

Abstract
Certain botanical dietary supplements have been associated with idiosyncratic organ-specific toxicity. Similar toxicological events, caused by drug-induced mitochondrial dysfunction, have forced the withdrawal or U.S. FDA “Black Box” warnings of major pharmaceuticals. To assess the potential mitochondrial liability of botanical dietary supplements, extracts from 352 authenticated plant samples used in traditional Chinese, Ayurvedic, and Western herbal medicine were evaluated for the ability to disrupt cellular respiration. Blue cohosh (Caulophyllum thalictroides) methanol extract exhibited mitochondriotoxic activity. Used by some U.S. midwives to help induce labor, blue cohosh has been associated with perinatal stroke, acute myocardial infarction, congestive heart failure, multiple organ injury, and neonatal shock. The potential link between mitochondrial disruption and idiosyncratic herbal intoxication prompted further examination. The C. thalictroides methanol extract and three saponins, cauloside A (1), saponin PE (2), and cauloside C (3) exhibited concentration- and time-dependent mitochondriotoxic activities. Upon treatment, cell respiration rate rapidly increased and then dramatically decreased within minutes. Mechanistic studies revealed that C. thalictroides constituents impair mitochondrial function by disrupting membrane integrity. These studies provide a potential etiological link between this mitochondria-sensitive form of cytotoxicity and idiosyncratic organ damage.
Drug-induced mitochondrial dysfunction only recently has become recognized as a serious limiting factor for drug development and has forced the withdrawal of major drugs used to treat diabetes (e.g., troglitazone) and hyperlipidemia (e.g., cerivastatin).1 Nearly half of the drugs with hepatotoxicity- and cardiotoxicity-associated U.S. FDA “Black Box” Warnings are known to interfere with mitochondrial function.1 These safety issues highlight the importance of assessing potential mitochondrial toxicity early in the drug development process.1 The use of certain botanical dietary supplements has been associated with a variety of similar, often idiosyncratic, organ-specific intoxication events.2 While the pharmaceutical industry has begun to evaluate the potential mitochondrial liability of new therapeutic agents, similar efforts have not been applied to the plethora of phytochemicals in dietary supplement products.3 According to the 2007 National Health Interview Survey, the number of Americans that use and/or have used Complementary Alternative Medicine (CAM) for health and wellness is in the millions (38% adults and 12% children).4 Thus, it is critical to identify potential mitochondria-disrupting botanical dietary supplement constituents to protect public health.
In an exploratory study, extracts from 352 species of plants and other organisms used in traditional Chinese, Ayurvedic, and Western herbal medicine were evaluated for their potential to disrupt mitochondrial function. Primary screening identified an extract of Caulophyllum thalictroides (L.) Michx. (Berberidaceae) that may contain mitochondrial toxins. A native plant grown in eastern United States, C. thalictroides (also known as blue cohosh) has a history of being used as herbal medicine. As a dietary supplement, blue cohosh is used as an antispasmodic, emenagogue (menstrual flow stimulator), parturifacient (labor inducer), and abortifacient.5 In 1999, it was estimated that 64% of American midwives used blue cohosh to induce labor.6 The side effects associated with blue cohosh administration include diarrhea, increases in blood pressure and blood sugar, and stomach cramps.5 Neonates born to women that have taken C. thalictroides tincture/dietary supplements may suffer perinatal stroke, acute myocardial infarction, congestive heart failure, shock, and multiple organ injury.5 While blue cohosh is known to produce a variety of bioactive natural products, toxic alkaloids (e.g., N-methylcytisine) have been considered the most likely cause of intoxication events.7 Based on the hypothesis that mitochondrial disruption may contribute to the toxic side effects exerted by blue cohosh, cell-based studies were conducted to examine the impact of blue cohosh extract and purified compounds on cellular respiration and to determine mechanisms of action.
