

Abstract
Five lipopeptides of the lyngbyabellin structure class, four cyclic (1-3, and 5) and one linear (4), were isolated from the extracts of two collections of filamentous marine cyanobacteria obtained from Palmyra Atoll in the Central Pacific Ocean. Their planar structures and absolute configurations were elucidated by combined spectroscopic and chromatographic analyses as well as chemical synthesis of fragments. In addition to structural features typical of the lyngbyabellins, such as two thiazole rings and a chlorinated 2-methyloctanoate residue, these new compounds possess several unique aspects. Of note, metabolites 2 and 3 possessed rare mono-chlorination on the 3-acyloxy-2-methyloctanoate residue while lyngbyabellin N (5) had an unusual N,N-dimethylvaline terminus. Lyngbyabellin N also possessed a leucine statine residue, and showed strong cytotoxic activity against HCT116 colon cancer cell line (IC50 = 40.9 ± 3.3 nM).
Keywords: Natural Products, Peptides, Structure Elucidation, Biological Activity
Introduction
Over the last 20 years, marine cyanobacteria have emerged as exceptionally prolific producers of biologically active secondary metabolites rivaling the metabolic richness of the actinobacteria.[1] Because they lack other more visible defense mechanisms, such as a hardened exterior or a cryptic habitat, and have an overall macroscopic structure, it is thought that cyanobacteria derive value from the biosynthesis of these structurally intriguing secondary metabolites for their chemical defense.[2] The genus Moorea (formally Lyngbya spp) is one of the most chemically prolific and has yielded such important metabolites as the apratoxins,[3] antillatoxin A,[4] lyngbyatoxin A,[5] curacin A,[6] barbamide,[7] the jamaicamides[8] and the malyngamides.[9] In general, these structurally diverse metabolites exhibit a range of interesting biological activities, such as anti-cancer,[10] anti-feedant,[11] molluscicidal,[12] anti-inflammatory[13] and neuromodulatory[14]. The lyngbyabellins are another family of metabolites produced by Moorea sp. which are NRPS/PKS derived peptides and have a recognizable architecture composed of thiazole rings, hydroxy acid residues, and an acyl group with distinctive chlorination at the penultimate carbon atom.[15] Several of the lyngbyabellins are reported to exhibit moderate to potent cytotoxicity to various cancer cell lines and to exert this activity through interference with the actin system.[15]
In the present work, a number of filamentous marine cyanobacteria were collected from Palmyra Atoll (approximately 1000 miles SSW of Hawaii) in 2008 and 2009, and their extracts were evaluated in several biological assays. A few of the reduced complexity fractions from two extracts, both subsequently identified as Moorea bouillonii, were found to be highly cytotoxic to H-460 human lung cancer cells in vitro [52% survival at 30 μg/mL (fraction F from the 2009 collection), and 20% survival at 3 μg/mL (fraction H from the 2008 collection)], and these were chosen for further investigation. Bioassay-guided fractionation of these extracts yielded five new peptides, lyngbyabellin K-N (1-5), and these were fully structurally defined by spectrochemical methods. Additionally, the known metabolites apratoxins F and G were also isolated from these fractions, and accounted for the majority of the remarkable cytotoxicity of the crude fractions.
Results and Discussion
Collection and isolation of lyngbyabellins K-N (1-5)
A collection of tube-like mats of the filamentous marine cyanobacterium M. bouillonii (PAL/3/09-1) was obtained via SCUBA in the north lagoon at Strawn Island, Palmyra Atoll, USA, in August 2009. The ethanol-preserved collection was subsequently repeatedly extracted with CH2Cl2/CH3OH (2:1) and then fractionated by silica gel vacuum column chromatography (VLC) to produce nine fractions (A–I). The F fraction was found to possess moderate anti-cancer activity against H-460 human lung cancer cells (52% survival at 30 μg/mL). Thus, this fraction was further chromatographed by RP HPLC to afford lyngbyabellins K (1, 10 mg, 0.26%) and M (4, 2 mg, 0.05%) as well as mixtures of two epimers of lyngbyabellin L (2 and 3). This latter mixture was subjected to chiral HPLC to ultimately yield pure lyngbyabellin L (2, 0.7 mg, 0.018%) and 7-epi-lyngbyabellin L (3. 0.6 mg, 0.015%).
