher exact test. Correlation between survivin and other molecular biomarkers was evaluated using Spearman's correlation coefficient analyses. Kaplan Meier (KM) analyses were used to determine the potential impact of survivin expression levels on MPNST disease specific survival (DSS). All computations were performed using SAS for Windows (release 9.2; SAS Institute, Cary, NC). Cell culture-based quantitative assays were repeated at least thrice; mean ± SD was calculated. Cell-lines were examined separately. For outcomes that were measured at a single time point, two-sample t-tests were used to assess the differences. Differences in xenograft growth in vivo were assessed using a two-tailed Student's t-test. Significance was set at p≤0.05.
Results
Human MPNST exhibit increased survivin levels; cytoplasmic expression is significantly enhanced in MPNST as compared to plexiform neurofibroma
Survivin over-expression has been previously demonstrated for many cancer types (9). Supported by previous publications suggesting BIRC5 gene amplification and enhanced survivin mRNA expression in MPNST (16-18), we examined survivin protein expression levels and intracellular localization in a cohort of human samples. A previously constructed clinically annotated TMA was used, containing human MPNST and plexiform neurofibroma samples (25). Survivin IHC analysis was conducted (Fig 1A). Both normal peripheral nerves were negative for survivin expression. All MPNSTs demonstrated survivin expression; both nuclear and cytoplasmic distribution was noted (Fig 1A). Weak nuclear expression levels were found in 31 (49%), moderate in 28 (45%), and high in 4 (6%); an average of 73% (±18%) of tumor cells per sample exhibited positive nuclear staining. Cytoplasmic expression levels were low in 3 (5%), moderate in 13 (21%), and high in 47 (74%); an average of 83% (±11%) of tumor cells per sample exhibited positive cytoplasmic staining. Similarly, all plexiform neurofibroma expressed survivin. Low nuclear levels were found in 9 (43%), moderate in 8 (38%), and high in 4 (19%); an average of 52% (±23%) of tumor cells per sample exhibited positive nuclear staining. Cytoplasmic expression levels were low in 8 (38%), moderate in 11 (52%), and high in 2 (10%); an average of 80% (±9%) of tumor cells per sample exhibited positive cytoplasmic staining. Notably, while no difference in nuclear survivin expression was found between MPNST and plexiform neurofibroma derived specimens, a statistically significant enhanced cytoplasmic survivin expression was identified in MPNST (p<0.0001; Spearman's rank test; Fig 1B). No correlation between survivin expression levels and MPNST disease status (i.e., primary, recurrent, or metastatic) or NF1 vs sporadic lesions was identified. KM analysis was further conducted to determine whether survivin expression levels correlate with MPNST disease outcome; only scoring results of evaluable localized MPNST samples (n=48) were included. No correlation was found between either cytoplasmic or nuclear survivin expression levels or MPNST patient disease specific survival. In summary, MPNST and their precursor lesions, plexiform neurofibroma both commonly express survivin; cytoplasmic survivin expression is markedly more pronounced in malignant lesions. In contrast to findings in other malignancies, we did not identify survivin expression to be a molecular prognosticator of MPNST disease outcome.
Figure 1. Survivin is highly expressed in human MPNST specimens; cytoplasmic survivin expression is more pronounced in MPNSTs as compared to plexiform neurofibroma.

A. Upper panel depicts the entire TMA stained for survivin; a range of expression levels can be observed. Representative photographs of survivin stained neurofibroma (left) and MPNST spots (middle and right) are shown in the lower panel. Neurofibroma image was captured in 20×, inset demonstrating normal peripheral nerve negative for survivin. MPNST images captured at 20× demonstrating both nuclear (best elucidated in middle picture; inset picture was taken at 40×, arrow marks nuclear staining) and cytoplasmic (best elucidated in right picture; inset picture was taken at 40×, arrow marks cytoplasmic staining) survivin expression (scale bars are included); B. Heat map representation of nuclear and cytoplasmic survivin expression levels in each of the evaluable TMA spots. Cytoplasmic survivin expression was found to be statistically significantly more pronounced in MPNST as compared to neurofibroma.
Next, we assessed whether there was a correlation (Spearman rank correlation analyses) between survivin expression levels (cytoplasmic and nuclear) and other biomarkers using our MPNST TMA, including Ki67 (as a marker of proliferation), p53 (commonly deregulated in MPNST; 32), VEGF (as a marker of angiogenesis), and Ras pathway signaling effectors including pMEK, pAKT, and the mTOR downstream target pS6RP. No association between Ki67 or p53 intensity and survivin was identified. However, enhanced cytoplasmic survivin intensity was found to directly and statistically significantly correlate with increased VEGF (r=0.28, p=0.013), pMEK (r=0.48, p<0.0001), pAKT (r=0.29, p=0.012), and pS6RP (r=0.33, p=0.007) expression levels. Interestingly, increased nuclear survivin expression was found to inversely correlate with VEGF (r=-0.33, p=0.004) and pAKT (r= -0.32, p=0.008) intensities.