RESULTS AND DISCUSSION
The transcription factor hypoxia-inducible factor-1 (HIF-1) promotes cellular adaptation and survival under hypoxic conditions by regulating gene expression.8 Our HIF-1 targeted natural product discovery effort has revealed that mitochondrial disruptors exhibited stimulus-dependent effects on HIF-1 activation. Inhibitors of mitochondrial respiration selectively suppress HIF-1 activation by hypoxia (decreased oxygen tension) relative to that induced by chemical hypoxia (i.e., iron chelators).9–12 Protonophores that uncouple mitochondrial electron transport from oxidative phosphorylation inhibit both hypoxia- and iron chelator-induced HIF-1 activation.13,14 Mitochondrial function has been monitored traditionally by polarographic electrode-based measurement of oxygen consumption.15 Although reliable at detecting mitochondria-disrupting substances, this method is low-throughput (one sample at a time) and not suitable for examining a large number of samples. Therefore, a T47D cell-based HIF-1 reporter assay was used to prescreen natural products for potential mitochondrial poisons. Extracts from 352 species of authenticated plants and other organisms used in traditional Chinese, Indian, African, and Western herbal medicine were evaluated at the concentration of 20 μg mL−1 for their effects on HIF-1 activation. Forty-six samples that inhibited hypoxia-induced HIF-1 activation by >70% were selected for further T47D cell-based respiration studies to assess their effects on mitochondria, using an Oxytherm Clarke-type electrode system.16 Derived from human breast ductal carcinoma, T47D cells are estrogen-dependent, utilize oxidative phosphorylation for ATP production, and respond predictably to standard mitochondrial substrates and inhibitors.16 Oxygen consumption rates in T47D cells were measured in the presence of extracts at the concentrations of 20, 40, and 100 μg mL−1 and presented as “% Inhibition” of the untreated control. In this system, the prototypical mitochondrial electron transport chain (ETC) complex I inhibitor rotenone suppressed cellular respiration by 24%, 46%, and 80% at 1, 10, and 100 nM, respectively. Extracts from 16 different plant species exhibited pronounced inhibition of cellular respiration (> 50% inhibition), samples from nine other species weakly suppressed respiration (25% – 50% inhibition), and extracts from five species increased the rate of oxygen consumption. Among the actives, an extract of blue cohosh displayed time- and concentration-dependent biphasic effects on cellular respiration and was selected for further evaluation.
Previous phytochemical studies of C. thalictroides afforded a small chemical library for biological evaluation. A panel of ten purified compounds was assembled and examined in the T47D cell-based HIF-1 reporter assay (Figure S1, Supporting Information). Based on the relatively high total saponin content of commercial dietary supplements (e.g., up to >300 mg/day suggested intake),17 these compounds were tested at low micromolar (1, 10, and 30 μM) concentrations. Cauloside A (1), saponin PE (2), and cauloside C (3) inhibited both hypoxia and 1,10-phenanthroline-induced HIF-1 activation. This form of stimulus-nonselective HIF-inhibitory effect was observed with protonophores that uncouple mitochondrial respiration,13,14 prompting further evaluation of 1–3 in cell-based respiration assays. T47D and human hepatocarcinoma-derived Hep3B cells were employed as in vitro models to determine the effects of the blue cohosh extract and purified compounds 1–3 on cellular respiration. Test samples were added to intact cells and oxygen consumption (cell respiration) rates were measured at 30, 115, and 295 s after sample addition. Respiration rates were compared to the rates of untreated cells. Both the C. thalictroides extract and pure compound samples exerted cell line-, concentration-, and time-dependent effects on cellular oxygen consumption (Figure 1). Hep3B cells were more sensitive than T47D cells to the respiration disruptive activity. At each of the active concentrations, a short burst in oxygen consumption was followed by an immediate decrease. Cell respiration was inhibited at higher concentrations. Compounds 1 and 2 exerted more pronounced effects on cellular respiration than 3. In previous studies, protonophores increased oxygen consumption at lower concentrations and decreased oxygen consumption at higher concentration.13 A time-course study was performed with a prototypical protonophore uncoupler FCCP [2-([4-(trifluoromethoxy)phenyl]hydrazinylidene)propanedinitrile]. At the concentration that stimulated oxygen consumption (0.3 μM),13 FCCP did not exhibit a time-dependent biphasic effect on respiration (Figure S2, Supporting Information) as those observed with the blue cohosh extract and compounds 1–3.
Figure 1.

Blue cohosh extract and purified saponins exert biphasic effects on cellular oxygen consumption. (A) Blue cohosh extract disrupted cellular oxygen consumption in a concentration- and time-dependent manner (left panel: Hep3B cells; right panel: T47D cells). Extract sample was tested at the concentrations of 10, 30, 50, and 100 μg mL−1. Oxygen consumption rates were recorded 30, 115, and 295 s (◢) after the addition of samples to intact cells and presented as “% Inhibition” of the untreated control. Negative values indicate stimulation of oxygen consumption. Data shown are average + SD from two independent experiments for Hep3B cells (n = 3), and average + SD of three independent experiments for T47D cell. (B) Effect of 1 on cellular respiration. Compound 1 was tested at 5.6, 10.0, 17.8 and 30.0 μM. Data presentation is the same as that described in (A) except that data shown are average + SD of three independent experiments. (C) Effect of 2 on cellular oxygen consumption. Experimental conditions, data collection, and presentation are the same as described in (B) except that n = 1 for Hep3B cells and n = 3 for T47D cells. (D) Effect of 3 on cellular respiration. Experimental conditions, data collection, and presentation are the same as described in (B). The uncoupler, FCCP (0.3 μM), was used as a positive control in the initial experiments. FCCP increased respiration at all time points [T47D cells: 30 s (108%, ± 9% SD); 115 s (114%, ± 9% SD); 295 s (103%, ± 14% SD); Hep3B cells: 30 s (149%, ± 13% SD); 115 s (157%, ± 24% SD); 295 s (148%, ± 30% SD)]; representative results from one independent experiment (n = 3), controls produced similar results in multiple independent experiments. Additionally, a separate digitonin (mechanistically similar) positive control experiment was performed, Figure 2).
