

Abstract
Objective
Adverse drug reaction (ADR) can have dire consequences. However, our current understanding of the causes of drug-induced toxicity is still limited. Hence it is of paramount importance to determine molecular factors of adverse drug responses so that safer therapies can be designed.
Methods
We propose a causality analysis model based on structure learning (CASTLE) for identifying factors that contribute significantly to ADRs from an integration of chemical and biological properties of drugs. This study aims to address two major limitations of the existing ADR prediction studies. First, ADR prediction is mostly performed by assessing the correlations between the input features and ADRs, and the identified associations may not indicate causal relations. Second, most predictive models lack biological interpretability.
Results
CASTLE was evaluated in terms of prediction accuracy on 12 organ-specific ADRs using 830 approved drugs. The prediction was carried out by first extracting causal features with structure learning and then applying them to a support vector machine (SVM) for classification. Through rigorous experimental analyses, we observed significant increases in both macro and micro F1 scores compared with the traditional SVM classifier, from 0.88 to 0.89 and 0.74 to 0.81, respectively. Most importantly, identified links between the biological factors and organ-specific drug toxicities were partially supported by evidence in Online Mendelian Inheritance in Man.
Conclusions
The proposed CASTLE model not only performed better in prediction than the baseline SVM but also produced more interpretable results (ie, biological factors responsible for ADRs), which is critical to discovering molecular activators of ADRs.
Introduction
The percentage of Americans consuming prescription medications is continuously rising because of the aging population and improved medication coverage. However, each medication intake is necessarily associated with side effects. Some side effects are minor, but many have dangerous consequences leading to patient morbidity, hospitalization, and other permanent or life-threatening conditions. Adverse drug reactions (ADRs) have become a major health problem, estimated to account for more than two million hospitalization incidents per year and more than 100 000 deaths in the USA annually.1 2
Although every new drug undergoes extensive safety screening before market approval, it is often difficult to characterize ADRs because of a number of limitations related to restrictive patient sampling in premarketing trials, with only a few expected adverse events included in the trials, usually for a short period of surveillance. The etiology of drug-induced adverse reactions is multifactorial. Our current understanding is that individual genetics are a major factor; therefore, much pharmacogenomic effort has been devoted to relating ADRs to genetic biomarkers.3–6 In a recent study, Pauwels et al7 suggested that ADRs may be modulated both by their chemical structures and their gene targets. Despite significant progress in identifying the causes of ADRs, the process remains challenging and our knowledge of causal factors contributing to ADRs is still limited.1 2 8–10
In this study, we propose a novel causality analysis model based on structure learning, called CASTLE, to discover molecular factors of ADRs from an integrated set of chemical and biological properties of drugs. CASTLE reassigns the causal feature selection problem to the equivalent parent-and-child (PC) discovery problem in Bayesian Network (BN) structure learning. The PC algorithm is a well-known methodology for learning the neighborhood of a node in a BN.11 12 This study is the first application of PC to discovering causal factors of ADRs. In addition, conflicts between neighborhoods are addressed to improve the significance of results in the high-dimensional ADR prediction problem. The main advantage of CASTLE is its ability to eliminate features that correlate with ADRs but do not directly affect them.
Background
Pharmacogenomic efforts to determine molecular predictors of ADRs have primarily focused on gene variants. Genes encoding drug-metabolizing enzymes have been studied the most in relation to ADRs.13 14 For instance, the highly polymorphic cytochrome P450 (CYP) enzymes have been closely studied because of their involvement in metabolizing ∼67% of all drugs.15 Both the candidate gene sequencing approach and recent genome-wide association studies have successfully uncovered many associations between genetic variants and ADRs.16–19 For example, specific alleles in CYP2C9 have been linked to hemorrhage in patients taking warfarin,20 and variants in CYP2D6 have been linked to tardive dyskinesia and bradycardia in patients taking antipsychotics and β-blockers.21
From the system biology perspective, the response of the human body to a drug is a complex phenomenological observation of the perturbations induced by drug molecules, reflecting not only favorable effects from interaction with their intended protein targets, but also side effects from off-target interactions. This concept has been illustrated in a number of computational methods aimed at predicting potential ADRs from preclinical characteristics of the compounds, and they can be categorized into protein target-based and chemical structure-based approaches.
The protein target-based approach is to relate ADRs to the drugs’ protein-binding profiles. It has been shown that drugs with similar in vitro protein-binding profiles tend to exhibit similar side effects.22 23 Scheiber et al24 illustrated the concept by comparing pathways involving proteins targeted by toxic compounds with those targeted by non-toxic compounds. Fuzuzaki et al25 proposed a method for linking ADRs to sub-pathways that share correlated modifications of gene-expression profiles in the presence of a drug. Xie et al26 identified off-targets of a drug by docking the drug into binding pockets of proteins that are structurally similar to its primary targets. Brouwers et al27 quantified the contribution of the protein interaction network neighborhood to the observed side effect similarity of drugs. Pouliot et al9 used screening data from the PubChem BioAssay28 database to determine the correlation of ADRs with drug bioactivities. Lounkine et al29 systematically evaluated the potential clinical relevance of protein targets with ADRs.
Alternatively, the chemical structure-based approach aims to link ADRs with the chemical structures of drugs. For instance, Bender et al30 explored the chemical spaces of drugs and established their correlation with ADRs. Scheiber et al31 presented a global analysis that identified chemical substructures associated with ADRs. Hammann et al32 used decision tree modeling to determine the chemical, physical, and structural properties of compounds that predispose them to causing organ-level toxicities. Pauwels et al7 proposed a sparse canonical correlation analysis (SCCA) method for predicting high-dimensional drug side effect profiles based on chemical structures.