Survivin is highly expressed in human MPNST cell lines
Next, we determined survivin expression levels in a panel of human MPNST cell lines to confirm that our experimental model recapitulated the findings in human samples and thus could be used to further evaluate the potential function of this protein in MPNST (Fig 2A). WT survivin mRNA levels were found to be markedly increased in all MPNST cell lines as compared to normal human Schwann cell (NHSC) and did not correlate with NF1 (S462, ST88, MPNST642) vs. sporadic (STS26T, MPNST724) disease background, cell growth (growth rates: STS26T>S462>MPNST724>ST88>MPNST642), or p53 mutational status (WT = ST88 and MPNST642, mutated/null = MPNST724, S462, and STS26T; 29). Similarly, WT survivin protein expression was enhanced in MPNST cell lines, and no other bands were identified to suggest expression of survivin splice variants. Of note, survivin protein expression and survivin mRNA levels did not entirely match, possibly suggesting that additional post-transcriptional factors may contribute to survivin protein levels, whereas a closer association with MPNST cell growth rate was noted. Finally, as observed in human MPNST specimens, survivin expression was noted in both nuclear and cytoplasmic cellular compartments of MPNST cells (Fig 2B).
Figure 2. Survivin is highly expressed in human MPNST cell lines.

A. qRTPCR (upper panel) and RTPCR (middle panel) demonstrating markedly increased survivin mRNA expression in MPNST cell lines as compared to normal human Schwann cells (NHSC). Western blot (WB) analysis (lower panel) further confirming WT survivin protein over-expression in MPNST cell lines; B. WB analyses and immunocytochemistry (scale bars are included) demonstrating both nuclear and cytoplasmic survivin expression in MPNST cells.
Survivin promotes MPNST cell growth, cell cycle progression, and survival
We next conducted loss of survivin function experiments to determine its contribution to MPNST cell growth. Anti-survivin siRNA specific constructs were used to achieve knockdown; a non-targeting construct was utilized as control. Marked survivin knockdown was noted 48h after transfection (Fig 3A). No decrease in the expression of the IAP proteins XIAP (Fig 3A), cIAP1, or cIAP2 (Fig S1) was noted in response to anti-survivin siRNA transfection. MTS assays demonstrated significant (p<0.05) decreases in tumor cell growth at 24 and 48h (experiments were initiated 24h after transfection). PI staining/FACS analyses demonstrated significant (p<0.05) G2/M cell cycle arrest 48h after transfection (Fig 3B). These assays also demonstrated increased sub-G1 fractions in MPNST cells secondary to survivin knockdown. PI/Annexin V FACS analyses confirmed a statistically significant (p<0.05) increase in apoptosis 72h post siRNA transfection and WB demonstrated increased cleaved PARP (85kDa fragment) expression in survivin knocked down cells (Fig 3C and Fig S2). Taken together, these studies demonstrated a role for survivin in MPNST cell growth, cell cycle progression, and survival, thereby supporting further investigation of anti-survivin therapeutic strategies in our MPNST experimental models.
Figure 3. Survivin knockdown abrogates MPNST cell growth, cell cycle progression, and survival.

A. Anti-survivin siRNA (20nM pool) resulted in pronounced survivin knockdown (KD) in MPNST cell lines 48h post transfection (non targeting [NT-siRNA] constructs were used as control; WB analyses, upper panel). No decrease in the expression of the IAP protein XIAP was noted. Survivin KD significantly inhibited MPNST cell growth. MTS assays were performed for 24 and 48h (48 and 72h post transfection, respectively); B. Survivin KD resulted in a statistically significant G2 cell cycle arrest in MPNST cells (48h post transfection; graphs represent at least three independent experiments). Increased sub-G1 fraction was also noted; C. Survivin KD induced marked apoptosis in MPNST cells (assays were conducted 72h post siRNA transfection) as is demonstrated via Annexin-V/PI staining FACS analyses (graphs represent at least three independent experiments) and increased cleaved PARP expression (using an antibody directed against the 85kDa fraction of PARP; WB analyses further demonstrate marked survivin KD at this time point). [* denotes statistically significant effects (p<0.05)]
YM155, a small molecule survivin inhibitor, exerts marked anti-MPNST effects in vitro and in vivo
The anti-MPNST effects of YM155, a small molecule inhibitor of survivin currently under clinical investigation, were next considered. YM155 was identified using a cell based chemical library screen to specifically inhibit survivin promoter activity (31). It is suggested to exert its anti-tumor effects through inhibition of survivin mRNA transcription resulting in decreased protein expression while not impacting the expression of other IAP proteins such as cIAP1, cIAP2, or XIAP (31). WB analyses confirmed that YM155 induced a dose- (in the nM range) and treatment time-dependent decrease in survivin expression in MPNST cell lines (Fig 4A). No decrease in the expression of the IAP proteins, XIAP and cIAP1 was observed in response to YM155 (Fig 4A). MTS assays likewise demonstrated a marked YM155 dose-dependent decrease in MPNST cell growth after 48 and 96h of treatment (Fig 4B). YM155 effects were found to correlate with cell line growth rates. Of note, a less pronounced effect was demonstrated in MPNST642 cells; among the human MPNST cell lines tested, this specific cell line exhibited the slowest growth rate and lowest survivin expression level. Based on these initial results, YM155 doses used for the remaining cell culture based assays ranged between 1-10nM; these doses are lower than the YM155 plasma levels achievable in humans (33, 34), hence clinically relevant. YM155 administration significantly (p<0.05) abrogated the colony forming capacity and anchorage independent growth of MPNST cells (Fig 4C&D). Compatible with the effects observed above in response to survivin knockdown, YM155 treatment resulted in abrogated G2/M cell cycle progression (p<0.05) and increased sub-G1 fraction in MPNST cells (Fig 5A). Finally, a statistically significant (p<0.05) increase in tumor cell apoptosis was noted in response to YM155 (evaluated after 48h of treatment; Fig 5B) and an increase in the expression of cleaved PARP (85kDa fraction) was observed (Fig 5B and Fig S2).