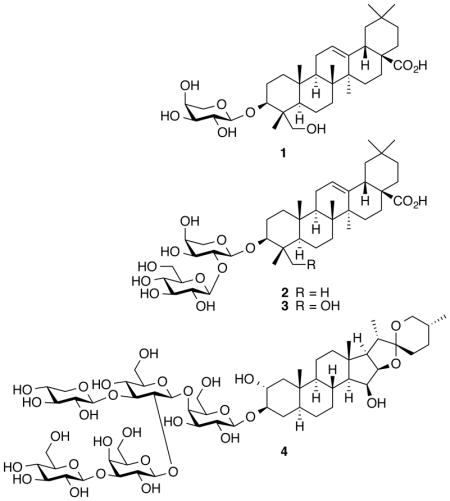
Structurally, 1–3 are plant saponins and certain saponins have been reported to interact with and solubilize biological membranes.18 To test the hypothesis that blue cohosh saponins disrupt respiration by permeabilizing plasma and mitochondrial membranes, a concentration-response and time-course respiration study was performed with digitonin (4), a plant saponin commonly used to disrupt cellular membranes in mitochondria mechanistic studies. Similar to the response observed with the blue cohosh saponins, compound 4 exerted a time-dependent biphasic effect on cellular oxygen consumption at higher concentrations (17.8 and 30 μM, Figure 2).
Figure 2.
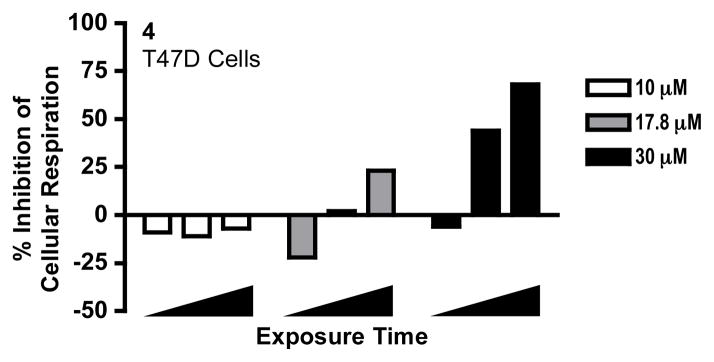
Digitonin (4) exerts concentration- and time-dependent biphasic effect on cellular respiration. Compound 4 was added at 10.0, 17.8 and 30.0 μM, respectively, to intact T47D cells. Oxygen consumption rates were recorded 30, 115 and 295 s (◢) after compound addition. The data were presented as percentage inhibition of cellular respiration in comparison to that of the untreated cells. Negative values indicate a relative stimulation of oxygen consumption. Data shown are from one experiment performed in triplicate.
Compound 4 is used routinely to selectively permeabilize the plasma membrane in cell-based mechanistic studies to manipulate substrate availability at specific mitochondrial complexes.19 Succinate, a substrate for mitochondrial ETC complex II, is anionic at physiological pH in solution and cannot freely penetrate the plasma membrane. The mitochondrial inner membrane dicarboxylate transporter that imports succinate into the mitochondrial matrix is not expressed on the plasma membrane. When the plasma membrane is permeabilized, exogenously added succinate acts as a complex II substrate that reinitiates cellular respiration blocked by complex I inhibitors. To determine if blue cohosh saponins can permeabilize cellular membranes, 1 was selected as a representative compound for mechanistic studies. Substrates and inhibitors of specific mitochondrial ETC complexes were added to T47D cells in a sequential manner to monitor their effects on cell respiration. Exogenously added pyruvate can be transported across plasma membrane by the monocarboxylate carrier to stimulate mitochondrial respiration at ETC complex I (reflected by a decrease in oxygen tension). The complex I inhibitor rotenone reduced the rate of cellular oxygen consumption. Under these conditions, the ability of extracellular succinate (ETC complex II substrate) to reinitiate rotenone-inhibited respiration depends on plasma membrane permeabilization to make substrate available (Figure 3). In the presence of the solvent control (DMSO), succinate failed to overcome rotenone-inhibited respiration (Figure 3A). However, addition of either 1 (10 μM) or the prototypical membrane disruptor 4 (4 μM) enabled succinate to reinitiate rotenone-suppressed respiration (Figures 3B and 3C). At these active concentrations, neither 1 nor 4 strongly affected T47D cell respiration fueled by glucose (Figures 1B and 2). At lower concentrations, 1 (3 and 5.6 μM) failed to make externally supplemented succinate available to the mitochondria (data not shown).