In addition, an emerging approach is to combine information sources for more effective and accurate prediction. Yamanishi et al33 integrated chemical, genomic, and pharmacological data to infer drug–target interactions. Cami et al34 combined network structures formed by known ADRs with chemical and ontological information to identify ADRs. In our previous study, we investigated the use of phenotypic information, together with chemical and biological properties of drugs, to predict their ADR profiles.35 Vilar et al36 proposed approaches for prioritizing ADRs generated from spontaneous reports and electronic medical records37 by chemical structure similarity.
Although machine learning algorithms such as the support vector machine (SVM) have shown high predictive power in ADR studies, biological interpretability of the outputs is poor, and the relationships between input features and output class (eg, side effects) are unknown.7 35 Hammann et al32 and Pauwels et al7 addressed the issue by using methods with higher interpretability such as decision tree learner and SCCA. However, a limitation of these methods is that they primarily measure correlations between variables, and the identified relations do not necessarily imply causation, which not only requires correlation but also a counterfactual dependence. Inferring a cause-and-effect relationship is intrinsically difficult, and researchers have developed algorithmic solutions to discover causal features,12 identify causal genes related to diseases,38–41 and identify causal factors of clinical outcomes to assist in better healthcare management.42 This study proposes a novel causality analysis model (CASTLE) based on BN structure learning to identify molecular factors responsible for ADRs. The core PC causality module has previously been shown to be superior to other methods in different domains.12 41 43
Methods
Data description
In this study, we used ADR data in SIDER (V.1), which contains information on 888 approved drugs and the corresponding 1385 unique side effect keywords extracted from public documents and package inserts.44 Since SIDER represents ADRs as Unified Medical Language System (UMLS) Concept Unique Identifiers (CUIs),45 one type of ADR may be represented by a group of CUIs. For example, myocardial infarction (C0027051), chest pain (C0008031), atrial fibrillation (C0004238), and congestive heart failure (C0018802) are related concepts but are represented as distinct concepts in SIDER. By adopting Pouliot et al's9 approach, we grouped the ADR concepts according to where they may be manifested in the body, also known as system organ class (SOC), defined by the Medical dictionary for regulatory activities (MedDRA).46 Hence the above-mentioned concepts would all be listed under the ‘heart diseases’ SOC. All 1385 side effect keywords in SIDER were mapped to 12 SOCs (table 1) either directly or indirectly through parent–child relations in UMLS.
Table 1.
KS significance analysis of model performance using integrated feature set
| Method 1 | Method 2 | p Value |
|---|---|---|
| CASTLE with robust PC | SVM with random FS | 7.91E-7 |
| CASTLE with robust PC | SVM | 0.02 |
| CASTLE with robust PC | LASSO | 0.34 |
| CASTLE with robust PC | CASTLE with PC | 0.06 |
CASTLE, causality analysis model based on structure learning; FS, feature selection; KS, Kolmogorov–Smirnov; LASSO, Least Absolute Shrinkage and Selection Operator; PC, parents-and-children; SVM, support vector machine.
Chemical structures of drugs were collected from PubChem,47 48 and biological properties were obtained from DrugBank49–51 and the Kyoto encyclopedia of genes and genomes (KEGG).52–54 To link these databases, we mapped drugs in SIDER to DrugBank.49–51 Of the 888 drugs in SIDER, 58 drug names could not be mapped to their respective DrugBank IDs, resulting in a final dataset of 830 drugs, each of which has a ‘yes’ or ‘no’ label for each of the 12 SOC-specific ADRs, indicating whether a drug has ADRs manifested in the SOC or not.
Data representation
Each drug is represented by its chemical and biological properties and is associated with a binary side effect profile, y, whose elements correspond to the presence or absence of each of the SOC-specific ADRs with 1 or 0, respectively. To encode drug chemical structures, we used fingerprints corresponding to 881 chemical substructures defined in PubChem.47 48 The biological properties of a given drug consisted of its intended targets, transporters (for drug transportation), enzymes (for drug metabolism), and derived pathway from the targets, which can be directly obtained from DrugBank.49–51 Pathway information is obtained by mapping each drug target to its corresponding KEGG pathway52–54 through its protein-coding gene symbol. For a particular SOC-specific ADR yi, each drug is represented by its chemical and biological properties as a 2023 (881 chemical+786 targets+72 transporters+111 enzymes+173 pathway) dimensional vector in which each element is either 1 or 0 for the presence or absence of the corresponding feature.
CASTLE
ADRs may arise from complex interactions between drugs’ chemical structures and patients’ biological systems; as such, most learning methods will identify covariates as good features in predicting ADRs. Taking the causal structure given in figure 1 as an example, features f1, f2, f3 and f4 may be good features identified as highly predictive of the state of the ADR, but, in reality, only the intervention of features f1 and f2 can change the state of the ADR and are thus factors responsible for the ADR.
Figure 1.