Figure 4. YM155, a small molecule survivin inhibitor, inhibits MPNST cell growth in vitro.

A. WB analyses confirm that YM155 induces a dose- (in the nM range) and treatment time-dependent decrease in survivin expression in MPNST cell lines. No decrease in the expression of the IAP proteins XIAP or cIAP1 was observed in response to YM155; B. MTS assays demonstrating a marked YM155 dose-dependent decrease in MPNST cell growth after 48 and 96h of treatment. A correlation between YM155 sensitivity and cell line growth rate was observed. Of note, a less pronounced effect was demonstrated in MPNST642 cells -- amongst the human MPNST cell lines tested, this cell line exhibits the slowest growth and lowest survivin expression; C. YM155 (pretreatment and continuous treatment) significantly inhibits the colony forming capacity of MPNST cells (graphs represent at least three independent experiments); D. YM155 significantly inhibits the anchorage independent growth of MPNST cells (graphs represent at least three independent experiments). [* denotes statistically significant effects (p<0.05)]
Figure 5. YM155 inhibits MPNST cell cycle progression and survival in vitro.

A. YM155 induces a statistically significant G2 cell cycle arrest and increases in sub-G1 fraction in MPNST cells (graphs represent at least three independent experiments); B. Moreover, YM155 (48h of treatment) exerts a statistically significant increase in tumor cell apoptosis (graphs represent at least three independent experiments). Increased cleaved PARP (85kDa fraction) expression can also be seen. [* denotes statistically significant effects (p<0.05)]
To determine whether these in vitro observations might be recapitulated in vivo, we conducted a series of therapeutic experiments using human MPNST xenograft mouse models. First, we examined the effect of YM155 on STS26T growth (Fig 6A). YM155 (6mg/kg/d) therapy was initiated after establishment of tumor (∼5mm in largest dimension). Therapy was administered using subcutaneously implanted micro-osmotic pumps delivering a three-day continuous infusion; pumps were implanted on day 1 and day 8 of treatment as per previously published reports (29, 30). Control mice were treated with saline (carrier) equivalently delivered. YM155 was well tolerated and no significant weight loss was observed. YM155 treatment markedly abrogated tumor growth; the average size of control-treated tumors at study termination was 1109mm3 (±167) vs. 251mm3 (±134) of YM155-treated tumors (p<0.0001; Fig 6A). Moreover, treatment with YM155 significantly reduced tumor weight compared to control (p=0.005); average tumor weights at study termination were 0.82g (±0.18) and 0.36g (±0.19) in control and YM155 groups, respectively (Fig 6A). Tumor sections from each experimental arm containing viable cells were selected for IHC studies. A marked decrease in survivin expression was observed in YM155-treated tumors (Fig 6A). Furthermore, a pronounced decrease in MPNST cell proliferation (evaluated via Ki67 staining) and a demonstrable increase in tumor cell apoptosis (evaluated using cleaved caspase 3 IHC) were also noted.
Figure 6. YM155 exerts marked anti-MPNST effects in vivo.
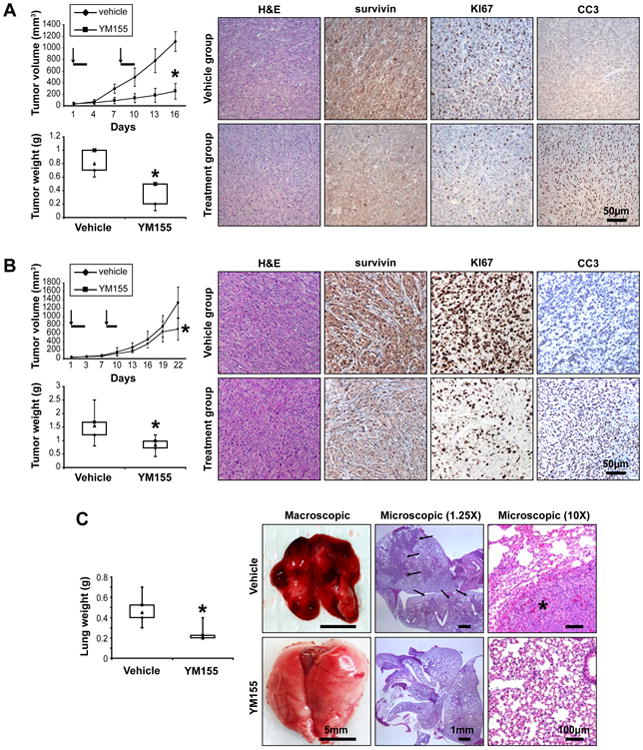
A. SCID mice bearing STS26T xenografts (∼4-5mm in average larger dimension) were implanted with a subcutaneous micro-osmotic pump delivering YM155 (6mg/kg/d) or vehicle only in a continuous fashion for three consecutive days. Pumps were replaced once (on day 8; arrows denote days of pump implantation and bolded line days of treatment). YM155 markedly abrogated tumor growth (p<0.0001; upper graph). Moreover, treatment with YM155 significantly reduced tumor weight compared to control (p=0.005; lower graph). IHC analyses demonstrated decreased survivin and Ki67 and increased cleaved caspase 3 (CC3) expression in YM155 treated xenografts (scale bars are included); B. Experiment was repeated as per above for MPNST724 xenografts: YM155 treatment was found to delay tumor growth, a statistically significant decrease in tumor size (p<0.01; upper graph) and tumor weight (p<0.05; lower graph) was observed at study termination. IHC analyses results (right panel) aligned with the STS26T findings above (scale bars are included); D. STS26T lung metastases bearing mice were treated with YM155. A significant (p<0.001; right panel) difference in average lung weight between control and YM155 treated mice was found at study termination. Pulmonary metastases were macroscopically observed in all control mice but not in YM155 treated mice. H+E staining further demonstrated large lung tumor deposits in control mice lungs but no (n=6) or only small microscopic lesions (n=2) in the YM155 treated group (scale bars are included). [* denotes statistically significant effects (p<0.05)]
We next evaluated the effects of YM155 in a second MPNST xenograft model derived from s.c. injection of MPNST 724 cells; YM155 doses and regimens were as per above. YM155-treated mice exhibited smaller tumors at study termination: the average size and weight of YM155-treated tumors were 702mm3 (±257) and 0.83g (±0.27) as compared to 1335mm3 (±362) and 1.53g (±0.58) for the control group (Fig 6B). While statistically significant (p<0.05), the effects of YM155 on MPNST724 xenografts were less profound as compared to its in vivo anti-STS26T effects, and are probably more consistent with cytostasis than cytolysis per se. This difference might be related to the inherently slower growth of MPNST724 xenografts as compared to STS26T which was also reflected in the lower baseline Ki67 expression in these tumors. Similar to the IHC results obtained from the STS26T therapeutic experiments, a decrease in survivin and Ki67 and an increase in cleaved caspase 3 expression was found in MPNST 724 YM155-treated xenografts (Fig 6B).