An additional sample of M. bouillonii (PAL 8/16/08-3) was collected by SCUBA in 2008 from reefs 9-15 m deep surrounding Palmyra Atoll. This latter ethanol-preserved material was repetitively extracted (CH2Cl2/CH3OH 2:1) and fractionated using normal-phase VLC to yield nine fractions. Three polar fractions (100% EtOAc, 25% EtOAc/CH3OH, and 75% EtOAc/CH3OH) were strongly cytotoxic to H-460 cancer cells (20% survival at 3 μg/mL) and were thus fractionated with reverse phase solid phase extraction (RP-SPE) followed by preparative thin layer chromatography (prepTLC) to yield 4.3 mg of highly purified lyngbyabellin N (5) as a pale yellow oil. These same fractions also contained several known apratoxins, and these latter compounds were responsible for a majority of their cytotoxicity.[3]
Structures of lyngbyabellins K-N (1-3, 5) and 7-epi-lyngbyabellin L (4)
Lyngbyabellin K (1), a pale yellow oil, showed an ion cluster at m/z 578.99/580.96/582.96 in a ratio of 100:85:25 by LR ESIMS, indicating the presence of two chlorine atoms in the molecule. The molecular formula of 1 was determined as C23H28Cl2N2O7S2 by interpretation of HR ESITOFMS data (m/z [M+Na]+ 601.0609). The IR spectrum of 1 displayed absorption bands at 3421, 1737, and 1616 cm−1, indicating the presence of hydroxy, ester and amide functionalities, respectively. The 13C NMR spectra of 1 in CDCl3 showed five downfield shifted quaternary carbons (δ = 174.3, 169.8, 169.1, 161.0, and 160.2) that made double bonds with O or N and two carbon-carbon double bonds (δ = 146.4, 145.8, 128.9, and 128.5) accounting for seven of the ten degrees of unsaturation. Two downfield shifted singlet protons at δ = 8.13, and 8.17 with attached carbon atoms at δ = 128.9 and 128.5, respectively, were reflective of two 2,4-disubstituted thiazole rings, accounting for two more degrees of unsaturation. The final degree of unsaturation was thus deduced to constitute an overall macrocyclic structure for 1.
The two thiazole ring structures were confirmed by HMBC correlations from 12-H (δ = 8.13) to C-11 (δ = 145.8) and C-13 (δ = 169.8), and 18-H (δ = 8.17) to C-17 (δ = 146.4) and C-19 (δ = 169.1), respectively (Table 1). The HMBC correlations from 12-H and 18-H of the two thiazole rings to carbonyl carbons C-10 (δ = 160.2) and C-16 (δ = 161.0) indicated that carboxylic acid derivatives were directly attached to the 4 position of each of thiazole ring. In addition, COSY correlations between 14-OH (δ = 5.73), 14-H (δ = 5.39), and 15-Ha (δ = 4.74) and 15-Hb (δ = 4.65), as well as HMBC correlations from 15-Ha and 15-Hb to C-13 and C-16, established that a 1,2-dihydroxyethyl moiety formed a linkage between the two thiazole-4-carboxylates (Figure 1).
Table 1.