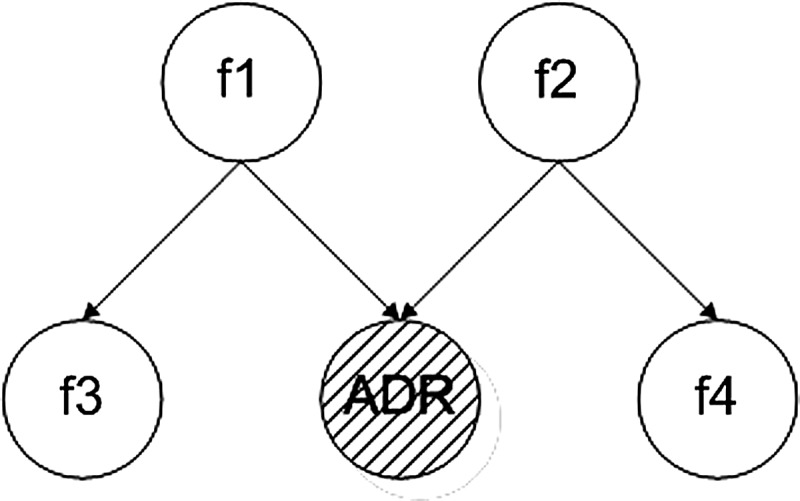
An example causal structure of an adverse drug reaction.
Here, we propose a method for distinguishing causal influences from spurious covariations based on the inductive causal model of Pearl and Verma.55 As defined by Pearl and Verma, causal nodes must be directly connected to the target node. Based on this idea, our method is designed to search for parent-and-child relations between features and ADRs. Additionally, with the asserted knowledge that only the chemical and biological features can mediate ADRs but not vice versa, the non-deterministic polynomial-time hard causality discovery problem can be reduced to the PC discovery problem in BN structure learning. In this study, we used the G2 conditional independence test to detect the separation of two nodes for the identification of parent-and-child relations.56
In the case of a high-dimensional feature space, the PC algorithm may produce many false-positive relations. For instance, consider a set of 20 000 features and a conditional independence threshold of 95%, 5%*20 000 features will be falsely discovered. To address this issue, a robust PC discovery algorithm is proposed to reduce the false discovery rate by first identifying a PC candidate set of ADRs, and then filtering the set with additional conditions. The number of robust PCs will always be a subset of the discovered PCs. A formal definition and formulation of our proposed method is provided in the online appendix.
Experimental design
CASTLE models were built for each of the 12 SOC-specific ADRs (table 1) using chemical and biological properties, which can predict whether a drug leads to a particular SOC-specific ADR or not, but most importantly, derive molecular factors mediating the ADR. First, we evaluated the effectiveness of CASTLE models on the prediction of 12 SOC-specific ADRs through simultaneous extractions of PCs and robust PCs formed by a set of chemical and biological features shared across drugs likely to have a particular ADR, and then using an SVM for the classification task using the extracted PCs and robust PCs. Second, we compared the performance of CASTLE against a traditional SVM trained on the full and randomly selected feature set and against Least Absolute Shrinkage and Selection Operator (LASSO) logistic regression.57 Third, we analyzed the biological factors identified by CASTLE.
Prediction evaluation
Performance of CASTLE was measured by precision, recall and F1 score. To summarize the global performance across 12 SOC-specific ADRs, we reported macro- and micro-averages of each metric. The following are formulas for the macro-average (Equation 1) and micro-average (Equation 2) of each metric.
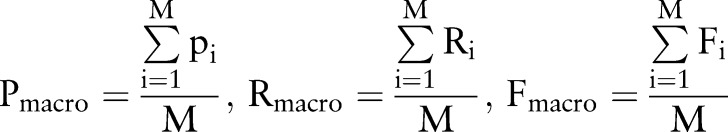 |
1 |
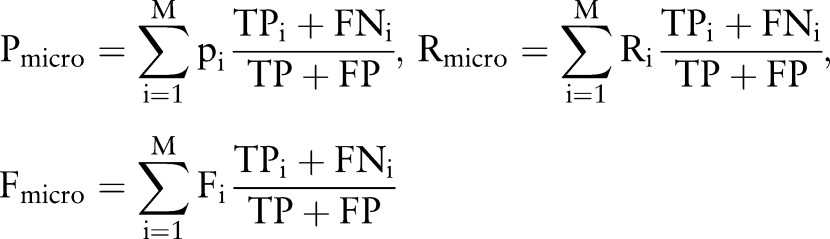 |
2 |
M is the number of SOC-specific ADRs and Pi, Ri, Fi, TPi, and FNi are precision, recall, F1 score, true positives, and false negatives for each ADR, respectively.
Biological evaluation
As the extracted factors in each robust PC are deemed to have cause-and-effect relationships with the SOC-specific ADRs, we assessed the predicted relationships using Online Mendelian Inheritance in Man (OMIM).58 We explored data from the morbid map, which provides gene symbol(s) associated with each disorder, the cytogenetic location of each disorder gene, and links to OMIM records. There was a total of 6284 records in the morbidmap file (March 3, 2013). After mapping the gene symbols to these records, we identified the related disorders. As the name of each disorder in OMIM is different from that of the SOC-specific ADR, a physician manually checked whether the disorder documented in OMIM was a clinical match to the ADR description used in this study.
Statistical significance test
To assess whether the prediction improvement between methods and feature spaces (ie, chemical vs chemical+biological) is statistically significant, we computed the two-sample Kolmogorov–Smirnov test (KS test),59 60 which is a general non-parametric method for comparing two samples in order to test whether the two underlying probability distributions differ. In this study, we calculated the KS test over the F1 scores generated by different methods and feature spaces for individual ADRs. For example, when the chemical space is compared with the combined set (chemical+biological), F1 scores are obtained for predicting each of the 12 ADRs using each feature set; then the KS test determines if the F1 scores generated by ‘chemical+biological’ features are stochastically larger than the F1 scores generated by chemical features alone. Because this study only focuses on 12 ADRs, the sample size may be too small for KS test comparison. To increase the number of samples, we performed five threefold cross-validations. Each cross-validation would result in 12 F1 scores for each of the 12 ADR classes, and repeating it five times would generate 60 samples.
