

Abstract
After traumatic brain injury (TBI), arachidonic acid (ArA) is released from damaged cell membranes and metabolized to many bioactive eicosanoids, including several epoxyeicosatrienoic acids (EETs). Soluble epoxide hydrolase (Ephx2, sEH) appears to be the predominant pathway for EET metabolism to less active dihydroxyeicosatrienoates (DHETs). Prior studies indicate that brain levels of EETs increase transiently after TBI and EETs have antiinflammatory and neuroprotective activities which may benefit the injured brain. If the net effect of increased EET levels in the injured brain is beneficial to recovery, then Ephx2 gene disruption would be expected to enhance elevated EET levels and improve recovery in the injured brain. Thus, Ephx2-KO (Ephx2−/− bred onto pure C57Bl/6 background) mice were compared to wild-type controls in a unilateral controlled cortical impact model of TBI.
Before injury, animals behaved comparably in open field activity and neurologic reflexes. Interestingly, the Ephx2-KO mice showed improved motor coordination on a beam walk task, yet showed indications of defective learning in a test of working spatial memory. After surgery, brain-injured Ephx2-KO mice again had less of a deficit in the beam walk than wild-type, and the difference in latency (post – pre) showed a trend of protection for Ephx2-KO mice after TBI. Brain-injured mice showed no genotype differences in working memory. Surprisingly, sham-operated Ephx2-KO mice exhibited an injured phenotype for working memory, compared to sham-operated wild-type mice.
Brain eicosanoid levels were measured using liquid chromatography with tandem mass spectrometry. Of the 20 eicosanoids evaluated, only 8,9-EET was elevated in the Ephx2-KO cerebral cortex (37d post-surgery, in both sham and injured). Tissue DHET levels were below the limit of quantification. These results reflect a significant contribution of sEH deficiency in coordination of ambulatory movements and working spatial memory in the mouse. Further investigation of differential sEH expression and EET levels at earlier time points and across other brain regions may shed light on these behavioral differences.
INTRODUCTION
Epoxyeicosatrienoic and hydroxyeicosatetraenoic acids (EETs, HETEs) comprise a group of arachidonic acid metabolites with potent bioactivity in numerous organisms, organ systems, and cells. In the brain, these eicosanoids normally are found in trace quantities, but brain EET levels increase transiently after injury [1]. EETs and HETEs exhibit a wide variety of neurophysiological activities. For example, EETs have been identified as endothelium-derived relaxation factors in cerebrovascular beds [2, 3]. EETs are involved in cerebrovascular coupling between neurons, astrocytes, and endothelial cells in response to mechanical or direct glutamatergic stimulation [4–7]. Moreover, some eicosanoids appear to benefit the injured central nervous system, including neuroprotection from excitotoxicity [8], anti-inflammatory [9], antipyretic [10, 11], and analgesic [12–14] effects.
EETs and HETEs are synthesized by cytochrome P450 enzymes (e.g., epoxygenases, hydroxylases) and selected HETEs also are formed, via hydroperoxy intermediates, through lipoxygenase-mediated catalysis. EETs are metabolized to less active dihydroxy-eicosanoids (DHETs) via soluble epoxide hydrolase [15], and this isoform (sEH or Ephx2) appears to be the predominant EET metabolizing enzyme in renal cells [16]. In the rat brain sEH is expressed predominantly in astrocytes and also appeared to be the dominant pathway for EET metabolism [17]. Soluble epoxide hydrolase deficiency has been implicated as an important contributor to neuroprotection after ischemic brain injury [18, 19]. Eicosanoid levels, including prostanoids, leukotrienes, EETs and HETEs, have been shown to increase in the brain after injuries. Thus, we hypothesized that sEH gene disruption might increase brain EET levels and prolong their potential beneficial activity after traumatic brain injury (TBI). Here we examine functional recovery after unilateral cortical contusion TBI in Ephx2−/− and wild-type mice, and measure brain eicosanoid levels in regions associated with functional deficits observed.
MATERIALS & METHODS
Animals
All experimental protocols complied with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. All animals were handled according to IACUC approved protocols (Michigan State University, National Institute of Environmental Health Science).
Breeding & Genotyping
The sEH-deficient (Ephx2−/−) mice on a pure C57BL/6 genetic background were obtained from Darryl Zeldin’s colony at the NIEHS (Research Triangle Park, NC). Mice were genotyped using PCR-based methods [20] using the following primers: Ephx2-Common (5′-TGG CAC GAC CCT AAT CTT AGG TTC-3′); Ephx2-WT (5′-TGC ACG CTG GCA TTT TAA CAC CAG-3′), and Ephx2-KO (5′-CCA ATG ACA AGA CGC TGG GCG-3′). Homozygous null males and heterozygous females were bred to generate Ephx2-KO mice (16 male, 4 female) for this study. Four animals per cage were housed in a temperature (25 ± 3°C) and humidity (50 ± 20%) controlled room with a 12-h light, 12-h dark cycles and access to normal chow ad libitum (9% fat, 20% protein, 53% starch, and 5% fiber) plus water. Wild-type C57Bl/6 mice (age- and sex- matched) were purchased (Charles River Labs, NC) from the same housing unit as those used for backbreeding the Ephx2-KO mice.