Figure 3.

Blue cohosh saponins exert their effects on cellular respiration by permeabilizing cellular membranes. (A) The solvent control DMSO was added to T47D cells, followed by a mixture of malate/pyruvate (5 mM each) to stimulate mitochondrial respiration at ETC complex I, rotenone (1 μM) to inhibit complex I, and the complex II substrate succinate (5 mM). Oxygen consumption rates were determined after each treatment and provided in the parentheses for each section. (B) Effect of digitonin (4, 4 μM)-induced membrane permeabilization on oxygen consumption in T47D cells. (C) Effect of cauloside A (1, 10 μM) on T47D cellular respiration. Data shown in A–C are recordings from one experiment, representative of at least two independent experiments. (D) T47D cells loaded with the TMRM+ dye were exposed to compounds and controls as specified and representative images are shown. For cauloside A (30 μM), saponin PE, and digitonin treatments, images before compound addition are shown as inserts inside each panel.
As a complimentary line of investigation, a microscopy-based method was used to assess the effects of blue cohosh saponins on cellular membranes. In order to achieve a mitochondrial membrane potential, membrane integrity must be maintained. The fluorescent dye tetramethylrhodamine methyl ester (TMRM+) accumulates in the mitochondrial matrix due to its cationic charge and is used as an indicator of the magnitude of the mitochondrial membrane potential.20 T47D cells loaded with TMRM+ dye were exposed to test compounds and controls, and examined by epifluorescence microscopy (Figure 3D). As expected, the uncoupler FCCP (control) reduced the fluorescent intensity by dissipating the mitochondrial membrane potential.20 Compound 1 displayed a concentration-dependent biphasic effect. At 10 μM, 1 augmented cellular fluorescence. While this effect is not clearly understood, it is possible that 1 may disrupt ion flux or alter proton gradient due to its slightly acidic nature, and thus affect mitochondrial membrane potential. However, at increased concentrations, compound 1 (30 μM) diminished TMRM+ fluorescence, consistent with cellular membrane disruption. While structurally related, compound 2 collapsed the membrane potential at 10 μM, and the less active 3 did not exert significant effect, even at 30 μM. Addition of the membrane permeabilizer 4, as a positive control, resulted in loss of fluorescence. In combination with the cell-based respiration study results (Figures 1, 2, and 3A–3C), these observations support the hypothesis that C. thalictroides saponins significantly disrupt oxygen consumption by membrane permeabilization. While the mechanism responsible for the observed transient increase in respiration rate is not clear, extended exposure and/or high concentration treatment leads to complete membrane disruption that correspondingly blocks oxygen consumption and interferes with oxidative phosphorylation. As a result, active concentrations of these non-selective mitochondrial inhibitors impose concentration- and time-dependent biphasic effects on cellular oxygen consumption.
To determine the effects of blue cohosh saponins on cell viability, concentration-response studies were performed in T47D and Hep3B cells. Exponentially grown cells were exposed to 1–3 and blue cohosh extract at a range of concentrations for 48 h and 6 d, respectively. Cell viability was measured using the sulforhodamine B method and the IC50 values shown in Table 1. Both the C. thalictroides methanol-soluble extract and purified saponins suppressed cell proliferation/viability. More pronounced inhibitory effects were observed in Hep3B cells, relative to the effects on T47D cells. This form of cell line-selective cytotoxicity was consistent with the results obtained in the respiration studies. Extended exposure (6 days) enhanced growth inhibitory effect relative to the 48 h exposure studies. This pattern of exposure time- and cell line-dependent cytotoxicity resembles the previously observed cytotoxicity profiles of compounds that interfere with mitochondrial function.16,21 In human umbilical vein endothelial cells (HUVEC), blue cohosh saponins exhibited antiproliferative activities comparable to those observed in T47D cells (HUVEC, 48 h, Figure 4). However, at concentrations up to 30 μg/mL, the blue cohosh extract did not affect HUVEC cell proliferation/viability (data not shown).
Table 1.