Results
Model performance
We compared abilities of various methods in predicting known side effects of drugs through a threefold cross-validation using either chemical properties or a combination of chemical and biological properties, which include SVM, SVM with random feature selection, LASSO logistic regression, CASTLE with PC (ie, PC+SVM), and CASTLE with robust PC (ie, robust PC+SVM). In the experiment, SVM parameters were tuned on the training set using tools provided by Lib-SVM 3.14.61 Parameters for LASSO are also optimized using threefold cross-validation on the training set. For random feature selection, we randomly selected the same number of features as the robust PCs. As shown in table 2, CASTLE with robust PC achieved the highest macro and micro F1 scores on both feature sets, which demonstrated its ability to identify a high-quality feature set for ADR prediction. Integration of causality analyses by PC and robust PC into the traditional SVM increased its performance in both macro and micro F1 scores. The success of PC and robust PC indicated that a representative set of features can be effectively learned from a high-dimensional dataset through CASTLE. Moreover, comparing the results of PC and robust PC, we can see that macro and micro precision of robust PC is always higher than that of PC, which implies that robust PC improved prediction precision by removing spurious factors.
Table 2.
Model performance on the feature spaces: (1) chemical; (2) chemical+biological
| Method | Macro precision | Macro recall | Macro F1 | Micro precision | Micro recall | Micro F1 |
|---|---|---|---|---|---|---|
| Chemical | ||||||
| SVM with random FS | 0.78 | 0.93 | 0.79 | 0.62 | 0.82 | 0.68 |
| SVM | 0.80 | 0.95 | 0.85 | 0.63 | 0.81 | 0.70 |
| CASTLE with PC | 0.80 | 1.0 | 0.88 | 0.65 | 0.80 | 0.71 |
| CASTLE with robust PC | 0.81 | 0.98 | 0.88 | 0.67 | 0.81 | 0.73 |
| Chemical+biological | ||||||
| SVM with random FS | 0.76 | 0.94 | 0.80 | 0.69 | 0.78 | 0.70 |
| SVM | 0.81 | 0.96 | 0.88 | 0.69 | 0.81 | 0.74 |
| LASSO | 0.80 | 0.98 | 0.88 | 0.67 | 0.81 | 0.73 |
| CASTLE with PC | 0.83 | 0.94 | 0.88 | 0.73 | 0.83 | 0.78 |
| CASTLE with robust PC | 0.85 | 0.94 | 0.89 | 0.77 | 0.85 | 0.81 |
The KS test-based statistical analysis of the F1 score improvement observed in CASTLE with robust PC over other methods (using chemical+biological features) is summarized.
CASTLE, Causality analysis model based on structure learning; FS, feature selection; KS, Kolmogorov–Smirnov; LASSO, Least Absolute Shrinkage and Selection Operator; PC, parents-and-children; SVM, support vector machine.
Table 3 shows the performance of CASTLE with robust PC and PC and LASSO on the integrated feature set for individual SOC-specific ADR prediction. As illustrated, with only a small number of features (ie, 10–21 from the original 2023), CASTLE with robust PC was able to achieve F1 scores over 0.90 for six organ-specific ADRs with the highest being 0.97 for nervous system disorder and for skin and subcutaneous tissue disorders. F1 scores between 0.81 and 0.90 were achieved for five SOC-specific ADRs, and the lowest F1 score achieved was 0.74 for neoplasms. Also, as shown in table 3, robust PC performed better than PC with only half the number of features, which further supported our assertion that some spurious features were filtered out by the constraints proposed in the robust PC algorithm. For almost every ADR class, LASSO extracted many more features than CASTLE, but achieved lower F1 scores.
Table 3.
Performance of CASTLE with robust PC, CASTLE with PC and LASSO for each SOC-specific ADR using both chemical and biological features
| MedDRA SOC-specific ADR | CASTLE with robust PC | CASTLE with PC | LASSO | |||
|---|---|---|---|---|---|---|
| No of features | F1 score | No of features | F1 score | No of features | F1 score | |
| C0014130; Endocrine system diseases | 21 | 0.83 | 35 | 0.83 | 147 | 0.79 |
| C0015397; Disorder of eye | 14 | 0.81 | 31 | 0.79 | 104 | 0.77 |
| C0009450; Communicable diseases | 11 | 0.93 | 22 | 0.92 | 0 | 0.92 |
| C0027651; Neoplasms | 14 | 0.74 | 34 | 0.71 | 110 | 0.73 |
| C0004936; Mental disorders | 18 | 0.90 | 38 | 0.85 | 106 | 0.88 |
| C0027765; Nervous system disorder | 11 | 0.97 | 21 | 0.98 | 0 | 0.97 |
| C0018799; Heart diseases | 19 | 0.88 | 30 | 0.85 | 173 | 0.87 |
| C0263660; Musculoskeletal and connective tissue disorders | 10 | 0.91 | 41 | 0.90 | 88 | 0.88 |
| C0017178; Gastrointestinal diseases | 17 | 0.96 | 28 | 0.96 | 25 | 0.95 |
| C0178298; Skin and subcutaneous tissue disorders | 12 | 0.97 | 23 | 0.97 | 0 | 0.98 |
| C0042075; Urologic diseases | 17 | 0.85 | 34 | 0.85 | 50 | 0.83 |
| C0042373; Vascular diseases | 11 | 0.94 | 35 | 0.94 | 0 | 0.94 |
ADR, adverse drug reaction; CASTLE, causality analysis model based on structure learning; LASSO, Least Absolute Shrinkage and Selection Operator; MedDRA, Medical dictionary for regulatory activities; PC, parents-and-children; SOC, system organ class.