Survival Surgery
Brain injury was induced with unilateral controlled cortical impact. Surgeries were performed exactly as described [21]. Briefly, mice were rapidly preanesthetized with 4% isoflurane, scalp plucked, and brought to a surgical plane of anesthesia with 2.5% isoflurane in oxygen via nose cone on a stereotactic platform. The eyes were lubricated and temperature was monitored by rectal thermometer and maintained at 37°C by an integrated heating mat (Harvard Instruments, Holliston, MA). The scalp was cleansed, incised, and a 3.5 mm craniectomy performed left of midline, equidistant from lambda, bregma, the midline, and the temporal ridge. Centering the piston over the somatosensory cortex, the exposed dura was subjected to impact (2 mm piston, 1 mm depth, for 100 msec at 4 m/s; AMS 201, AmScien Instruments, Richmond, VA). For sham mice, a craniectomy was performed but the animal was removed from the platform before activating the piston. Hemostasis was achieved, avoiding direct contact with the exposed cortex, and the incision was closed over the open craniectomy. Mice became ambulatory (≤ 9 min) and recovered in a warmed cage for at least 20 min. The survival rate after surgery was 100%.
Functional Measures
Upon arrival at MSU, at least one week was allowed for acclimation in the new environment. Mice were housed as above, except chow and water was witheld during behavioral and surgical procedures. Animals were 14–16 weeks of age at the initiation, at which time mice were handled daily to acclimate them to the investigator. For all tests, the examiner performing the neurological assessments was blinded to the injury status and genotype of the individual mice. There were four groups (n = 10 per group): wild-type sham, wild-type injured, Ephx2-KO sham, Ephx2-KO injured. Behavioral tests were carried out before (to establish baseline function) and after brain injury/sham surgery. Mice were evaluated in open field exploration (distance travelled, number of rearings), neurologic reflexes (SNAP score), a beam walk task (complex motor coordination), and in the Morris water maze (working spatial memory), as described [21]. Videomonitoring and data analysis were accomplished using hardware and Anymaze software from San Diego Instruments (San Diego, CA)
SNAP scoring was performed exactly as described [21]. Briefly, the SNAP score comprised 8 tests, rated on a scale of 0 (no impairment) to 5 (severely impaired): initial interaction with the handler; strength and coordination of forelimb grasp; visually stimulated forelimb placement; ambulatory circling/pacing; gait and posture; head tilt; visual fields; strength and coordination in grasping a thin applicator stick with both fore- and hindlimbs while suspended by the tail.
In the beam walk mice learned to ambulate along a narrow beam (1.25 cm × 1 m) past vertical rods on alternating sides of the beam. An aversive stimulus (bright light) was used to motivate the mice to reach a dark box at the far end of the beam. Training consisted of 4 blocks of 3 trials each over two days before surgery. Latency to reach the box was the outcome measure. Performance was reassessed at 3 days after surgery.
The Morris water maze was adapted for mice using a 1 m diameter pool with 24°C water and a 15 cm diameter submerged platform. Animals were trained to find the platform through 2 blocks of 4 swim trials per day for two days. Each mouse was started at a different point (compass points at the perimeter) for each trial in a block; if after 1 min it did not find the platform, it was placed there and allowed to orient for 15 s before removal to a warmed cage between trials. The first block was performed with a visible flag on the platform. After training was completed, at the end of the fourth block, a 1 min videotaped probe trial (with the platform removed) was run. Three probe trials were performed at 6 days after surgery to test for retention. At 30 days after surgery, mice were tested again for retention. Starting at 31 days, mice retrained with the platform in a new location, using the same paradigm, and tested for retention at day 37 after surgery. Outcome measures were latency to reach the platform (or in probe trials, a zone equal to 2 platform diameters centered on the platform), frequency and duration of visits to the platform zone, and average swim speed.
Tissue Preparation
Following the last behavioral tests on day 37, mice were euthanized by cervical dislocation and the brain rapidly dissected on ice. Cerebral hemispheres were separated then cortex, hippocampus, cerebellum, and midbrain were rapidly dissected and immediately frozen on powdered dry ice. Tissue was stored at −80°C until analyzed.
Eicosanoid Quantification
Eicosanoids levels were determined as described [22], with modifications. Organics (LC-MS grade) were purchased from Thermo-Fisher (Waltham, MA), and eicosanoid standards from Cayman Chemical (Ann Arbor, MI). Methanol, acetonitrile, and deionized water solutions were sparged with argon. Briefly, solid tissue was homogenized by high power sonication in ≥5 volumes of methanol extraction buffer containing 0.1% formic acid and 0.01% butylated hydroxytoluene (BHT, an antioxidant) and internal standards. The deuterated internal standards (4 ng each) used were: 20-HETE-d4, 15-HETE-d8, 12-HETE-d8, 5-HETE-d8; 14,15-EET-d11; 11,12-EET-d11; 8,9-EET-d11; 5,6-EET-d11; 8-isoPGF2a-d4; PGD2-d4; PGE2-d9. Homogenates of cortex were sampled for total protein (modified Bradford assay, Bio-Rad) and clarified for 10 min, 14,000 ×g, 0°C. The rest of sample preparation took place in a glove box under argon gas. Supernatants were transferred to fresh tubes on ice, diluted 10-fold with chilled water and loaded onto preconditioned Oasis HLB columns (3 cc, 60 mg, Waters Corp., Milford, MA) for solid phase extraction. Cartridges were washed with water, then with 5% methanol (2 mL each) and dried under vacuum for 20 min. Specimens were eluted with 3.0 mL acetonitrile, dried in a vacuum centrifuge and resuspended in chilled acetonitrile:water (1:1) containing 0.01% formic acid, 0.01% BHT. If not analyzed immediately, samples were stored at or below −20°C.