NMR Spectral Data for Lyngbyabellin K (1) in CDCl3 at 500 MHz (1H) and 125 MHz (13C)
| 1 | 2 | 3 | 4 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Position | δC [ppm] | δH [ppm] multi (J in Hz) | COSY | HMBC | δ C | δ H | δ C | δ H | δ C | δ H |
| 1 | 174.3 | 174.6 | 174.6 | 174.4 | ||||||
| 2 | 43.2 | 3.18 dt (9.4, 7.3) | 3, 9 | 1, 3, 9 | 42.7 | 3.31 | 42.6 | 3.31 | 44.4 | 2.84 |
| 3 | 75.1 | 5.36 brs | 2, 4a, 4b | 1, 10 | 75.6 | 5.33 | 75.4 | 5.34 | 75.4 | 3.80 |
| 3-OH | 5.51 | |||||||||
| 4a | 30.7 | 1.92 m | 3, 4b, 5 | 29.9 | 1.91 | 30.3 | 1.95 | 34.4 | 1.93 | |
| 4b | 1.73 m | 3, 4a, 5 | 3, 5, 6 | 1.72 | 1.67 | |||||
| 5a | 20.5 | 1.75 m | 4, 6a, 6b | 3, 4, 6, 7 | 20.5 | 1.61 | 21.0 | 1.63 | 21.5 | 1.86 |
| 5b | 1.55 | 1.52 | ||||||||
| 6a | 49.5 | 2.30 m | 5, 6b | 4, 5, 7, 8 | 40.1 | 1.77 | 40.3 | 1.77 | 50.0 | 2.24 |
| 6b | 2.15 m | 5, 6a | 4, 5, 7, 8 | 1.66 | 1.66 | |||||
| 7 | 90.4 | 58.2 | 4.02 | 58.5 | 3.98 | 90.8 | ||||
| 8 | 37.6 | 2.12 s | 6, 7 | 25.5 | 1.49 | 25.4 | 1.49 | 37.7 | 2.16 | |
| 9 | 15.3 | 1.30 d (7.3) | 2 | 1, 2, 3 | 15.3 | 1.30 | 15.1 | 1.30 | 15.0 | 1.26 |
| 10 | 160.2 | 160.0 | 160.4 | 161.8 | ||||||
| 11 | 145.8 | 145.7 | 145.9 | 147.6 | ||||||
| 12 | 128.5 | 8.13 s | 10, 11, 13 | 128.3 | 8.15 | 128.2 | 8.14 | 128.2 | 8.16 | |
| 13 | 169.8 | 169.7 | 169.2 | 171.2 | ||||||
| 14 | 70.1 | 5.39 brs | 14-OH, 15a, 15b | 70.1 | 5.35 | 70.0 | 5.33 | 70.30 | 5.41 | |
| 14-OH | 5.73 brs | 14 | 6.25 | 6.24 | 6.68 | |||||
| 15a | 69.1 | 4.74 dd (11.3, 2.3) | 14 | 13, 16 | 69.9 | 4.76 | 69.4 | 4.76 | 70.26 | 4.95 |
| 15b | 4.65 dd (11.3, 3.0) | 14 | 13, 14, 16 | 4.58 | 4.57 | 4.30 | ||||
| 16 | 161.0 | 160.8 | 160.7 | 161.7 | ||||||
| 17 | 146.4 | 146.1 | 145.5 | 145.9 | ||||||
| 18 | 128.9 | 8.17 s | 16, 17, 19 | 128.9 | 8.16 | 128.9 | 8.16 | 129.2 | 8.30 | |
| 19 | 169.1 | 169.9 | 169.7 | 170.5 | ||||||
| 20 | 77.5 | 5.53 d (6.6) | 21 | 1, 19, 21, 22, 23 | 77.6 | 5.51 | 77.4 | 5.51 | 77.5 | 5.99 |
| 21 | 33.6 | 2.37 m | 20, 22, 23 | 19, 20, 22, 23 | 34.3 | 2.30 | 34.8 | 2.32 | 34.4 | 2.33 |
| 22 | 19.3 | 0.99 d (6.5) | 21 | 20, 21, 23 | 19.2 | 1.04 | 19.0 | 1.03 | 19.5 | 1.13 |
| 23 | 17.9 | 1.07 d (6.5) | 21 | 20, 21, 22 | 17.4 | 1.06 | 17.3 | 1.07 | 16.8 | 0.98 |
| 24 | 61.8 | 4.42 | ||||||||
| 25 | 14.7 | 1.41 | ||||||||
Figure 1.

Structures of lyngbyabellin K-N (1-5) and lyngbabellin H (6).