Extracted biological factors responsible for ADRs
For each SOC-specific ADR, CASTLE extracts robust PCs that consist of a small number of factors identified as responsible for the ADR. We analyzed these robust PCs to investigate whether the predicted links to a specific ADR are supported by any evidence. Since OMIM was used in this analysis, we only examined the identified targets, transporters, and enzymes. Table 4 presents the set of genes that were identified as having causal relationships with specific ADRs which are clinical matches to the disorders in OMIM. All potential molecular determinants identified by robust PC (including both biological and chemical features) are listed in online supplementary table S1.
Table 4.
Set of biological factors identified as having a causal relationship with SOC-specific ADRs and supported by evidence in OMIM
| Gene | SOC-specific ADR | OMIM |
|---|---|---|
| ARMD1 | Neoplasms | Colorectal cancer, susceptibility to |
| BAAT | Vascular diseases | Hypercholanemia, familial |
| BCR | Disorder of eye | Retinal dystrophy, early-onset severe retinitis pigmentosa |
| BRCA1 | Endocrine system diseases | Pancreatic cancer |
| Gastrointestinal diseases | Pancreatic cancer | |
| COX1 | Gastrointestinal diseases | Peroxisomal acyl-CoA oxidase deficiency |
| DIH1 | Musculoskeletal and connective tissue disorders | Hernia, congenital diaphragmatic |
| MAL | Mental disorders | Mental retardation, autosomal dominant |
| Nervous system disorder | Hereditary motor and sensory neuropathy | |
| Skin and subcutaneous tissue disorders | Digital arthropathy-brachydactyly, familial | |
| NEP | Mental disorders | Mental retardation, autosomal dominant |
| RIT1 | Nervous system disorder | Cavernous malformations of central nervous system and retina |
| RLF | Mental disorders | Cold-induced sweating syndrome |
| SERPINC1 | Endocrine system diseases | Thrombophilia due to antithrombin III deficiency |
| SERPINH2 | Heart diseases | Osteogenesis imperfecta, type X |
ADR, adverse drug reaction; OMIM, Online Mendelian Inheritance in Man; SOC, system organ class.
Discussion
This study proposed a novel causality analysis model based on BN structure learning called CASTLE for determining molecular factors of ADRs from an integrated set of chemical and biological properties of drugs. Evaluation of CASTLE on the prediction of 12 organ-specific ADRs showed that the highest macro and micro F1 scores were 0.89 and 0.81, respectively.
Compared with SVM and LASSO, use of the integrated feature set, CASTLE with robust PC, achieved a higher micro F1 score: 0.81 vs 0.74 for SVM and 0.73 for LASSO. In terms of macro F1 score, CASTLE with robust PC (0.89) had a slight gain over SVM (0.87) and LASSO (0.88). The slight gain over SVM was determined to be statistically significant (p=0.02), but the gain over LASSO was not statistically significant (p=0.34). In a further analysis, we observed that, for the four ADRs where the positive to negative sample ratio was larger than 3.8, LASSO assigned labels of the majority class to all samples, thus yielding recalls of 1.0. This phenomenon is illustrated in table 3, where zero features were extracted by LASSO for those four ADR classes. Under the same conditions, CASTLE was able to extract a small set of meaningful features. Moreover, LASSO is fundamentally different from CASTLE. LASSO aims to find a good way to predict ADR within the carefully designed regularization factors. CASTLE, on the other hand, attempts to identify causal factors of an ADR. The following example illustrates the basic difference between LASSO and CASTLE. Considering the mechanism, F1 →(F2, F3)→ADR; LASSO would use F1 to predict the state of ADR because it can achieve similar prediction results to F2 and F3, but with fewer variables, while CASTLE would use F2 and F3 because F2 and F3 are more direct causes of ADR. Essentially, through BN structure learning, CASTLE considers both degree and separation of the associations between features and outcome.
Generally, the integration of chemical and biological features achieved higher performance than chemical structures alone. Compared with SVM using chemical structures only, CASTLE with PC resulted in a 4% increase in macro and micro F1 scores and yielded an increase of 1% in macro F1 score and 3% in micro F1 score with robust PC. Using the integrated features, CASTLE with PC did not improve the macro F1 score, but improved the micro F1 score by 3%, while CASTLE with robust PC improved macro and micro F1 scores by 2% and 6%, respectively. The performance improvement from PC discovery was not as significant as from robust PC and this may be due to high dimensionality. For instance, the chemical feature is a smaller set, with only 881 variables in which PC performed relatively well. However, feature space grew to 2023 after the addition of drug biological properties, and the PC performance was close to that of SVM. Furthermore, macro and micro precisions of robust PC are always higher than that of PC, which indicate the success of robust PC in eliminating spurious factors. Lastly, statistical analysis by KS test showed that, when CASTLE with robust PC was used, the improvement in F1 score was significant for the addition of biological features to the chemical features (p=0.04).