Finally, we utilized the STS26T experimental lung metastasis model to evaluate whether YM155 can affect the growth of MPNST pulmonary metastases. Pumps were implanted two weeks after tumor cell tail-vein injection and then replaced one time seven days later. Mice were sacrificed three weeks after treatment initiation. A significant difference in average lung weight was found between control (0.47g ± 0.13) and treated mice (0.24g ± 0.07, p=0.0004; Fig 6C). Macroscopic lung metastasis were observed in all (n=8) control mice but not in YM155-treated mice (n=8; Fig 6C). H+E staining identified large lung tumor deposits in all control mice whereas only two of the YM155-treated mouse lungs exhibited microscopic lesions (Fig 6C).
Discussion
Driven by the critical need to identify MPNST molecular deregulations amenable to therapeutic targeting, studies here focused on the multifunctional protein survivin. More than a decade of intensive research has illuminated the fundamental role survivin plays across a broad range of cancer histologies (9). Multiple molecular mechanisms have been identified that drive aberrant survivin expression in cancer, including amplification of the BIRC5 genetic locus (on chromosome 17q25), BIRC5 demethylation, enhanced transcription, and deregulated cellular signaling pathways for which survivin acts as a convergence point (35-37). While not previously extensively studied in MPNST, published data suggest that BIRC5 amplification is a common molecular event in these devastating malignancies (17).
The current study further expands these initial observations, demonstrating enhanced survivin protein expression in human MPNST specimens. Studies presented here did not identify survivin expression as a predictor of MPNST patient disease specific survival. This is in contrast to observations made in many other solid malignancies including non–small cell lung, gastric cancer, colorectal cancer, breast carcinomas, neuroblastoma, and osteosarcoma where survivin expression levels were found to correlate with poor patient outcome (10-13). However, our negative results are potentially confounded by the relatively small number of evaluable localized MPNST samples and should perhaps be re-examined using a larger cohort of specimens.
In alignment with its diverse molecular functions, survivin has been shown a dynamic intracellular expression pattern, localizing to both the cytoplasm and the nucleus of tumor cells (38). Interestingly, nuclear survivin has been shown to be a predictor of favorable outcome in non-small cell lung cancer and osteosarcoma (18, 39), while portending a poor prognosis in mantle cell lymphoma and squamous cell carcinomas of the esophagus (39). Enhanced cytoplasmic survivin expression has been shown to correlate with shorter disease free survival in patients with oral squamous cell carcinoma (40). Possibly related to the above observations, it has been previously suggested that the anti-apoptotic effects of survivin are greatest when the protein is localized in the cytoplasm, whereas nuclear survivin may play a more critical role in mitosis (41). Together, it is clear that the prognostic and functional significance of these distinct collections of sub-cellular survivin are yet to be fully appreciated and delineated (15). In our study we noted survivin expression in both the nuclear and cytoplasmic cellular compartments of MPNST. Interestingly, while equivalent nuclear survivin expression levels could be observed when comparing MPNSTs to their pre-malignant plexiform neurofibroma precursors, cytoplasmic expression levels were markedly increased in the malignant lesions. It is well established that progression of neurofibromas to MPNST mandates accumulation of genetic and epigenetic alterations that drive transformation and subsequent tumor progression (4); our findings possibly justify evaluating the role of cytoplasmic survivin in this process. Of note, PI3K/AKT/mTOR and MEK/ERK signaling have previously been identified to regulate survivin expression (42). Deregulation of these pathways is commonly observed in MPNST (43); published data from our laboratory has identified enhanced AKT/mTOR and MEK activation in human MPNST samples compared to plexiform neurofibroma tissues (25). Moreover, in the current study we found a direct correlation between cytoplasmic survivin expression levels and those of the activated forms of MEK, AKT, and S6RP. These results offer at least one possible molecular mechanism underlying enhanced cytoplasmic survivin expression in MPNST.