IC50 Values of C. thalictroides Methanol Extract and Purified Saponins on Cell Proliferation/Viability after 48 hours and 6 day Treatment (n = 3).a,b
| number | T47D | Hep3B | ||
|---|---|---|---|---|
|
| ||||
| 48 h | 6 d | 48 h | 6 d | |
| 1 (μM) | 13.0 (12.2 – 13.8) | 11.5 (11.2 – 11.8) | 7.5 (7.11 – 7.98) | 6.6 (6.41 – 6.87) |
| 2 (μM) | 12.6 (12.2 – 13.0) | 10.2 (10.1 – 10.4) | 7.0 (6.63 – 7.39) | 3.5 (3.32 – 3.58) |
| 3 (μM) | 30.1 (29.3 – 30.9) | 25.6 (25.0 – 26.2) | 27.8 (22.3 – 34.7) | 9.0 (8.46 – 9.56) |
| 4 (μM) | 9.5 (8.7 – 10.4) | 10.5 (9.8 – 11.3) | 7.4 (6.5 – 8.5) | 8.8 (7.4 – 10.4) |
| C. thalictroides extract (μg/mL) | 55.6 (52.7 – 58.7) | 32.0 (30.9 – 33.2) | 30.8 (29.9 – 31.6) | 24.7 (23.9 – 25.4) |
| emetinec (1 μM) | 60% (+/− 5%)c | 86% (+/− 11%)c | 66% (+/− 10%)c | 93% (+/−6%)c |
95% confidence interval values (95% CI) were provided inside the parentheses.
The Digitalis saponin, digitonin (4), and protein translation inhibitor, emetine, were used as positive controls.
Emetine data were expressed as % inhibition of the untreated control (average +/− SD).
Figure 4.
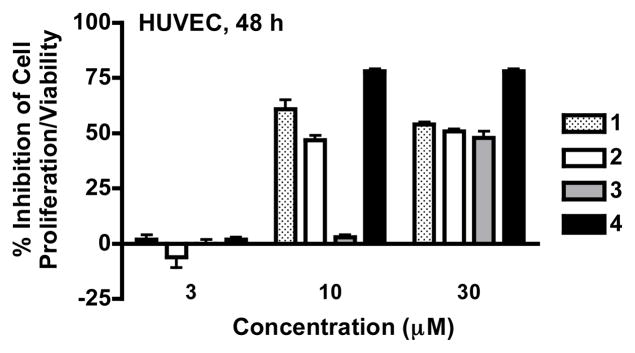
Concentration-response results of 1–4 on HUVEC proliferation/viability. HUVEC cells were exposed to compounds at 3, 10 and 30 μM for 48 h, cell viability was determined by the SRB method, and presented as ‘% Inhibition’ of the untreated control. Digitonin (4) was used as a positive control for a cytotoxic saponin that disrupts cell membranes. Data shown are average ± standard deviation (n = 3).
Saponins are ubiquitous plant secondary metabolites with surfactant properties.22 Multiple dietary supplements that contain saponin glycosides are marketed in the United States for various applications.27,23 Among the bioactivities attributed to this class of compounds, hemolytic or membrane permeabilizing effects are prominent.24 Due to their surfactant properties, saponins interact with the lipid bilayers of cellular membranes and alter membrane permeability by irreversibly forming pores. Proposed saponin mechanisms of action range from cholesterol sequestration to aquaporin disruption-associated unregulated water transport.25 Numerous saponin intoxication events have been recorded in animals, although limited bioavailability make some saponins less toxic when ingested orally in comparison to peritoneal administration.26 Recent studies indicate that saponin absorption, bioactivity, and potential toxicity may be enhanced significantly when consumed with other plant extract components.28 In cultured cells, plant saponins such as avicins and OSW-1 were found to activate apoptosis by directly permeabilizing mitochondrial membranes.27 Saponins have been reported to affect a number of cellular pathways that result in cytotoxicity.29 However, it is unclear whether the observed effects on cellular signaling are secondary to a toxic mitochondrial insult. These results suggest that the ability of saponins to induce biphasic concentration- and time-dependent alterations in mitochondrial function may contribute to their antiproliferative activity.
Mitochondrial toxicity has recently been recognized as a major contributor of drug-induced toxicity that has led to the withdrawn of major drugs,1 yet its relevance to botanical dietary supplement intoxication is not known. Annonaceous acetogenins found in antitumor dietary supplement products (e.g., Paw Paw Cell-Reg®, Graviola Max®, Royal Graviola®, Graviola Liquid Extract®) disrupt mitochondrial ETC at complex I30 and interfere with hypoxic signaling.31 Prenylated coumarins from Mammea americana produce mitochondriotoxic effects by functioning as anionic protonophores that potently uncouple the mitochondrial ETC.13,32 Protoliminoids and other constituents found in Bael tree (Aegle marmelos) products act as mitochondrial poisons that disrupt the ETC and interfere with global protein translation through an endoplasmic reticulum (ER)-stress activated signaling pathway.21,33 These A. marmelos studies further indicate that even auraptene (a purported cancer chemopreventative agent) potentially may act as a mitochondrial poison. One of the major challenges for the identification of botanical dietary supplement-induced mitochondrial toxicity is that these products are often consumed in conjunction with clinically approved drugs, which may also produce off-target effects on the mitochondria.1 In addition, consumption of botanical dietary supplements is perceived as safe and often goes unreported. This study represents an initial systematic effort to evaluate these materials for mitochondrial toxins.