Separations were performed on a Xevo TQ-S (Waters) with H-class ultrahigh pressure liquid chromatography with tandem double quadrupole mass detectors in negative ionization mode (UPLC/MS/MS). A BEH column (100 × 2.1 mm, 1.7 μm resin, Waters) was used with initial mobile phase of 1:1 acetonitrile:water (each containing 0.01% formic acid) at 0.61 mL/min. The 8 min separation method was: from 0 to 5 min the acetonitrile went from 50% to 66.7% at constant rate, then immediately changed to 100% acetonitrile for 2 min, then immediately changed to initial conditions for 0.7 min. The mass spectrometric method was developed using MassLynx software (Waters) to analyze standards and analytes in separate “multiple reaction mode” functions as they exited the column, in order to maximize sensitivity and reproducibility. Eicosanoids were unambiguously resolved by ensuring minimal temporal overlap (≥ 0.3 min) for appearance of any similar mass transition fragment ions.
Testing for ion suppression by spiking 2H-standards into resuspended brain extracts showed minimal to no suppression with injections of up to 5 mg tissue equivalents (data not shown). Thus, 4 mg tissue equivalent was injected for quanification of brain eicosanoids. Recoveries (gauged by 2H-standards) ranged between 25–50% (data not shown). Standard curves of increasing analytes spiked into either mobile phase or tissue extracts gave comparable linear responses (r2 ≥ 0.998, slopes not significantly different) over the range 1 – 125 pg (mobile phase) or 2.5 – 125 pg (extracts, difference likely due to interference by endogenous levels of the analytes). Quantification of eicosanoid levels was achieved by software integration of mass transition (daughter ion) peaks at predetermined retention times, using the ratio of area under the curve for analyte (AUC, signal:noise >10) to analogous 2H-standard (or a closely related 2H-standard with previously characterized response factor) and extrapolation to mass within the linear range of standard curves constructed with constant 2H-standard concentration and increasing analyte levels. This mass was corrected to total in the original tissue specimen, and normalized by total protein in the original specimen (pg/mg protein in the original brain tissue).
Statistics
The statistical software package JMP (SAS Institute, Cary, NC) was used for analysis of variance, and Microsoft Excel was used for linear regression analysis. Group means were compared by two-way analysis of variance using Tukey’s HSD test for multiple comparisons. For post-hoc comparisons where only two groups were compared, Dunnett’s test was used. A P-value less than 0.05 was required to reject the null hypothesis that group means were equivalent.
RESULTS & DISCUSSION
Behavioral Studies
No differences were seen between wild-type and Ephx2-KO mice in control measures of exploratory activity or swim speed in the water maze (1.69±0.04 cm/s vs. 1.72±0.05 cm/s, respectively) before or after surgery. As expected, TBI induced significant deficits in both wild-type and Ephx2-KO mice in neurologic reflexes (Table 1), however, no genotype effect was observed. TBI also increased beam walk latency and induced memory deficits in the water maze. Interestingly, some genotype-specific differences were observed in both these measures before and after TBI.
Table 1. Neurologic deficits after traumatic brain injury.
Mice were examined before and 3d after surgery using a validated panel of 8 neurologic exams for interaction, limb reflexes, and coordination of movement (aSNAP score[21]). No genotype differences in SNAP score were observed before surgery.
| SNAP Scorea | ||
|---|---|---|
| Genotype | Sham | Injured |
| Wild-Type | 1.75 ± 0.50 | 4.91 ± 0.52* |
| Ephx2-KO | 1.00 ± 0.48 | 6.00 ± 0.58* |
Results are mean ± standard error, with n = 20 for training, and n = 10 animals per post-surgery group.
p<0.05 vs. Shams, Tukey HSD.
Beam Walk
During training (before surgery), Ephx2-KO outperformed wild-type mice in the beam walk with lower latency to reach the target (4.16±0.27 s vs. 6.01±0.53 s, p<0.008, Fig. 1A). This indicated an improvement in motor coordination ambulating down the narrow beam. At 3 days after surgery, sham-operated mice maintained the genotype-related effect (see Fig 1 legend). Because of this behavioral phenotype, it was necessary to analyze individual differences in performance (ΔLatency, Fig. 1B) as the outcome measure for the treatment effect after surgery/TBI.
Figure 1.
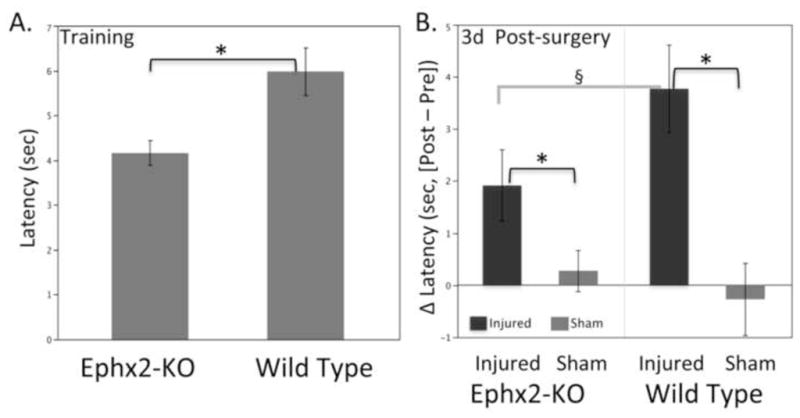
Genotype differences in beam walk performance. (A) Ephx2-KO mice showed improved ability in the beam walk task during training, before surgery/injury. (B) Difference in beam walk latency 3 days after surgery (Δ = post − pre). As expected from the genotype effect before surgery, both sham and brain-injured Ephx2-KO mice showed better latencies than respective wild-type mice (sham: 4.70±0.92 s vs. 6.74±0.80 s, p<0.05;TBI: 5.80±0.87 s vs. 8.64±0.79 s, p<0.03). Results are mean ± standard error, with n = 20 for training, and n = 10 animals per post-surgery group. *p<0.05, Tukey HSD; §p<0.10, Genotype•Treatment effect, Tukey HSD.