Further inspection of the 1H NMR spectrum of 1 revealed a series of upfield and highly coupled resonances reflective of an aliphatic chain. Additionally, a downfield methyl singlet signal at δ = 2.12 (8-H3) showed HMBC correlations to a quaternary carbon at δ = 90.4 (C-7) as well as a methylene carbon at δ = 49.5 (C-6). The chemical shift of C-7 (δ = 90.4) was indicative of a gem-dichloro substituent, as observed in dolabellin[16], hectochlorin[17] and the lyngbyabellins,[15] and thus accounted for the two chlorine atoms in the molecular formula of 1. This moiety was extended to include an additional six carbons (C-1 to C-5, and C-9, see Table 1) by integrated reasoning of COSY, HSQC and HMBC data, and identified this moiety as 7,7-dichloro-3-acyloxy-2-methyloctanoate (DCAMO). Additional HMBC correlations from 3-H of the DCAMO residue to a carbonyl carbon, C-10 of the first thiazole-4-carboxylate unit, allowed connection between these atoms via an ester bond.
Sequential COSY correlations between adjacent methine protons 20-H (δ = 5.53) and 21-H (δ = 2.24) to both doublet methyl groups 22-H3 and 23-H3, along with HMBC correlations from these two methyl groups back to C-20 and C-21, defined the side chain of a 2-hydroxyisovaleric acid (HIVA) residue. The HMBC correlations from 20-H to C-1 (δ = 174.3 ) and C-19 (δ = 169.1) supported positioning this HIVA-derived residue between C-19 and C-1, as shown in Figure 1, and completed the assignment of all atoms from the molecular formula of lyngbyabellin K (1). Additionally, from the above discussion, the sequence of residues in the macrocyclic ring was defined as DCAMO – HIVA – thiazole-1 carboxylate – glyceric acid – thiazole-2 carboxylate – DCAMO.
To define the absolute configuration at each stereocenter, lyngbyabellin K (1) was crystallized from CH3CN. A needle shaped crystal (monoclinic, 0.21 × 0.11 × 0.05 mm) were subjected to single-crystal X-ray diffraction analysis. Resulting from the presence of heavy atoms (chlorine and sulfur) in compound 1, the absolute configuration could be deduced as 2S, 3S, 14R, 20S [final R(F2) = −0.02(2), see Supporting Information].
Lyngbyabellin L (2) and 7-epi-lyngbyabellin L (3) showed essentially identical sodiated parent ion clusters by HR ESITOF MS at m/z values 567.1000 and 569.0980 in a ratio of 78:32, indicating that both possessed the same molecular formula of C23H28ClN2O7S2 containing a single chlorine atom each. The 1H and 13C NMR spectra of 2 possessed several resonances similar to those of 1 (Table 1), thus confirming the presence of two extended 2,4-disubstituted thiazole units, a HIVA unit and a 1,2-dihydroxyethyl moiety (Figure 1). However, in the 1H and 13C NMR spectra of 2, a new doublet methyl signal appeared at δ = 1.49 (8-H3) and a downfield shifted methine was observed at δ 4.02 (7-H) and δ 58.2 (C-7). These new features were accompanied by several missing resonances relative to 1, namely the distinctive methyl singlet methyl at δ = 2.12 (8-H3) and the quaternary gemdichloro carbon at δ = 90.4 (C-7). Interpretation of these data in combination with the altered molecular formula suggested that 2 was the mono-chloro species at C-7. The spectroscopic and 2D NMR linkage data as well as the HR ESITOFMS of compound 3 were almost identical to those of 2, and thus 3 was deduced to be stereoisomeric with compound 2 at C-7.
Because the planar structures of 2 and 3 are closely related to that of 1, except for the alterations at C-7, the similar 13C NMR shifts, optical rotations and CD absorption measurements for all three compounds indicate that are of the same enantiomeric series at all comparable centers (see Supporting Information). Therefore, the absolute configurations of C-2, C-3, C-14, and C-20 of 2 and 3 were assigned as for lyngbyabellin K (1), namely 2S, 3S, 14R, and 20S.