In addition to aberrant survivin expression in human MPNST, our data further suggest that survivin plays an important role in the MPNST pro-tumorigenic phenotype enhancing tumor cell growth, cell cycle progression, and survival. These survivin-induced effects are not unique to MPNST and are similar to those previously demonstrated in other solid and hematological malignancies (6, 31, 36, 42). As such, our study expands previous pre-clinical observations to include the devastating malignancy MPNST as a cancer type in which survivin might play a role and further highlights the potential utility of this protein as an attractive and possibly universal target for molecularly-based anti-cancer therapies. A wide variety of anti-survivin therapeutic strategies, including antisense oligonucleotides, ribozymes, siRNA-based approaches, immunotherapy, and small molecular weight inhibitors are currently in various stages of development (8). YM155 is one such extensively tested inhibitor and has already received clinical attention (15). An imidazolium-based compound originally identified by Nakahara et al (31) through a high-throughput compound screen to specifically inhibit survivin transcription, the exact mechanisms of YM155 action are yet to be elucidated. This compound induces marked anti-tumor effects in multiple preclinical cancer models including prostate cancer, non-small cell lung cancer, melanoma, and non-Hodgkin lymphoma (31, 44, 45). These encouraging results have led to the development of initial phase I/II YM155 human clinical studies which have demonstrated favorable toxicity and tolerability profiles (33, 34, 46). Furthermore, signs of efficacy, albeit modest, have been noted including partial responses in non-Hodgkin's lymphoma and in heavily pre-treated refractory non-small cell lung carcinoma patients. Our studies confirm that human MPNST cells are highly sensitive to survivin knockdown, resulting in marked cell cycle arrest and apoptosis. Seeking to establish translational applicability, we examined the potential efficacy of YM155 in our preclinical model. These studies demonstrated promising YM155-induced anti-MPNST effects in vitro where a dose dependent decrease in survivin expression, tumor cell growth, cell cycle progression and survival were observed at low nanomolar concentrations. Furthermore, YM155 inhibited the local and metastatic growth of human MPNST xenografts in SCID mice; an especially pronounced effect was noted in the STS26T rapid growth in vivo model. While these results are very encouraging (and possibly support the inclusion of MPNST patients in anti-survivin based clinical trials), it is of note that the YM155-induced complete regression reported in other tumor models using similar (or even lower) drug dose and regimen (44, 45) was not observed in our MPNST xenografts. In that these latter observations may more closely resemble the effects observed in YM155 monotherapy clinical trials, a more efficacious therapeutic strategy will mandate combining anti-survivin strategies with other MPNST-relevant conventional and/or molecularly-targeted agents. Clinical studies combining YM155 with conventional chemotherapies (e.g. taxanes, carboplatinum) or biological agents (e.g. rituximab) for the treatment of several cancer types are currently ongoing (ClinicalTrials.gov), buttressed by recent evidence for enhanced anti-tumor activity of such combinations in preclinical models (47, 48). Future preclinical studies combining YM155 with agents previously showing promising effects in MPNST such as tyrosine kinase inhibitors (e.g. MET; 49) or inhibitors of cellular signaling (e.g. AKT/mTOR, MEK; 25, 50) and others are currently being planned.
Supplementary Material
Statement of translational relevance.
Malignant peripheral nerve sheath tumors (MPNSTs) are characterized by aggressive local growth, propensity for systemic spread, and marked resistance to conventional chemo- and radiotherapy. Consequently, afflicted patients have guarded prognoses and desperately need efficacious treatments. This past decade has witnessed remarkable interests in novel, molecularly targeted therapeutic regimens, ultimately mandating comprehensive knowledge of molecular deregulations driving tumor progression. Studies reported here demonstrate a potential role for the protein survivin in MPNST biology. Survivin was found to be highly expressed in human MPNSTs; enhanced cytoplasmic sub-cellular localization differentiated MPNSTs from their pre-malignant plexiform neurofibroma counterparts. Survivin knockdown abrogated MPNST cell growth, inducing G2 cell cycle arrest and apoptosis. Most importantly, YM155, a small molecule survivin inhibitor, significantly reduced local and metastatic human MPNST xenograft growth. Taken together, these data offer new insights into MPNST molecular deregulations and support further development of anti-survivin-based therapeutic strategies for MPNST patients.
Acknowledgments
We appreciate the expert assistance in Figure preparation provided by Ms. Kim Vu. This manuscript was supported in part by a NIH/NCI RO1CA138345 (to DL), an Amschwand Foundation Seed Grant (to DL), a Deutsche Forschungsgemeinschaft Fellowship grant (supporting MPG), an MD Anderson Physician-Scientist Grant (to AJL), and a NIH/NCI 5T32CA009599-21 training grant (supporting KL). MDACC cell line characterization Core Facility was further supported by an NCI Cancer Center Support Grant (CA#16672).