Among other factors, inherently elevated metabolic status and the ability to enzymatically activate toxic metabolites predispose liver cells to mitochondrial impairment and drug-induced hepatotoxicity.34 In this study, Hep3B cells were more sensitive to the inhibitory effects exerted by blue cohosh compounds, relative to T47D cells (Figures 1A–1D). As Hep3B cells are of neoplasmic origin, evaluation of these C. thalictroides metabolites in primary hepatocytes will be required to distinguish some form of tumor cell-selective, rather than liver cell-selective, cytotoxicity. Such primary hepatocyte-based studies may also provide additional information regarding the potential hepatotoxicity of C. thalictroides saponins. In summary, our studies suggest that the relatively broad distribution of mitochondriotoxic substances in botanical dietary supplements may pose a potential health hazard, and that it is important to identify and characterize such molecules to better understand, and prevent, potential idiosyncratic adverse reactions associated with dietary supplement consumption.
EXPERIMENTAL SECTION
Sample Acquisition and Preparation
Preparation of a methanol extract from authenticated C. thalictroides sample (NCNPR code #2973), isolation and chemical characterization of the purified compounds 1–3, N-methylcytisine, cauloside H, cauloside D, cauloside B, cauloside G, leonticin D, and ciwujianoside A1 (Figure S1, Supporting Information) were previously described.17 The ground roots of C. thalictroides (NCNPR code #2973) were purchased from Mountain Rose HerbsTM (www.mountainherbs.com) in 2006. A detailed analytical characterization/authentication of the major alkaloid and triterpene saponins found in this extract were reported (with HPLC, UPLC, HPTLC chromatograms).17b The material was deposited at the NCNPR repository. Digitonin (4) and prototypical mitochondrial respiration inhibitors were from Sigma. The samples were dissolved in either DMSO or isopropanol as 10 mM stock solutions and stored at −20 °C. Unless specified, all reagents and solvents were from Sigma and prepared following manufacturer’s instructions.
Cell-Based Reporter and Proliferation/Viability Assays
Human breast tumor T47D and hepatoma Hep3B cells were from ATCC, HUVEC cells were from Lonza, and the cells were maintained as previously described.16 The T47D cell-based reporter assay with the pHRE3-TK-Luc construct to monitor HIF-1 activity and the cell-based proliferation/viability assay were the same as previously described.13,16 Cycloheximide (protein synthesis inhibitor) and rotenone (mitochondrial respiration inhibitor) were used as positive controls for the reporter assays (results shown in Figure S2, Supporting Information). Emetine and digitonin (4) were used as positive controls for the T47D and Hep3B cell proliferation/viability studies (Table 1) and 4 was used as a positive control for the HUVEC viability study (Figure 4). Cell viability was determined by the sulforhodamine B method and presented as ‘% Inhibition’ of the untreated control using the formula: . IC50 and 95% CI values were calculated from concentration-response studies that examined test compounds at concentrations in quarter-log increments, using GraphPad Prism 5.
Cell-Based Respiration Assay
An Oxytherm Clark electrode system (Hansatech) was used to monitor oxygen consumption by T47D and Hep3B cells. The experimental procedure was similar to that previously reported.13,16 The prototypical complex I inhibitor rotenone was added from an EtOH stock solution to achieve a final concentration of 1 μM where indicated. The final concentration of solvent was less than 0.3% (v/v). The following formula was used for data presentation:
Mitochondrial Membrane Potential Assay
The fluorescent dye TMRM was employed as an indicator for mitochondrial membrane potential as described.13,16 MDA-MB-231 and T47D cells loaded with TMRM for 2 h at 37 °C were treated with compounds for 30 min and live cell imaging was performed with an Axiovert 200M epifluorescence microscope (Zeiss).
Statistical Analysis
Data analyses were performed with GraphPad Prism 5. Differences between data sets were considered statistically significant when p < 0.05.
Supplementary Material
Acknowledgments
The authors thank Dr. S. L. McKnight (University of Texas Southwestern Medical Center at Dallas) for providing the pHRE3-TK-luc construct. This work was supported in part by the National Cancer Institute, NIH (grant CA98787). This investigation was conducted in a facility constructed with Research Facilities Improvement Grant C06 RR-14503 from the National Institutes of Health.