Several different approaches were employed to determine the absolute configuration of C-7 in lyngbyabellin L (2) and its C-7 epimer (3). First, a detailed analysis of the 1H NMR data for compound 2 using the DQF-COSY experiment revealed all of the vicinal 1H-1H coupling constants in the 7-chloro-3-acyloxy-2-methyloctanoate (CAMO) residue (Figure 2a). The large vicinal coupling constants (> 7 Hz) of 2-H–3-H and 3-H–4-Hb in 2 indicated that these protons were anti to one another. Conversely, the relatively small vicinal coupling constant of 3-H–4-Ha (J = 3 Hz) showed that they were in a gauche relationship. NOESY correlations between 2-H/9-H3–4-Ha indicated 4-Ha and 4-Hb to be pro-S and pro-R, respectively. The large vicinal coupling constants between 4-Ha and 5-Hb, as well as the NOESY correlation between 5-Ha and 3-H, allowed assignment of 5-Ha and 5-Hb as pro-S and pro-R, respectively. Similarly, a large vicinal coupling constant observed between 5-Ha and 6-Hb and NOESY correlation between 4-Hb and 6-Hb indicated 6-Ha and 6-Hb to be pro-R and pro-S. Finally, a large coupling constant between 6-Ha and 7-H, small vicinal coupling constant between 6-Hb and 7-H, and NOESY correlations between 7-H with 5-Ha and 5-Hb and 8-H3 with 6-Ha and 6-Hb, revealed that the absolute configuration at C-7 of 2 was R. In a similar fashion, a detailed coupling constant analysis for the DCAMO residue of 3 revealed that the absolute configuration at C-7 to be S (Figure 2a).
Figure 2.

Approaches to determination of relative configuration of the C-7 stereocenter in the DCAMO residue of lyngbyabellin L (2) and 7-epi-lyngbyabellin L (3): a) homonuclear coupling constant and nOe-based determination of relative configuration b) J-based configuration analysis on lyngbyabellin L (2), and c) deshielding effects to non-equivalent protons from the chlorine atom at C-7 and oxygen atom at C-3 of lyngbyabellin L (2) and 7-epi-lyngbyabellin L (3). [a] The coupling constant is not observed due to overlap. [b] The coupling constant is not detected due to weak signal.
Finally, the prochirality of each proton attached to C-4, C-5 and C-6 as well as the absolute configuration at C-7 in 2 was confirmed by J-based configurational analysis as shown in Figure 2c. [18] A large homonuclear coupling constant, as measured by the DQF-COSY experiment, between 2-H and 3-H (3J2-H, 3-H = 11.0 Hz) indicated an anti relationship between these protons. The HETLOC experiment was used to measure a large heteronuclear coupling constant between H-2 and C-3 (2J2-H,C-3 = − 6.4 Hz), and in conjunction with NOESY correlations between 9-H3 and 3-H/4-Ha, led to the assignment of the relative stereochemistry of C-12–C-13 as the erythro rotamer B-3. Heteronuclear coupling constants between C-3/4-Ha (2JC-3,4-Ha = 0 Hz) and C-3/4-Hb (2JC-3,4-Hb = − 5.9 Hz) also indicated that 4-Ha and the oxygen substituent at C-3 were in an anti relationship whereas 4-Hb and the C-3 oxygen atom were gauche. The small heteronuclear coupling constants between 4-Ha/C-6, 5-Ha/C-3, 5-Hb/C-3 (3J4-Ha,C-6 = 1.5 Hz, 3JC-3,5-Ha = 1.0 Hz, 3JC-3,5-Hb = 0.8 Hz) revealed that the paired protons and carbons were gauche to one another (Figure 2b). Additional small heteronuclear coupling constants between 5-Ha/C-7 and 5-Hb/C-7 (3J5-Ha,C-7 = 2.1 Hz, 3J5-Hb,C-7 = 1.7 Hz) confirmed that C-7 was gauche to both 5-Ha and 5-Hb. Further small heteronuclear coupling constants between 7-H/C-5, 6-Ha/C-8, and 6-Hb/C-8 indicated that these paired atoms were in gauche relationships. Thus, based on this J-based configuration analysis and the absolute configuration of C-2 and C-3, the absolute configuration at C-7 in 2 was deduced to be R, consistent with the previous analysis of homonuclear coupling constants and NOESY correlations. These assignments were supported by a reversal in chemical shifts for the pro-R and pro-S protons attached to C-5 in 2 (δ = 1.55 and 1.61) as versus compound 3 (δ = 1.63 and 1.52), which we interpret to reflect deshielding effects to these non- equivalent protons from the chlorine atom at C-7 and oxygen atom at C-3[19] (Figure 2c).