Funding Support: This manuscript was supported in part by a NIH/NCI RO1CA138345 (to DL), an Amschwand Foundation Seed Grant (to DL), a Deutsche Forschungsgemeinschaft Fellowship grant (supporting MPHG), and a NIH/NCI 5T32CA009599-21 training grant (supporting KL)
Footnotes
Conflicts of Interest: None
References
- 1.Anghileri M, Miceli R, Fiore M, Mariani L, Ferrari A, Mussi C, et al. Malignant peripheral nerve sheath tumors: prognostic factors and survival in a series of patients treated at a single institution. Cancer. 2006;107:1065–74. doi: 10.1002/cncr.22098. [DOI] [PubMed] [Google Scholar]
- 2.Ducatman BS, Scheithauer BW, Piepgras DG, Reiman HM, Ilstrup DM. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer. 1986;57:2006–21. doi: 10.1002/1097-0142(19860515)57:10<2006::aid-cncr2820571022>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 3.Lau N, Feldkamp MM, Roncari L, Loehr AH, Shannon P, Gutmann DH, et al. Loss of neurofibromin is associated with activation of RAS/MAPK and PI3-K/AKT signaling in a neurofibromatosis 1 astrocytoma. J Neuropathol Exp Neurol. 2000;59:759–67. doi: 10.1093/jnen/59.9.759. [DOI] [PubMed] [Google Scholar]
- 4.Mawrin C, Kirches E, Boltze C, Dietzmann K, Roessner A, Schneider-Stock R. Immunohistochemical and molecular analysis of p53, RB, and PTEN in malignant peripheral nerve sheath tumors. Virchows Arch. 2002;440:610–5. doi: 10.1007/s00428-001-0550-4. [DOI] [PubMed] [Google Scholar]
- 5.Maki RG, D'Adamo DR, Keohan ML, Saulle M, Schuetze SM, Undevia SD, et al. Phase II study of sorafenib in patients with metastatic or recurrent sarcomas. J Clin Oncol. 2009;27:3133–40. doi: 10.1200/JCO.2008.20.4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;3:917–21. doi: 10.1038/nm0897-917. [DOI] [PubMed] [Google Scholar]
- 7.Li F, Ambrosini G, Chu EY, Plescia J, Tognin S, Marchisio PC, et al. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature. 1998;396:580–4. doi: 10.1038/25141. [DOI] [PubMed] [Google Scholar]
- 8.Altieri DC. Survivin, cancer networks and pathway-directed drug discovery. Nat Rev Cancer. 2008;8:61–70. doi: 10.1038/nrc2293. [DOI] [PubMed] [Google Scholar]
- 9.Velculescu VE, Madden SL, Zhang L, Lash AE, Yu J, Rago C, et al. Analysis of human transcriptomes. Nat Genet. 1999;23:387–8. doi: 10.1038/70487. [DOI] [PubMed] [Google Scholar]
- 10.Kawasaki H, Altieri DC, Lu CD, Toyoda M, Tenjo T, Tanigawa N. Inhibition of apoptosis by survivin predicts shorter survival rates in colorectal cancer. Cancer Res. 1998;58:5071–4. [PubMed] [Google Scholar]
- 11.Adida C, Haioun C, Gaulard P, Lepage E, Morel P, Briere J, et al. Prognostic significance of survivin expression in diffuse large B-cell lymphomas. Blood. 2000;96:1921–5. [PubMed] [Google Scholar]
- 12.Islam A, Kageyama H, Takada N, Kawamoto T, Takayasu H, Isogai E, et al. High expression of Survivin, mapped to 17q25, is significantly associated with poor prognostic factors and promotes cell survival in human neuroblastoma. Oncogene. 2000;19:617–23. doi: 10.1038/sj.onc.1203358. [DOI] [PubMed] [Google Scholar]
- 13.Vischioni B, van der Valk P, Span SW, Kruyt FA, Rodriguez JA, Giaccone G. Nuclear localization of survivin is a positive prognostic factor for survival in advanced non-small-cell lung cancer. Ann Oncol. 2004;15:1654–60. doi: 10.1093/annonc/mdh436. [DOI] [PubMed] [Google Scholar]
- 14.Khanna N, Dalby R, Tan M, Arnold S, Stern J, Frazer N. Phase I/II clinical safety studies of terameprocol vaginal ointment. Gynecol Oncol. 2007;107:554–62. doi: 10.1016/j.ygyno.2007.08.074. [DOI] [PubMed] [Google Scholar]
- 15.Pennati M, Folini M, Zaffaroni N. Targeting survivin in cancer therapy: fulfilled promises and open questions. Carcinogenesis. 2007;28:1133–9. doi: 10.1093/carcin/bgm047. [DOI] [PubMed] [Google Scholar]
- 16.Karube K, Nabeshima K, Ishiguro M, Harada M, Iwasaki H. cDNA microarray analysis of cancer associated gene expression profiles in malignant peripheral nerve sheath tumours. J Clin Pathol. 2006;59:160–5. doi: 10.1136/jcp.2004.023598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Storlazzi CT, Brekke HR, Mandahl N, Brosjö O, Smeland S, Lothe RA, et al. Identification of a novel amplicon at distal 17q containing the BIRC5/SURVIVIN gene in malignant peripheral nerve sheath tumours. J Pathol. 2006;209:492–500. doi: 10.1002/path.1998. [DOI] [PubMed] [Google Scholar]
- 18.Lévy P, Vidaud D, Leroy K, Laurendeau I, Wechsler J, Bolasco G, et al. Molecular profiling of malignant peripheral nerve sheath tumors associated with neurofibromatosis type 1, based on large-scale real-time RT-PCR. Mol Cancer. 2004;3:20. doi: 10.1186/1476-4598-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Katoh M, Wilmotte R, Belkouch MC, de Tribolet N, Pizzolato G, Dietrich PY. Survivin in brain tumors: an attractive target for immunotherapy. J Neurooncol. 2003;64:71–6. doi: 10.1007/BF02700022. [DOI] [PubMed] [Google Scholar]