At 3d post-surgery, brain-injured mice showed deficits compared to their respective sham groups on the beam walk, demonstrating a brain injury effect (p<0.0003, Fig 1B). Moreover, brain-injured Ephx2-KO mice showed less of a change in latency compared to wild-type (genotype treatment effect, p<0.10, Fig 1B), suggesting some protection from TBI on this complex motor coordination task.
Water Maze
During pre-training and training in the water maze (Fig. 2), there were indications that wild-type mice learned to reach the submerged platform faster than Ephx2-KO mice (mean latencies of 6.16 ±0.39 s vs. 8.02 ±0.97 s, respectively; p = 0.06). However, in a 1 min probe trial after training was completed, there were no significant differences in latency to find the target (nor in latency to enter the target quadrant) between Ephx2-KO and wild-type mice (Table 2A). Interestingly, two secondary probe trial measures showed genotype-related differences (Table 2B,C). The frequency of visits to the target (and target quadrant) was lower for Ephx2-KO mice. Wild-type mice visited the target 8.4±0.5 times and Ephx2-KO mice did so only 6.6±0.5 (p<0.01). Duration of each visit was also reduced for Ephx2-KO compared to wild-type (p<0.02). Taking this result a step further, there were no differences in the time per visit (the quotient of frequency and duration for each mouse, Table 2D) immediately after training or post-surgery. Mice spent ~0.7 s per visit after training, then about half that time during subsequent probe trials. The decreased duration and frequency after training, with a lack of difference in latency or time per visit may have implications for soluble epoxide hydrolase in the rate of learning and retention. However, there were no apparent differences in the learning curves (training blocks 1–4, data not shown); it would likely require a much larger sample size to distinguish an effect of this sort.
Figure 2.
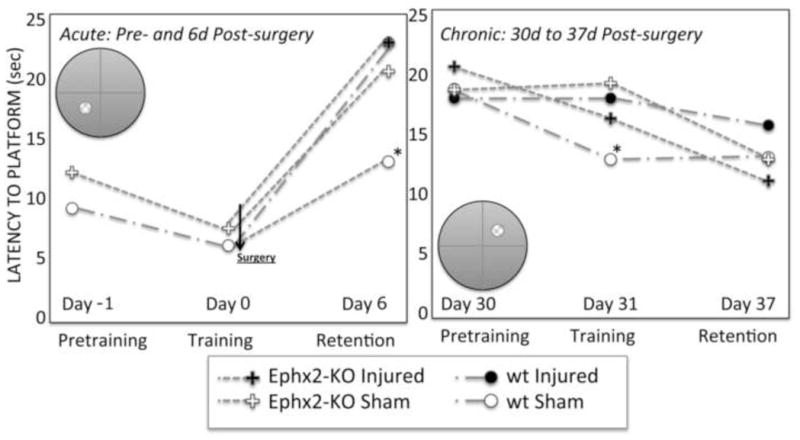
Summary of water maze learning and memory results before and after TBI. Ephx2-KO mice had impaired performance in the Morris water maze independent of TBI. Sham-operated Ephx2-KO mice exhibited an injury-like phenotype for working spatial memory at 6 days, with a suggestion of continued impairment at 37d, compared to wild-type mice. Inset diagrams show the position of the submerged platform in the training portion of each phase of the study. See Table 2, text and Methods for details. *p<0.05, Tukey HSD.
Table 2.
Genotype-Related Differences in Memory. Performance in the Morris water maze probe trials was used to test working spatial memory. A single probe trial (with the platform removed) was employed immediately after completion of training, and 3 probe trials at 6 days post-surgery to assess memory function.
| A. Platform Latency (sec) | Training | Post-surgery Retention (6 d) | New Target Retention (31 d) | ||
|---|---|---|---|---|---|
| Genotype | Sham | Injured | Sham | Injured | |
| Wild-Type | 4.34±0.66 | 13.0 ±3.4§ | 22.6 ±3.5 | 13.1 ±2.7 | 18.3 ±2.8 |
| Ephx2-KO | 5.11±0.65 | 23.0 ±3.9 | 20.4 ±3.8 | 18.5 ±3.0 | 17.1 ±3.4 |
| B. Platform Frequency (#) | Sham | Injured | Sham | Injured | |
| Wild-Type | 8.43±0.45 | 3.53 ±0.38* | 1.76 ±0.40 | 4.82 ±0.84 | 3.94 ±0.80 |
| Ephx2-KO | 6.59±0.47¥ | 2.15 ±0.44 | 2.22 ±0.44 | 3.67 ±0.93 | 3.59 ±0.88 |
| C. Platform Duration (sec) | Sham | Injured | Sham | Injured | |
| Wild-Type | 6.32±0.50 | 1.43 ±0.16* | 0.65 ±0.16 | 1.52 ±0.23 | 1.41 ±0.20 |
| Ephx2-KO | 4.57±0.36¥ | 0.74 ±0.18 | 0.79 ±0.18 | 1.39 ±0.19 | 0.93 ±0.25 |
| D. Time per Visita | Sham | Injured | Sham | Injured | |
| Wild-Type | 0.75±0.06 | 0.40 ±0.02 | 0.38 ±0.02 | 0.31 ±0.03 | 0.36 ±0.03 |
| Ephx2-KO | 0.74±0.07 | 0.34 ±0.02 | 0.34 ±0.02 | 0.42 ±0.04 | 0.26 ±0.04 |
p<0.05 vs. all other post-surgery groups;
p<0.02 vs. wild-type;
p<0.05 vs. wild-type injured, Tukey HSD.
The time per visit was calculated as (platform duration/platform frequency).
Results are mean ± standard error, with n = 20 for training, and n = 10 animals per post-surgery group.