The LR ESIMS of lyngbyabellin M (4) showed a parent ion cluster at m/z 624.95/626.95/628.93 in the ratio of 100:80:20, indicating the presence of two chlorine atoms as found in metabolite 1. The molecular formula of 4 was determined as C25H34N2O8Cl2S2 by HR ESITOFMS m/z [M+Na]+ 647.1029 (calcd for C25H34N2O8Cl2S2Na 647.1026). Both the 1H and 13C NMR spectra of 4 showed many similar features to those of 1. However, as a point of difference, 3-H in lyngbyabellin K (1) was at δ = 5.36 whereas this proton in 4 was observed at δ = 3.80. This latter proton in 4 showed a unique COSY correlation with an additional exchangeable proton found at δ = 5.51. Furthermore, in lyngbyabellin M a new methylene quartet (24-H2) and methyl triplet (25-H3) were observed at δ = 4.42 and 1.41, respectively, and these were connected to one another by COSY. An HMBC correlation from 24-H2 to C-10 confirmed that the ester linkage between C-3 and C-10 of 1 had been cleaved and the carboxylic acid at C-10 in 4 was esterified with an ethyl moiety. By the close comparability in 13C NMR shifts at most carbon positions and similar optical rotation, we propose lyngbyabellin M (4) to be of the same enantiomeric series as lyngbyabellin K (1).
The relative configuration at C-2 and C-3 of the DCAMO residue in lyngbyabellin M (4) was assigned as erythro by comparing 3J2-H,3-H of 4 (6.2 Hz) to the coupling constants of model diastereomers of methyl 3-hydroxy-2-methyl-octanoate (3J2-H,3-H for erythro isomer = 6.3 Hz; 3J2-H,3-H for threo isomer = 3.6 Hz).16 The absolute configuration at these two centers in compound 4 were revealed by NMR analyses of the di-Mosher esters produced by esterification of the C-3 and C-14 hydroxy groups. Calculation of ΔδS-R values for protons near to C-3 and C-14 allowed assignment of the absolute configuration as 3S and 14R, respectively (Figure 3). Finally, the absolute configuration of the HIVA residue in 4 was determined as l by GC-MS analysis of the methyl ester derivative following acid hydrolysis and comparison with standards, and thus the complete absolute configuration of 4 was determined as 2S, 3S, 14R, 20S.
Figure 3.
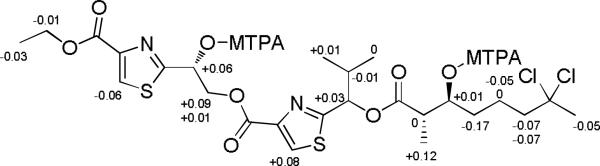
ΔδS-R values of the di-Mosher esters of lyngbyabellin M (4).
The HR ESITOFMS of lyngbyabellin N (5) showed a nearly identical parent ion cluster as the other lyngbyabellin analogs with an [M+H]+ at m/z 905.2997/907.2977/909.2950 (calcd for C40H59Cl2N4O11S2 905.2993) in a ratio of 100:80:20, indicating the presence of two chlorine atoms and 13 degrees of unsaturation, three more than compound 1. The 1H and 13C NMR spectra of 5 again showed similar features to those of 1; however, there was a significant increase in new signals, suggesting the addition of several more residues. In the1H NMR spectrum, the broad singlet at 5.73 ppm was no longer present, thus suggesting that 5 was similar to other known lyngbyabellins that contain attached residues esterified to C-14.[15] Furthermore, there were three additional downfield methyl groups (δ = 2.77, 2.72, and 1.91) and two amide protons (δ = 9.38, and 8.81). From the 13C NMR, there were an additional three ester/amide carbonyls at δ = 165.8, 168.9, and 169.4, accounting for the remaining degrees of unsaturation. COSY and HMBC correlations established the presence of an N,N-dimethylvaline and an O-acetylated leucine statine. HMBC correlations from 14-H (δ = 6.27) to C-24 (δ = 168.9) and NH-1(δ=8.81) to C-32 (δ = 165.8) completed the planar structure of 5 as depicted in Figure 1.