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.(a) Chan K, Truong D, Shangari N, O’Brien PJ. Expert Opin Drug Metab Toxicol. 2005;1:655–669. doi: 10.1517/17425255.1.4.655. [DOI] [PubMed] [Google Scholar]; (b) Dykens JA, Will Y. Drug Discov Today. 2007;12:777–785. doi: 10.1016/j.drudis.2007.07.013. [DOI] [PubMed] [Google Scholar]; (c) Dykens JA, Will Y. Int Drug Discov. 2010;5:32–36. [Google Scholar]
- 2.(a) Gunawan B, Kaplowitz N. Drug Metab Rev. 2004;36:301–312. doi: 10.1081/dmr-120034148. [DOI] [PubMed] [Google Scholar]; (b) Routledge PA. Drug Saf. 2008;31:416–418. doi: 10.2165/00002018-200831050-00006. [DOI] [PubMed] [Google Scholar]; (c) Teschke R, Glass X, Schulze J. Regul Toxicol Pharmacol. 2011;61:282–291. doi: 10.1016/j.yrtph.2011.08.008. [DOI] [PubMed] [Google Scholar]; (d) Teschke R, Wolff A, Frenzel C, Schulze J, Eickhoff A. Liver Int. 2012;32:1543–1556. doi: 10.1111/j.1478-3231.2012.02864.x. [DOI] [PubMed] [Google Scholar]
- 3.Smillie TJ, Khan IA. Clin Pharmacol Ther. 2010;87:175–186. doi: 10.1038/clpt.2009.287. [DOI] [PubMed] [Google Scholar]
- 4.Barnes PM, Bloom B, Nahin RL. Natl Health Stat Report. 2008;10:1–23. [PubMed] [Google Scholar]
- 5.Dugoua JJ, Perri D, Seely D, Mills E, Koren G. Can J Clin Pharmacol. 2008;15:e66–e73. [PubMed] [Google Scholar]
- 6.McFarlin BL, Gibson MH, O’Rear J, Harman P. J Nurse Midwifery. 1999;44:205–216. doi: 10.1016/s0091-2182(99)00037-3. [DOI] [PubMed] [Google Scholar]
- 7.Rader JI, Pawar RS. Anal Bioanal Chem. 2013;405:4409–4417. doi: 10.1007/s00216-013-6783-7. [DOI] [PubMed] [Google Scholar]
- 8.Semenza GL. Wiley Interdiscip Rev Syst Biol Med. 2010;2:336–361. doi: 10.1002/wsbm.69. [DOI] [PubMed] [Google Scholar]
- 9.Hodges TW, Hossain CF, Kim YP, Zhou YD, Nagle DG. J Nat Prod. 2004;67:767–771. doi: 10.1021/np030514m. [DOI] [PubMed] [Google Scholar]
- 10.Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Proc Natl Acad Sci USA. 1998;95:11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin X, David CA, Donnelly JB, Michaelides M, Chandel NS, Huang X, Warrior U, Weinberg F, Tormos KV, Fesik SW, Shen Y. Proc Natl Acad Sci USA. 2008;105:174–179. doi: 10.1073/pnas.0706585104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagle DG, Zhou Y-D. In: Comprehensive Natural Products Chemistry II. Mander LN, editor. Vol. 2. Elsevier; Oxford, UK: 2010. pp. 651–683. Chapter 2.23. [Google Scholar]
- 13.Du L, Mahdi F, Jekabsons MB, Nagle DG, Zhou YD. J Nat Prod. 2010;73:1868–1872. doi: 10.1021/np100501n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Du L, Mahdi F, Datta S, Jekabsons MB, Zhou YD, Nagle DG. J Nat Prod. 2012;75:1553–1559. doi: 10.1021/np3002892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gnaiger E. In: Drug-Induced Mitochondrial Dysfunction. Dykens JA, Will Y, editors. Chapter 12. John Wiley & Sons, Inc; Hoboken, NJ: 2008. pp. 327–352. [Google Scholar]
- 16.Liu Y, Veena CK, Morgan JB, Mohammed KA, Jekabsons MB, Nagle DG, Zhou YD. J Biol Chem. 2009;284:5859–5868. doi: 10.1074/jbc.M806744200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Ali Z, Khan IA. Phytochemistry. 2008;69:1037–1042. doi: 10.1016/j.phytochem.2007.10.011. [DOI] [PubMed] [Google Scholar]; (b) Avula B, Wang YH, Rumalla CS, Ali Z, Smillie TJ, Khan IA. J Pharm Biomed Anal. 2011;56:895–903. doi: 10.1016/j.jpba.2011.07.028. [DOI] [PubMed] [Google Scholar]
- 18.Wassler M, Jonasson I, Persson R, Fries E. Biochem J. 1987;247:407–415. doi: 10.1042/bj2470407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Boschmann M, Halangk W, Bohnensack R. Biomed Biochim Acta. 1989;48:645–652. [PubMed] [Google Scholar]; (b) Vercesi AE, Bernardes CF, Hoffmann ME, Gadelha FR, Docampo R. J Biol Chem. 1991;266:14431–14434. [PubMed] [Google Scholar]; (c) Ng H, Smith DJ, Nagley P. PLoS One. 2012;7:e42298. doi: 10.1371/journal.pone.0042298. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Gao M, Cheung KL, Lau IP, Yu WS, Fung KP, Yu B, Loo JF, Kong SK. Arch Toxicol. 2012;86:741–752. doi: 10.1007/s00204-012-0808-4. [DOI] [PubMed] [Google Scholar]