After surgery, there were significant effects in the injured wild-type mice (increased latency, reduced frequency and duration near the target). Surprisingly, both sham and brain injured Ephx2-KO mice performed equally poorly on primary and secondary measures (Table 2, Fig. 2). Thus, both sham and injured Ephx2-KO mice exhibited what appeared to be an injured phenotype. As mentioned above, there were no genotype- or treatment-related differences in swim speed.
In a follow-up study, initiated 30 days after surgery, the same mice were trained (using the same protocol) to find the submerged platform in a new location (Fig. 2, right). Sham-operated wild-type mice acquired the new platform location better than the other groups after 2 days of training (Table 2, 31d results), but there were no significant differences in latency to target in retention studies 6 days later. There was a suggestion of differences in quadrant latencies at 37d. In the second target location, the Ephx2-KO shams showed longer quadrant latencies than wild-type shams (wild-type-sham: 6.0 ±1.9 s, Ephx2-KO-sham: 11.6 ±2.0 s*, wild-type-injured: 8.4 ±1.8 s, Ephx2-KO-injured: 11.0 ±1.9 s; *p<0.05, wt-sham vs. Ephx2-KO-sham, Tukey HSD). Though commonly used as the primary outcome measure [23, 24], the quadrant data include a larger area of the pool (25%), thus the chance of false-positive results are increased compared to the smaller area (~10%) immediately surrounding the platform. In the initial maze, both target platform and quadrant data showed corresponding results in all measures; however, the 37d quadrant data did not match up with the target platform data (Fig. 2), shedding some doubt on its robustness.
That Ephx2-KO sham-operated mice showed an injury-like phenotype was unexpected. This indicated that either the anesthesia, the craniotomy, or a combination of the surgical procedures affected their working memory. However, a review of the surgical records did not reveal any differences in the time taken for these mice to regain their pinna or cornea reflexes, or righting reflex after isoflurane anesthesia (data not shown). It is expected that complex interactions between the global genetic mutation, local compensatory responses (e.g., expression of other epoxide hydrolases), and downstream physiologic changes (perhaps an inability to adequately regulate local microcirculation in brain regions subserving memory consolidation, such as hippocampus) were responsible for the apparent susceptibility to cognitive deficits with control surgical interventions.
Brain Eicosanoid Levels
Soluble epoxide hydrolase deficient mice have been previously shown to exhibit elevated EET levels in a variety of tissues [25–28]. The levels of EETs, DHETs, and several HETEs in cerebral cortex were quantified by UPLC/MS/MS in cerebral cortex extracts (Table 3). Several prostanoids in the cyclooxygenase pathway (PGE2, PGD2, PGF2α, TXB2) were also analyzed but no differences were observed (data not shown).
Table 3.
Cerebral Cortex Eicosanoid Levels (37 d postsurgery). Levels in the contralateral cortex, expressed as pg/mg protein, corrected for recovery. Dihydroxyeicosatrienoic acid (DHET) levels, corresponding to the 4 EETs above, were below the limit of quantification in these studies. Assessments of injured tissue, even weeks after TBI, can be confounded by wound necrosis, tissue cavitation and remodelling at the site of injury (at dissection, only cortex that was neither bloody nor patently necrotic was used). Likely due to these issues, eicosanoid levels in the injured cortex had greater variability than on the opposite side of the brain (data not shown), and no genotype- or injury-related differences were detected in ipsilateral cortex.
| (pg/mg protein)
|
|||
|---|---|---|---|
| Ephx2-KO | Wild Type | ||
| 5,6-EET | Sham | 6.07 ±0.98 | 9.20 ±3.65 |
| Injured | 6.52 ±1.85 | 7.75 ±2.49 | |
|
| |||
| 8,9-EET | Sham | 5.81 ±0.45* | 2.94 ±0.73 |
| Injured | 1.68 ±0.69 | 1.78 ±0.82 | |
|
| |||
| 11,12-EET | Sham | 1.27 ±0.48 | 1.45 ±0.15 |
| Injured | 1.57 ±0.36 | 1.63 ±0.43 | |
|
| |||
| 14,15-EET | Sham | 6.36 ±1.74 | 3.56 ±1.40 |
| Injured | 6.19 ±1.32 | 6.97 ±2.65 | |
|
| |||
| 15-HETE | Sham | 39.3 ±8.3 | 39.5 ±15.0 |
| Injured | 40.2 ±6.0 | 39.6 ±11.5 | |
|
| |||
| 12-HETE | Sham | 41.6 ±14.1 | 22.8 ±4.6 |
| Injured | 41.5 ±11.1 | 48.5 ±10.7 | |
|
| |||
| 11-HETE | Sham | 48.4 ±9.9 | 45.0 ±17.0 |
| Injured | 45.3 ±7.3 | 47.0 ±17.4 | |
|
| |||
| 8-HETE | Sham | 3.82 ±0.80 | 3.07 ±1.03 |
| Injured | 3.34 ±0.68 | 4.44 ±1.38 | |
|
| |||
| 5-HETE | Sham | 8.82 ±1.75 | 8.61 ±2.42 |
| Injured | 10.3 ±1.6 | 9.44 ±2.18 | |
Mean ± standard error, with n = 5 mice per group.
p<0.05, vs. all other groups, Tukey HSD.