Besides prediction accuracy, size of the extracted set is also an important criterion for evaluating model effectiveness because it typically reflects the redundancy of feature extractions. As illustrated in table 3, CASTLE was effective in minimizing the redundancy where the average number of extracted PCs from 2023 features is 31 and robust PCs is only 14. Although, CASTLE with robust PC did not result in a significant improvement in the F1 score over PC (p=0.06), it minimized the number of extracted PCs in the half with similar F1 score, which implied a smaller number of false discoveries. Compared with LASSO logistic regression, the number of features extracted by the CASTLE model is much smaller: average 77 features for LASSO versus 14 for robust PC. The highest F1 score of 0.97 was achieved for predicting toxicities in nervous system and skin and subcutaneous tissues. In contrast, the lowest F1 score of 0.74 was achieved for neoplasms, reflecting the complexity of the disorder.
Most importantly, the main advantage of CASTLE is its ability to produce interpretable outputs, leading to identification of factors that are responsible for ADRs. Interestingly, we were able to find evidence in OMIM supporting some of the identified links (table 4). For instance, the ARMD1 gene was identified to be linked to neoplasms and was indicated in OMIM to contribute to the susceptibility of colorectal cancer. In addition to the OMIM evidence, one of the authors, JH who is a geneticist, performed a literature search and found evidence for links not documented in OMIM. For example, the BAAT gene was identified to be related to both vascular and gastrointestinal diseases. No evidence in OMIM indicated such connections. In a recent study, Hadzic et al62 suggested that defects in BAAT can cause intrahepatic cholestasis. Furthermore, aromatase, CYP19A1, was found by our method to contribute to endocrine system and heart diseases. The aromatase is involved in androgen-to-estrogen conversion by regulating conversion of testosterone to estradiol. It has also been shown to be expressed in immature hearts/cardiomyocytes, suggesting intracardiac androgen–estrogen conversion.63 64 Moreover, Bell et al65 indicated that estrogen suppression may offer inotropic benefit in acute ischemia.
However, there are limitations and challenges to address in the future. For instance, the conditional independence test used in this method may be designed specifically for this problem to achieve more significant results. Moreover, ADR is a complex problem involving various contributing factors. Future development would involve incorporation of more detailed biological features such as the protein–protein interaction network, drug bioactivities, and use of other fingerprints for representing drug chemical structures. Furthermore, we will explore additional external validation sources such as OMOP66 and EU-ADR67 to increase the confidence of findings.
Conclusion
In this paper, we proposed CASTLE, a structure-learning-based causality analysis model, to determine factors that play essential roles in organ-specific ADRs from the chemical and biological profiles of drugs. Evaluation of CASTLE showed an increased prediction performance over traditional SVMs in terms of both macro and micro F1 scores, from 0.88 to 0.89 and 0.74 to 0.81, respectively. Moreover, integration of chemical and biological properties achieved a higher predictive value than using chemical structures alone. Most importantly, CASTLE can produce interpretable results by extracting a set of factors that are considered responsible for the ADRs of interest. Finally, the identified relationships were partially supported by evidence in OMIM, which further demonstrated the effectiveness of CASTLE.
Supplementary Material
Footnotes
Contributors: ML, RC, and YH were responsible for the overall design, development, and evaluation of this study. ML and YH prepared the dataset and designed the experiment for this study. RC implemented the method and ran the experiments. JS, JH, and MEM assisted in the biological evaluation of the results. ML, RC, and YH wrote the first draft and JS, JH, MEM, and HX contributed to editing of the manuscript. All authors reviewed the manuscript critically for scientific content, and all authors gave final approval of the manuscript for publication.
Funding: RC is supported by the Natural Science Foundation of China (61100148) and the Natural Science Foundation of Guangdong Province, China (S2011040004804). YH is supported by the National Science Foundation of China (71271061, 70801020), Science and Technology Planning Project of Guangdong Province, China (2010B010600034, 2012B091100192), and Business Intelligence Key Team of Guangdong University of Foreign Studies (TD1202). MEM is supported by the Veterans Administration HSR&D Career Development Award (CDA-08-020). HX is supported in part by National Cancer Institute grant R01CA141307.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA 1998;279:1200–5 [DOI] [PubMed] [Google Scholar]
- 2.Jha AK, Kuperman GJ, Rittenberg E, et al. Identifying hospital admissions due to adverse drug events using a computer-based monitor. Pharmacoepidemiol Drug Saf 2001;10:113–19 [DOI] [PubMed] [Google Scholar]
- 3.Daly AK. Pharmacogenomics of adverse drug reactions. Genome Med 2013;5:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ingelman-Sundberg M. Pharmacogenomic biomarkers for prediction of severe adverse drug reactions. N Engl J Med 2008;358:637–9 [DOI] [PubMed] [Google Scholar]