- 20.(a) Scaduto RC, Jr, Grotyohann LW. Biophys J. 1999;76:469–477. doi: 10.1016/S0006-3495(99)77214-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Joshi DC, Bakowska JC. J Vis Exp. 2011;51:2704. doi: 10.3791/2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J, Mahdi F, Du L, Jekabsons MB, Zhou Y-D, Nagle DG. Bioorg Med Chem. 2013;21:1795–1803. doi: 10.1016/j.bmc.2013.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sparg SG, Light ME, van Staden J. J Ethnopharmacol. 2004;94:219–243. doi: 10.1016/j.jep.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 23.(a) Yeh GY, Eisenberg DM, Kaptchuk TJ, Phillips RS. Diabetes Care. 2003;26:1277–1294. doi: 10.2337/diacare.26.4.1277. [DOI] [PubMed] [Google Scholar]; (b) Kozlova OI, Perederiaev OI, Ramenskaia GV. Vopr Pitan. 2011;80:67–71. [PubMed] [Google Scholar]
- 24.Francis G, Kerem Z, Makkar HP, Becker K. Br J Nutr. 2002;88:587–605. doi: 10.1079/BJN2002725. [DOI] [PubMed] [Google Scholar]
- 25.Melzig MF, Bader G, Loose R. Planta Med. 2001;67:43–48. doi: 10.1055/s-2001-10632. [DOI] [PubMed] [Google Scholar]
- 26.Hostettmann KM, Marston A. In: Chemistry and Pharmacology of Natural Products: Saponins. Hostettmann KM, Marston A, editors. Chapter 5. Cambridge University Press; New York, U.S.A: 1995. pp. 232–286. [Google Scholar]
- 27.(a) Haridas V, Higuchi M, Jayatilake GS, Bailey D, Mujoo K, Blake ME, Arntzen CJ, Gutterman JU. Proc Natl Acad Sci USA. 2001;98:5821–5826. doi: 10.1073/pnas.101619098. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhou Y, Garcia-Prieto C, Carney DA, Xu RH, Pelicano H, Kang Y, Yu W, Lou C, Kondo S, Liu J, Harris DM, Estrov Z, Keating MJ, Jin Z, Huang P. J Natl Cancer Inst. 2005;97:1781–1785. doi: 10.1093/jnci/dji404. [DOI] [PubMed] [Google Scholar]; (c) Lemeshko VV, Haridas V, Quijano Perez JC, Gutterman JU. Arch Biochem Biophys. 2006;454:114–122. doi: 10.1016/j.abb.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 28.(a) Zhou Y, Li W, Chen L, Ma S, Ping L, Yang Z. Environ Toxicol Pharmacol. 2010;29:229–234. doi: 10.1016/j.etap.2010.01.004. [DOI] [PubMed] [Google Scholar]; (b) Man S, Li Y, Fan W, Gao W, Liu Z, Li N, Zhang Y, Liu C. Int J Pharm. 2013;454:296–301. doi: 10.1016/j.ijpharm.2013.06.079. [DOI] [PubMed] [Google Scholar]
- 29.Man S, Gao W, Zhang Y, Huang L, Liu C. Fitoterapia. 2010;81:703–714. doi: 10.1016/j.fitote.2010.06.004. [DOI] [PubMed] [Google Scholar]; b) Podolak I, Galanty A, Sobolewska D. Phytochem Rev. 2010;9:425–474. doi: 10.1007/s11101-010-9183-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.(a) Ahammadsahib KI, Hollingworth RM, McGovren JP, Hui YH, McLaughlin JL. Life Sci. 1993;53:1113–1120. doi: 10.1016/0024-3205(93)90547-g. [DOI] [PubMed] [Google Scholar]; (b) McLaughlin JL. J Nat Prod. 2008;71:1311–1321. doi: 10.1021/np800191t. [DOI] [PubMed] [Google Scholar]
- 31.Veena CK, Liu Y, Mao SC, Morgan JB, Mahdi F, Jekabsons M, Nagle DG, Zhou YD. J Nat Prod. 2010;73:956–961. doi: 10.1021/np100228d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Du L, Mahdi F, Jekabsons MB, Nagle DG, Zhou YD. J Nat Prod. 2011;74:240–248. doi: 10.1021/np100762s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li J, Mahdi F, Du L, Datta S, Nagle DG, Zhou YD. J Nat Prod. 2011;74:1894–1901. doi: 10.1021/np200370z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pessayre D, Berson A, Fromenty B. In: Drug-Induced Mitochondrial Dysfunction. Dykens JA, Will Y, editors. Chapter 5. John Wiley & Sons, Inc; Hoboken, NJ: 2008. pp. 143–202. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.