Except for 8,9-EET, the levels of EETs and HETEs in contralateral cortex were similar between wild-type and Ephx2-KO mice (Table 3). In sham-operated Ephx2-KO, 8,9-EET levels were increased 2-fold compared to wild-type shams and 4-fold versus either injured cortex (p < 0.05 genotype treatment effect). Post-hoc analyses showed 8,9-EET levels increased in Ephx2-KO compared to wild-type cortex (genotype effect, p < 0.05), and decreased with injury (treatment effect, p < 0.002). In Ephx2-KO, the accumulation of 8,9-EET (compared with other EETs) suggests that active synthesis occurs in mouse cerebral cortex, above the rate of degradation/diffusion. The decreased levels observed in both injured Ephx2-KO and wild-type cerebral cortex also suggests a long-lasting regulation of 8,9-EET synthesis, even 37 days after TBI. It may be important in future studies to identify the source of 8,9-EET and its suppression in the injured brain.
Soluble epoxide hydrolase hydrolyzes EETs to form dihydroxyeicosatrienoates (DHETs). Although detectable, none of the corresponding DHETs were present above the limit of quantification in the cortex. In all mice, however, levels of 14,15-DHET appeared to be several-fold greater than those of the other DHETs. Levels of DHETs were also barely detectable in rat cortex [22], so it remains to be determined whether, as in other studies [25, 27, 28], the ratios of brain EETs to their respective vicinal diols are altered in the global Ephx2-KO. In addition to EET hydrolysis, liver sEH appeared to hydrolyze hepoxylins and trioxylins that may have opposite (i.e., proinflammatory) effects to EETs [26]. Thus, local administration of sEH inhibitors or brain-specific Ephx2-KO mice may be required to elucidate the effects of sEH deficiency in the injured brain, e.g., by reducing peripheral side effects due to increased hepoxylin or trioxylin levels.
Soluble epoxide hydrolase is the presumed major metabolic pathway for EETs [29, 30]. The findings that only 8,9-EET levels were increased in shams (Ephx2-KO > wild-type), and that TBI decreased 8,9-EET levels in both Ephx2-KO and wild-type cerebral cortex indicated that, at least for what were expected to be steady-state conditions (37 d after surgery/injury), the rate of synthesis may be less than or equal to the rate of diffusion/degradation for EETs, other than for 8,9-EET. De novo production of eicosanoids increases in the brain immediately after TBI. Studies of eicosanoid-related mRNA levels in the rat cerebral cortex (using real-time quantitative polymerase chain reaction) revealed a number of cyp epoxygenases that were transiently regulated in the 7d after TBI, but Ephx2 was unchanged (K. Strauss, manuscript in preparation). Thus, regulation of the synthesis of these eicosanoids (e.g., by inducing epoxygenase, hydroxylase and/or phospholipase activities) may be more critical to EET brain levels than their (lack of) degradation. Examination of P450 eicosanoid biosynthetic activities in these brain regions, comparing naïve and operated mice would be the natural next step.
This study focused on functional outcomes and did not look at early time points, when transient changes in EETs and HETEs might have been detected. Acute P450 eicosanoid changes have been detected in a rat model of TBI at multiple acute and later time points (e.g., increased levels of 12-HETE and 14,15-EET; K. Strauss, manuscript in preparation). Another possible explanation for the predominant lack of differences between genotypes is that EETs and HETEs can be incorporated in membrane phospholipids, enabling phospholipase-mediated release of these activities [31–33]. If this was the case in mouse brain, it would be necessary to study the total epoxyeicosanoid content after phospholipid hydrolysis.
Another important consideration in the analyses is that the Ephx2 enzyme has been characterized to comprise both epoxide hydrolase and lipid phosphatase activities [34]. This activity has recently been shown to contribute significantly to cytosolic lysophosphatidic acid hydrolysis in mouse liver and lung tissue extracts [35]. It would be difficult to speculate on the physiological implications of this second pathway in the brain, however, deficiency of this activity due to Ephx2 gene disruption is a potential source of the behavioral phenotypes noted in this study. This might be further elucidated, for example, by comparing the behavior of wild-type mice treated or untreated with selective sEH inhibitors that have no effect on the lipid phosphatase activity. It will be important for changes in the lipid phosphatase activity to be assessed in future studies.
CONCLUSIONS
Soluble epoxide hydrolase deficiency interacted both with neurologic and cognitive performance, independent of brain injury. Before treatment, Ephx2-KO mice outperformed wild-type in a complex motor coordination task on the beam walk. In the Morris water maze, minor learning and no memory differences were seen in Ephx2-KO mice before treatment. After surgical treatment, brain-injured Ephx2-KO mice showed a reduced deficit in complex motor coordination compared to wild-type. Unexpectedly, sham-operated Ephx2-KO mice exhibited impaired working memory in the water maze, expressing an injury-like phenotype. Brain-injured mice showed similar deficits, i.e., no genotype differences in memory function. Further investigation of these behavioral differences may shed light on the contribution of Ephx2 in coordination of ambulatory movement, retention of memories, as well as functional recovery after TBI. Moreover, studies of other brain regions (e.g., hippocampus, critical in consolidation of short term into long term memory) and more acute time points after TBI will shed light on the importance of Ephx2 (including both epoxide hydrolase and lipid phosphatase activities) in these neurophysiologic processes. Examination of conditional knock-out mice, in which Ephx2 null mutation is induced in subsets of brain cells would help to clarify whether the present findings are dependent on local or systemic changes in the expression of this epoxide hydrolase activity.
Nevertheless, these results are consistent with an important role for soluble epoxide hydrolase and its substrates in memory consolidation, coordination of movements (from what is known about sEH and the analgesic effects of its inhibitors [12, 13, 36], possibly at the level of proprioception), and in neuroprotection in the dynamic context of brain injury.
Highlights.
Ephx2-KO had better beam walk performance than wild-type mice before and after TBI
Ephx2-KO mice had impaired performance in the Morris water maze independent of TBI
Sham-operated Ephx2-KO mice had an injured phenotype for working spatial memory
Ephx2-KO mice exhibited elevated levels of 8,9-EET in the cerebral cortex compared to wild-type mice
DHET metabolites were below the level of quantification in mouse cerebral cortex.