- 20.Tabone-Eglinger S, Bahleda R, Côté JF, Terrier P, Vidaud D, Cayre A, et al. Frequent EGFR Positivity and Overexpression in High-Grade Areas of Human MPNSTs. Sarcoma. 2008;2008:849156. doi: 10.1155/2008/849156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lopez G, Torres K, Liu J, Hernandez B, Young E, Belousov R, et al. Autophagic survival in resistance to histone deacetylase inhibitors: novel strategies to treat malignant peripheral nerve sheath tumors. Cancer Res. 2011;71:185–96. doi: 10.1158/0008-5472.CAN-10-2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller SJ, Rangwala F, Williams J, Ackerman P, Kong S, Jegga AG, et al. Large-scale molecular comparison of human schwann cells to malignant peripheral nerve sheath tumor cell lines and tissues. Cancer Res. 2006;66:2584–91. doi: 10.1158/0008-5472.CAN-05-3330. [DOI] [PubMed] [Google Scholar]
- 23.Casella GT, Wieser R, Bunge RP, Margitich IS, Katz J, Olson L, et al. Density dependent regulation of human Schwann cell proliferation. Glia. 2000;30:165–77. doi: 10.1002/(sici)1098-1136(200004)30:2<165::aid-glia6>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 24.Lopez G, Torres K, Liu J, Hernandez B, Young E, Belousov R, et al. Autophagic survival in resistance to histone deacetylase inhibitors: novel strategies to treat malignant peripheral nerve sheath tumors. Cancer Res. 2011;71:185–96. doi: 10.1158/0008-5472.CAN-10-2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zou CY, Smith KD, Zhu QS, Liu J, McCutcheon IE, Slopis JM, et al. Dual targeting of AKT and mammalian target of rapamycin: a potential therapeutic approach for malignant peripheral nerve sheath tumor. Mol Cancer Ther. 2009;8:1157–68. doi: 10.1158/1535-7163.MCT-08-1008. [DOI] [PubMed] [Google Scholar]
- 26.Zou C, Smith KD, Liu J, Lahat G, Myers S, Wang WL, et al. Clinical, pathological, and molecular variables predictive of malignant peripheral nerve sheath tumor outcome. Ann Surg. 2009;249:1014–22. doi: 10.1097/SLA.0b013e3181a77e9a. [DOI] [PubMed] [Google Scholar]
- 27.Zhu QS, Rosenblatt K, Huang KL, Lahat G, Brobey R, Bolshakov S, et al. Vimentin is a novel AKT1 target mediating motility and invasion. Oncogene. 2011;30:457–70. doi: 10.1038/onc.2010.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jin Z, Lahat G, Korchin B, Nguyen T, Zhu QS, Wang X, et al. Midkine enhances soft-tissue sarcoma growth: a possible novel therapeutic target. Clin Cancer Res. 2008;14:5033–42. doi: 10.1158/1078-0432.CCR-08-0092. [DOI] [PubMed] [Google Scholar]
- 29.Iwasa T, Okamoto I, Takezawa K, Yamanaka K, Nakahara T, Kita A, et al. Marked anti-tumour activity of the combination of YM155, a novel survivin suppressant, and platinum-based drugs. Br J Cancer. 2010;103:36–42. doi: 10.1038/sj.bjc.6605713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakahara T, Kita A, Yamanaka K, Mori M, Amino N, Takeuchi M, et al. Broad spectrum and potent antitumor activities of YM155, a novel small-molecule survivin suppressant, in a wide variety of human cancer cell lines and xenograft models. Cancer Sci. 2011;102:614–21. doi: 10.1111/j.1349-7006.2010.01834.x. [DOI] [PubMed] [Google Scholar]
- 31.Nakahara T, Takeuchi M, Kinoyama I, Minematsu T, Shirasuna K, Matsuhisa A, et al. YM155, a novel small-molecule survivin suppressant, induces regression of established human hormone-refractory prostate tumor xenografts. Cancer Res. 2007;67:8014–21. doi: 10.1158/0008-5472.CAN-07-1343. [DOI] [PubMed] [Google Scholar]
- 32.Menon AG, Anderson KM, Riccardi VM, Chung RY, Whaley JM, Yandell DW, et al. Chromosome 17p deletions and p53 gene mutations associated with the formation of malignant neurofibrosarcomas in von Recklinghausen neurofibromatosis. Proc Natl Acad Sci U S A. 1990;87:5435–9. doi: 10.1073/pnas.87.14.5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giaccone G, Zatloukal P, Roubec J, Floor K, Musil J, Kuta M, et al. Multicenter phase II trial of YM155, a small-molecule suppressor of survivin, in patients with advanced, refractory, non-small-cell lung cancer. J Clin Oncol. 2009;27:4481–6. doi: 10.1200/JCO.2008.21.1862. [DOI] [PubMed] [Google Scholar]
- 34.Tolcher AW, Quinn DI, Ferrari A, Ahmann F, Giaccone G, Drake T, et al. A phase II study of YM155, a novel small-molecule suppressor of survivin, in castration-resistant taxane-pretreated prostate cancer. Ann Oncol. 2011 Aug 22; doi: 10.1093/annonc/mdr353. [DOI] [PubMed] [Google Scholar]