- 5.Roden DM, Altman RB, Benowitz NL, et al. Pharmacogenomics: challenges and opportunities. Ann Intern Med 2006;145:749–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilke RA, Lin DW, Roden DM, et al. Identifying genetic risk factors for serious adverse drug reactions: current progress and challenges. Nat Rev Drug Discov 2007;6:904–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pauwels E, Stoven V, Yamanishi Y. Predicting drug side-effect profiles: a chemical fragment-based approach. BMC Bioinformatics 2011;12:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woosley RL, Cossman J. Drug development and the FDA's Critical Path Initiative. Clin Pharmacol Ther 2007;81:129–33 [DOI] [PubMed] [Google Scholar]
- 9.Pouliot Y, Chiang AP, Butte AJ. Predicting adverse drug reactions using publicly available PubChem BioAssay data. Clin Pharmacol Ther 2011;90:90–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiang AP, Butte AJ. Data-driven methods to discover molecular determinants of serious adverse drug events. Clin Pharmacol Ther 2009;85:259–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kalisch M, Buhlmann P. Estimating high-dimensional direced acyclic graphs with the PC-algorithm. J Mac Learn Res 2007;8:613–36 [Google Scholar]
- 12.Cai R, Zhang Z, Hao Z. BASSUM: a Bayesian semi-supervised method for classification feature selection. Pattern Recognit 2011;44:811–20 [Google Scholar]
- 13.Evans WE, Johnson JA. Pharmacogenomics: the inherited basis for interindividual differences in drug response. Annu Rev Genomics Hum Genet 2001;2:9–39 [DOI] [PubMed] [Google Scholar]
- 14.Pirmohamed M, Park BK. Genetic susceptibility to adverse drug reactions. Trends Pharmacol Sci 2001;22:298–305 [DOI] [PubMed] [Google Scholar]
- 15.Williams JA, Hyland R, Jones BC, et al. Drug-drug interactions for UDP-glucuronosyltransferase substrates: a pharmacokinetic explanation for typically observed low exposure (AUCi/AUC) ratios. Drug Metab Dispos 2004;32:1201–8 [DOI] [PubMed] [Google Scholar]
- 16.Sharma SK, Balamurugan A, Saha PK, et al. Evaluation of clinical and immunogenetic risk factors for the development of hepatotoxicity during antituberculosis treatment. Am J Respir Crit Care Med 2002;166:916–19 [DOI] [PubMed] [Google Scholar]
- 17.Cooper GM, Johnson JA, Langaee TY, et al. A genome-wide scan for common genetic variants with a large influence on warfarin maintenance dose. Blood 2008;112:1022–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O'Donohue J, Oien KA, Donaldson P, et al. Co-amoxiclav jaundice: clinical and histological features and HLA class II association. Gut 2000;47:717–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008;359:789–99 [DOI] [PubMed] [Google Scholar]
- 20.Higashi MK, Veenstra DL, Kondo LM, et al. Association between CYP2C9 genetic variants and anticoagulation-related outcomes during warfarin therapy. JAMA 2002;287:1690–8 [DOI] [PubMed] [Google Scholar]
- 21.Meyer UA. Pharmacogenetics and adverse drug reactions. Lancet 2000;356:1667–71 [DOI] [PubMed] [Google Scholar]
- 22.Fliri AF, Loging WT, Thadeio PF, et al. Analysis of drug-induced effect patterns to link structure and side effects of medicines. Nat Chem Biol 2005;1:389–97 [DOI] [PubMed] [Google Scholar]
- 23.Campillos M, Kuhn M, Gavin AC, et al. Drug target identification using side-effect similarity. Science 2008;321:263–6 [DOI] [PubMed] [Google Scholar]
- 24.Scheiber J, Chen B, Milik M, et al. Gaining insight into off-target mediated effects of drug candidates with a comprehensive systems chemical biology analysis. J Chem Inf Model 2009;49:308–17 [DOI] [PubMed] [Google Scholar]
- 25.Fuzuzaki M, Seki M, Kashima H, et al. Side effect prediction using cooperative pathways. IEEE International Conference on Bioinformatics and Biomedicine; Washington DC; 2009:142–7 [Google Scholar]
- 26.Xie L, Li J, Bourne PE. Drug discovery using chemical systems biology: identification of the protein-ligand binding network to explain the side effects of CETP inhibitors. PLoS Comput Biol 2009;5:e1000387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brouwers L, Iskar M, Zeller G, et al. Network neighbors of drug targets contribute to drug side-effect similarity. PLoS ONE 2011;6:e22187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y, Bolton E, Dracheva S, et al. An overview of the PubChem BioAssay resource. Nucleic Acids Res 2010;38(Database issue):D255–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lounkine E, Keiser MJ, Whitebread S, et al. Large-scale prediction and testing of drug activity on side-effect targets. Nature 2012;486:361–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bender A, Scheiber J, Glick M, et al. Analysis of pharmacology data and the prediction of adverse drug reactions and off-target effects from chemical structure. ChemMedChem 2007;2:861–73 [DOI] [PubMed] [Google Scholar]
- 31.Scheiber J, Jenkins JL, Sukuru SC, et al. Mapping adverse drug reactions in chemical space. J Med Chem 2009;52:3103–7 [DOI] [PubMed] [Google Scholar]
- 32.Hammann F, Gutmann H, Vogt N, et al. Prediction of adverse drug reactions using decision tree modeling. Clin Pharmacol Ther 2010;88:52–9 [DOI] [PubMed] [Google Scholar]
- 33.Yamanishi Y, Kotera M, Kanehisa M, et al. Drug-target interaction prediction from chemical, genomic and pharmacological data in an integrated framework. Bioinformatics 2010;26:i246–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cami A, Arnold A, Manzi S, et al. Predicting adverse drug events using pharmacological network models. Sci Transl Med 2011;3:114ra27. [DOI] [PubMed] [Google Scholar]