Acknowledgments
This work was funded in part by NINDS Grant NS054890 (KIS). The authors would like to thank Laura Miller, J. Alyce Bradbury and Joan P. Graves for managing the animal colony, confirming genotypes and coordinating shipments. Thanks also to Analise Sauro and Jaimie Pineda for excellent technical assistance in the completion of this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Kenneth I. Strauss, Email: kenneth.strauss@hc.msu.edu.
Artiom Gruzdev, Email: artiom.gruzdev@nih.gov.
Darryl C. Zeldin, Email: zeldin@niehs.nih.gov.
References
- 1.Gopez JJ, Yue H, Vasudevan R, et al. Cyclooxygenase-2-specific inhibitor improves functional outcomes, provides neuroprotection, and reduces inflammation in a rat model of traumatic brain injury. Neurosurgery. 2005;56:590–604. doi: 10.1227/01.NEU.0000154060.14900.8F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Campbell WB. New role for epoxyeicosatrienoic acids as anti-inflammatory mediators. Trends Pharmacol Sci. 2000;21:125–7. doi: 10.1016/s0165-6147(00)01472-3. [DOI] [PubMed] [Google Scholar]
- 3.Higashimori H, Blanco VM, Tuniki VR, Falck JR, Filosa JA. Role of epoxyeicosatrienoic acids as autocrine metabolites in glutamate-mediated K+ signaling in perivascular astrocytes. Am J Physiol Cell Physiol. 2010;299:C1068–78. doi: 10.1152/ajpcell.00225.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alkayed NJ, Narayanan J, Gebremedhin D, Medhora M, Roman RJ, Harder DR. Molecular characterization of an arachidonic acid epoxygenase in rat brain astrocytes. Stroke. 1996;27:971–9. doi: 10.1161/01.str.27.5.971. [DOI] [PubMed] [Google Scholar]
- 5.Harder DR, Alkayed NJ, Lange AR, Gebremedhin D, Roman RJ. Functional hyperemia in the brain: hypothesis for astrocyte-derived vasodilator metabolites. Stroke. 1998;29:229–34. doi: 10.1161/01.str.29.1.229. [DOI] [PubMed] [Google Scholar]
- 6.Iliff JJ, Wang R, Zeldin DC, Alkayed NJ. Epoxyeicosanoids as mediators of neurogenic vasodilation in cerebral vessels. Am J Physiol Heart Circ Physiol. 2009;296:H1352–63. doi: 10.1152/ajpheart.00950.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peng X, Carhuapoma JR, Bhardwaj A, et al. Suppression of cortical functional hyperemia to vibrissal stimulation in the rat by epoxygenase inhibitors. American Journal of Physiology - Heart & Circulatory Physiology. 2002;283:H2029–37. doi: 10.1152/ajpheart.01130.2000. [DOI] [PubMed] [Google Scholar]
- 8.Hampson AJ, Grimaldi M. 12-hydroxyeicosatetrenoate (12-HETE) attenuates AMPA receptor-mediated neurotoxicity: evidence for a G-protein-coupled HETE receptor. Journal of Neuroscience. 2002;22:257–64. doi: 10.1523/JNEUROSCI.22-01-00257.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Node K, Huo YQ, Ruan XL, et al. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999;285:1276–9. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kozak W, Kluger MJ, Kozak A, Wachulec M, Dokladny K. Role of cytochrome P-450 in endogenous antipyresis. American Journal of Physiology - Regulatory Integrative & Comparative Physiology. 2000;279:R455–60. doi: 10.1152/ajpregu.2000.279.2.R455. [DOI] [PubMed] [Google Scholar]
- 11.Nakashima T, Harada Y, Miyata S, Kiyohara T. Inhibitors of cytochrome P-450 augment fever induced by interleukin-1 beta. American Journal of Physiology. 1996;271:R1274–9. doi: 10.1152/ajpregu.1996.271.5.R1274. [DOI] [PubMed] [Google Scholar]
- 12.Inceoglu B, Schmelzer KR, Morisseau C, Jinks SL, Hammock BD. Soluble epoxide hydrolase inhibition reveals novel biological functions of epoxyeicosatrienoic acids (EETs) Prostaglandins Other Lipid Mediat. 2007;82:42–9. doi: 10.1016/j.prostaglandins.2006.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Inceoglu B, Wagner K, Schebb NH, et al. Analgesia mediated by soluble epoxide hydrolase inhibitors is dependent on cAMP. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:5093–7. doi: 10.1073/pnas.1101073108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christie MJ, Vaughan CW, Ingram SL. Opioids, NSAIDs and 5-lipoxygenase inhibitors act synergistically in brain via arachidonic acid metabolism. Inflammation Research. 1999;48:1–4. doi: 10.1007/s000110050367. [DOI] [PubMed] [Google Scholar]
- 15.Chacos N, Capdevila J, Falck JR, et al. The reaction of arachidonic acid epoxides (epoxyeicosatrienoic acids) with a cytosolic epoxide hydrolase. Arch Biochem Biophys. 1983;223:639–48. doi: 10.1016/0003-9861(83)90628-8. [DOI] [PubMed] [Google Scholar]
- 16.Yu Z, Xu F, Huse LM, et al. Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ Res. 2000;87:992–8. doi: 10.1161/01.res.87.11.992. [DOI] [PubMed] [Google Scholar]
- 17.Marowsky A, Burgener J, Falck JR, Fritschy JM, Arand M. Distribution of soluble and microsomal epoxide hydrolase in the mouse brain and its contribution to cerebral epoxyeicosatrienoic acid metabolism. Neuroscience. 2009;163:646–61. doi: 10.1016/j.neuroscience.2009.06.033. [DOI] [PubMed] [Google Scholar]