- 35.Lens SM, Rodriguez JA, Vader G, Span SW, Giaccone G, Medema RH. Uncoupling the central spindle-associated function of the chromosomal passenger complex from its role at centromeres. Mol Biol Cell. 2006;17:1897–909. doi: 10.1091/mbc.E05-08-0727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aoki Y, Feldman GM, Tosato G. Inhibition of STAT3 signaling induces apoptosis and decreases survivin expression in primary effusion lymphoma. Blood. 2003;101:1535–42. doi: 10.1182/blood-2002-07-2130. [DOI] [PubMed] [Google Scholar]
- 37.Vaira V, Lee CW, Goel HL, Bosari S, Languino LR, Altieri DC. Regulation of survivin expression by IGF-1/mTOR signaling. Oncogene. 2007;26:2678–84. doi: 10.1038/sj.onc.1210094. [DOI] [PubMed] [Google Scholar]
- 38.Fortugno P, Wall NR, Giodini A, O'Connor DS, Plescia J, Padgett KM, et al. Survivin exists in immunochemically distinct subcellular pools and is involved in spindle microtubule function. J Cell Sci. 2002;115:575–85. doi: 10.1242/jcs.115.3.575. [DOI] [PubMed] [Google Scholar]
- 39.Li F, Yang J, Ramnath N, Javle MM, Tan D. Nuclear or cytoplasmic expression of survivin: What is the significance? Int J Cancer. 2005;114:509–512. doi: 10.1002/ijc.20768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Engels K, Knauer SK, Metzler D, Simf C, Struschka O, Bier C, et al. Dynamic intracellular survivin in oral squamous cell carcinoma: underlying molecular mechanism and potential as an early prognostic marker. J Pathol. 2007;211:532–540. doi: 10.1002/path.2134. [DOI] [PubMed] [Google Scholar]
- 41.Stauber RH, Mann W, Knauer SK. Nuclear and Cytoplasmic Survivin: Molecular Mechanism, Prognostic, and Therapeutic Potential. Cancer Res. 2007;67:5999–6002. doi: 10.1158/0008-5472.CAN-07-0494. [DOI] [PubMed] [Google Scholar]
- 42.Asanuma H, Torigoe T, Kamiguchi K, Hirohashi Y, Ohmura T, Hirata K, et al. Survivin expression is regulated by coexpression of human epidermal growth factor receptor 2 and epidermal growth factor receptor via phosphatidylinositol 3-kinase/AKT signaling pathway in breast cancer cells. Cancer Res. 2005;65:11018–25. doi: 10.1158/0008-5472.CAN-05-0491. [DOI] [PubMed] [Google Scholar]
- 43.Dasgupta B, Yi Y, Chen DY, Weber JD, Gutmann DH. Proteomic analysis reveals hyperactivation of the mammalian target of rapamycin pathway in neurofibromatosis 1-associated human and mouse brain tumors. Cancer Res. 2005;65:2755–60. doi: 10.1158/0008-5472.CAN-04-4058. [DOI] [PubMed] [Google Scholar]
- 44.Iwasa T, Okamoto I, Suzuki M, Nakahara T, Yamanaka K, Hatashita E, et al. Radiosensitizing effect of YM155, a novel small-molecule survivin suppressant, in non-small cell lung cancer cell lines. Clin Cancer Res. 2008;14:6496–504. doi: 10.1158/1078-0432.CCR-08-0468. [DOI] [PubMed] [Google Scholar]
- 45.Kita A, Nakahara T, Yamanaka K, Nakano K, Nakata M, Mori M, et al. Antitumor effects of YM155, a novel survivin suppressant, against human aggressive non-Hodgkin lymphoma. Leuk Res. 2011;35:787–92. doi: 10.1016/j.leukres.2010.11.016. [DOI] [PubMed] [Google Scholar]
- 46.Lewis KD, Samlowski W, Ward J, Catlett J, Cranmer L, Kirkwood J, et al. A multi-center phase II evaluation of the small molecule survivin suppressor YM155 in patients with unresectable stage III or IV melanoma. Invest New Drugs. 2011;29:161–6. doi: 10.1007/s10637-009-9333-6. [DOI] [PubMed] [Google Scholar]
- 47.Yamanaka K, Nakahara T, Yamauchi T, Kita A, Takeuchi M, Kiyonaga F, et al. Antitumor activity of YM155, a selective small-molecule survivin suppressant, alone and in combination with docetaxel in human malignant melanoma models. Clin Cancer Res. 2011;17:5423–31. doi: 10.1158/1078-0432.CCR-10-3410. [DOI] [PubMed] [Google Scholar]
- 48.Nakahara T, Yamanaka K, Hatakeyama S, Kita A, Takeuchi M, Kinoyama I, et al. YM155, a novel survivin suppressant, enhances taxane-induced apoptosis and tumor regression in a human Calu 6 lung cancer xenograft model. Anticancer Drugs. 2011;22:454–62. doi: 10.1097/CAD.0b013e328344ac68. [DOI] [PubMed] [Google Scholar]
- 49.Torres KE, Zhu QS, Bill K, Lopez G, Ghadimi MP, Xie X, et al. Activated MET is a molecular prognosticator and potential therapeutic target for malignant peripheral nerve sheath tumors. Clin Cancer Res. 2011 Jun 15;17(12):3943–55. doi: 10.1158/1078-0432.CCR-11-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ambrosini G, Cheema HS, Seelman S, Teed A, Sambol EB, Singer S, Schwartz GK. Sorafenib inhibits growth and mitogen-activated protein kinase signaling in malignant peripheral nerve sheath cells. Mol Cancer Ther. 2008 Apr;7(4):890–6. doi: 10.1158/1535-7163.MCT-07-0518. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.