- 35.Liu M, Wu Y, Chen Y, et al. Large-scale prediction of adverse drug reactions using chemical, biological, and phenotypic properties of drugs. J Am Med Inform Assoc 2012;19(e1):e28–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vilar S, Harpaz R, Chase HS, et al. Facilitating adverse drug event detection in pharmacovigilance databases using molecular structure similarity: application to rhabdomyolysis. J Am Med Inform Assoc 2011;18(Suppl 1):i73–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vilar S, Harpaz R, Santana L, et al. Enhancing adverse drug event detection in electronic health records using molecular structure similarity: application to pancreatitis. PLoS ONE 2012;7:e41471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mukhopadhyay ND, Chatterjee S. Causality and pathway search in microarray time series experiment. Bioinformatics 2007;23:442–9 [DOI] [PubMed] [Google Scholar]
- 39.Kim YA, Wuchty S, Przytycka TM. Identifying causal genes and dysregulated pathways in complex diseases. PLoS Comput Biol 2011;7:e1001095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schadt EE, Lamb J, Yang X, et al. An integrative genomics approach to infer causal associations between gene expression and disease. Nat Genet 2005;37:710–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cai R, Zhang Z, Hao Z. Causal gene identification using combinatorial V-structure search. Neural Networks 2013; 43:63–71 [DOI] [PubMed] [Google Scholar]
- 42.Mani S, Cooper GF. Causal discovery from medical textual data. Proceedings/AMIA Annual Symposium AMIA Symposium 2000:542–6 [PMC free article] [PubMed] [Google Scholar]
- 43.Hu Y, Zhang X, Ngai EWT, et al. Software project risk analysis using Bayesian networks with causality constraints. Decis Support Syst 2013;56:439–49 [Google Scholar]
- 44.Kuhn M, Campillos M, Letunic I, et al. A side effect resource to capture phenotypic effects of drugs. Mol Syst Biol 2010;6:343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bodenreider O. The Unified Medical Language System (UMLS): integrating biomedical terminology. Nucleic Acids Res 2004;32(Database issue):D267–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brown EG, Wood L, Wood S. The medical dictionary for regulatory activities (MedDRA). Drug Safety 1999;20:109–17 [DOI] [PubMed] [Google Scholar]
- 47.Chen B, Wild D, Guha R. PubChem as a source of polypharmacology. J Chem Inf Model 2009;49:2044–55 [DOI] [PubMed] [Google Scholar]
- 48.Bolton E, Wang Y, Thiessen PA, et al. PubChem: integrated platform of small molecules and biological activities. Chapter 12 in Annual Reports in Computational Chemistry. Washington, DC: American Chemical Society, 2008 [Google Scholar]
- 49.Knox C, Law V, Jewison T, et al. DrugBank 3.0: a comprehensive resource for 'omics’ research on drugs. Nucleic Acids Res 2010;39(Database issue):D1035–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wishart DS, Knox C, Guo AC, et al. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res 2008;36(Database issue):D901–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wishart DS, Knox C, Guo AC, et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res 2006;34(Database issue):D668–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000;28:27–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kanehisa M, Goto S, Furumichi M, et al. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res 2010;38(Database issue):D355–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kanehisa M, Goto S, Hattori M, et al. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res 2006;34(Database issue):D354–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pearl J, Verma TS. A theory of inferred causation. Proceedings of the 2nd International Conference on Principles of Knowledge Representation and Reasoning; April 1991; Cambridge, Massachusetts, 1991:441–52 [Google Scholar]
- 56.Spirtes P, Glymour C, Scheines R. Causation, prediction, and search. 2nd edn The MIT Press, 2001 [Google Scholar]
- 57.Friedman JH, Hastie T, Tibshirani R. RegularizationPaths for generalized linear models via coordinate descent. J Stat Software 2010;33:1–22 [PMC free article] [PubMed] [Google Scholar]
- 58.Online Mendelian Inheritance in Man, OMIM. http://omim.org/
- 59.Birnbaum ZW, Tingey FH. One-sided confidence contours for probability distribution functions. Ann Math Stat 1951;22:592–6 [Google Scholar]
- 60.Conover WJ. Practical nonparametric statistics. New York: John Wiley & Sons, 1971 [Google Scholar]
- 61.Chang C-C, Lin C-J. LIBSVM: a library for support vector machines. ACM Trans Intell Syst Technol 2011;2:1–27 [Google Scholar]
- 62.Hadzic N, Bull LN, Clayton PT, et al. Diagnosis in bile acid-CoA: amino acid N-acyltransferase deficiency. World J Gastroenterol 2012;18:3322–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Price T, Aitken J, Simpson ER. Relative expression of aromatase cytochrome P450 in human fetal tissues as determined by competitive polymerase chain reaction amplification. J Clin Endocrinol Metab 1992;74:879–83 [DOI] [PubMed] [Google Scholar]
- 64.Grohe C, Kahlert S, Lobbert K, et al. Expression of oestrogen receptor alpha and beta in rat heart: role of local oestrogen synthesis. J Endocrinol 1998;156:R1–7 [DOI] [PubMed] [Google Scholar]
- 65.Bell JR, Mellor KM, Wollermann AC, et al. Aromatase deficiency confers paradoxical postischemic cardioprotection. Endocrinology 2011;152:4937–47 [DOI] [PubMed] [Google Scholar]
- 66.Stang PE, Ryan PB, Racoosin JA, et al. Advancing the science for active surveillance: rationale and design for the Observational Medical Outcomes Partnership. Ann Intern Med 2010;153:600–6 [DOI] [PubMed] [Google Scholar]
- 67.Coloma PM, Schuemie MJ, Trifiro G, et al. Combining electronic healthcare databases in Europe to allow for large-scale drug safety monitoring: the EU-ADR Project. Pharmacoepidemiol Drug Saf 2011;20:1–11 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.