- 18.Koerner IP, Jacks R, DeBarber AE, et al. Polymorphisms in the human soluble epoxide hydrolase gene EPHX2 linked to neuronal survival after ischemic injury. J Neurosci. 2007;27:4642–9. doi: 10.1523/JNEUROSCI.0056-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang W, Otsuka T, Sugo N, et al. Soluble epoxide hydrolase gene deletion is protective against experimental cerebral ischemia. Stroke. 2008;39:2073–8. doi: 10.1161/STROKEAHA.107.508325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sinal CJ, Miyata M, Tohkin M, Nagata K, Bend JR, Gonzalez FJ. Targeted disruption of soluble epoxide hydrolase reveals a role in blood pressure regulation. J Biol Chem. 2000;275:40504–10. doi: 10.1074/jbc.M008106200. [DOI] [PubMed] [Google Scholar]
- 21.Shelton SB, Pettigrew DB, Hermann AD, et al. A simple, efficient tool for assessment of mice after unilateral cortex injury. J Neurosci Methods. 2008 doi: 10.1016/j.jneumeth.2007.11.003. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yue H, Jansen SA, Strauss KI, et al. A liquid chromatography/mass spectrometric method for simultaneous analysis of arachidonic acid and its endogenous eicosanoid metabolites prostaglandins, dihydroxyeicosatrienoic acids, hydroxyeicosatetraenoic acids, and epoxyeicosatrienoic acids in rat brain tissue. Journal of Pharmaceutical and Biomedical Analysis. 2007;43:1122–34. doi: 10.1016/j.jpba.2006.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McAuliffe JJ, Miles L, Vorhees CV. Adult neurological function following neonatal hypoxia-ischemia in a mouse model of the term neonate: water maze performance is dependent on separable cognitive and motor components. Brain Res. 2006;1118:208–21. doi: 10.1016/j.brainres.2006.08.030. [DOI] [PubMed] [Google Scholar]
- 24.Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 25.Seubert JM, Sinal CJ, Graves J, et al. Role of soluble epoxide hydrolase in postischemic recovery of heart contractile function. Circ Res. 2006;99:442–50. doi: 10.1161/01.RES.0000237390.92932.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cronin A, Decker M, Arand M. Mammalian soluble epoxide hydrolase is identical to liver hepoxilin hydrolase. J Lipid Res. 2011;52:712–9. doi: 10.1194/jlr.M009639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Revermann M, Schloss M, Barbosa-Sicard E, et al. Soluble epoxide hydrolase deficiency attenuates neointima formation in the femoral cuff model of hyperlipidemic mice. Arterioscler Thromb Vasc Biol. 2010;30:909–14. doi: 10.1161/ATVBAHA.110.204099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luria A, Weldon SM, Kabcenell AK, et al. Compensatory mechanism for homeostatic blood pressure regulation in Ephx2 gene-disrupted mice. The Journal of biological chemistry. 2007;282:2891–8. doi: 10.1074/jbc.M608057200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zeldin DC, Kobayashi J, Falck JR, et al. Regio- and enantiofacial selectivity of epoxyeicosatrienoic acid hydration by cytosolic epoxide hydrolase. The Journal of biological chemistry. 1993;268:6402–7. [PubMed] [Google Scholar]
- 30.Fang X, Kaduce TL, Weintraub NL, et al. Pathways of epoxyeicosatrienoic acid metabolism in endothelial cells Implications for the vascular effects of soluble epoxide hydrolase inhibition. Journal of Biological Chemistry. 2001;276:14867–74. doi: 10.1074/jbc.M011761200. [DOI] [PubMed] [Google Scholar]
- 31.Capdevila JH, Kishore V, Dishman E, Blair IA, Falck JR. A novel pool of rat liver inositol and ethanolamine phospholipids contains epoxyeicosatrienoic acids (EETs) Biochemical & Biophysical Research Communications. 1987;146:638–44. doi: 10.1016/0006-291x(87)90576-6. [DOI] [PubMed] [Google Scholar]
- 32.Karara A, Dishman E, Falck JR, Capdevila JH. Endogenous epoxyeicosatrienoyl-phospholipids A novel class of cellular glycerolipids containing epoxidized arachidonate moieties. Journal of Biological Chemistry. 1991;266:7561–9. [PubMed] [Google Scholar]
- 33.Brezinski ME, Serhan CN. Selective incorporation of (15S)-hydroxyeicosatetraenoic acid in phosphatidylinositol of human neutrophils: agonist-induced deacylation and transformation of stored hydroxyeicosanoids. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:6248–52. doi: 10.1073/pnas.87.16.6248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Newman JW, Morisseau C, Harris TR, Hammock BD. The soluble epoxide hydrolase encoded by EPXH2 is a bifunctional enzyme with novel lipid phosphate phosphatase activity. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:1558–63. doi: 10.1073/pnas.0437724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morisseau C, Schebb NH, Dong H, Ulu A, Aronov PA, Hammock BD. Role of soluble epoxide hydrolase phosphatase activity in the metabolism of lysophosphatidic acids. Biochemical and biophysical research communications. 2012;419:796–800. doi: 10.1016/j.bbrc.2012.02.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmelzer KR, Inceoglu B, Kubala L, et al. Enhancement of antinociception by coadministration of nonsteroidal anti-inflammatory drugs and soluble epoxide hydrolase inhibitors. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:13646–51. doi: 10.1073/pnas.0605908103. [DOI] [PMC free article] [PubMed] [Google Scholar]