The planar structure of lyngbyabellin N (5) is closely related to that of lyngbyabellin H (6) except for the replacement of the polyketide portion with a N,N-dimethylvaline residue. The comparison of optical rotations and carbon chemical shifts (macrocyclic lactone portion) strongly supported that the absolute configuration of the macrocyclic lactone in 5 and 6 were identical, and thus we assign the stereoconfiguration of lyngbyabellin N as 2S, 3S, 14R, 20S. Further support of this configuration was obtained by comparison of the CD absorption curve of 5 with that of 1, 2, and 3. All of these CD curves were nearly identical, confirming that 5 has the 2S, 3S, 14R, 20S configuration.
The absolute configuration of the leucine statine in 5 was determined by LC-MS analysis of the acid hydrolysate appropriately derivatized with Marfey's reagent (d-FDAA). The four standards, 3S,4S-statine (Sta), 3R,4S-Sta, 3S,4R-Sta, and 3R,4R-Sta, were all synthesized beginning with their corresponding amino acids (l- and d-leucine, respectively), and following literature procedures, converted to their respective N-benzyl protected aldehydes.[20] Each standard was then reacted with tert-butyl 2-bromoacetate in the presence of N-BuLi to yield a mixture of diastereomeric protected leucine statines. However, these diastereomers were inseparable by HPLC and were thus converted to Boc protected analogs, which were readily purified by RP HPLC.[21] Hydrolysis, followed by the derivatization with Marfey's reagent, yielded the four standards which each possessed a distinct retention time by LC-MS [(3R,4R)-Sta-d-FDAA (78.2 min), (3S,4R)-Sta-d-FDAA (80.9 min), (3S,4S)-Sta-d-FDAA (92.2 min), and (3R,4S)-Sta-d-FDAA (93.1 min)]. From the retention time of the natural product statine derivative (93.29 min), it was clear that this residue was of the 3R, 4S configuration.
The absolute configuration of the N,N-dimethylvaline (DiMeVal) residue in compound 5 was determined by comparing the chiral GC-MS retention time of the methylated residue liberated by acid hydrolysis with authentic standards. The two standards, l-N,N-DiMeVal and d-N,N-DiMeVal, were synthesized from l-Val, and d-Val, respectively, following literature procedures.[22] The two standards each possessed distinctly different retention times by GC-MS [l-DiMeVal (63.7 min) and (d-DiMeVal (64.2 min)]. The methylated residue from the acid hydrolysate gave a single peak at 63.8 min, thus indicating its configuration as l.
The lyngbyabellin family of compounds are known to exhibit moderate to potent cytotoxicity against a number of different cancer cell types through the promotion of actin polymerization.[15] Thus, after the completion of the structural analysis, compounds 1-5 were evaluated in a H-460 human lung carcinoma cell cytotoxicity assay. Compound 5 showed strong yet variable cytotoxicity (IC50 0.0048-1.8 μM, perhaps due to solubility problems), while compounds 1-4 were inactive. However, in the HCT116 colon cancer cell line, reproducible IC50 values were obtained for lyngbyabellin N of 40.9 ± 3.3 nM, confirming the potent cytotoxic effect of this new member of the lyngbyabellin class, and suggesting that the side chain of lyngbyabellin N is an essential structural feature for this potent activity. However, this trend is not entirely consistent within this structure class as other lyngbyabellin analogs lacking the side chain exhibit submicromolar activity against HT29 and HeLa cells.[15]
It is interesting to note the increasing structural complexity in the lyngbyabellin family of metabolites, with that of lyngbyabelling N (5) being the most intricate to date. While it has the recognizable core of the lygbyabellins, the side chain and N,N-dimethylvaline terminus resembles that of the dolastatin 10 and coibacin A families of metabolites, and in this regard, it has a hybrid structure between these cyanobacterial natural product classes. Additionally, from a biosynthetic logic perspective, these more complex lyngbyabellins are perplexing as they possess two logical points for the initiation of molecule construction: the polyketide chain represents one such point and the N,N-dimethyl valine a second. Thus, these complex lyngbyabellins may indeed represent the hybridization and co-joining of two natural product structure classes. An additional point of interest is the occurrence of monochlorinated carbon atoms in the polyketide section (compounds 2 and 3). Previously, the Walsh group[24]and ourselves[25] independently showed that the monochloro species was not encountered in the process of chlorination of the methyl group of barbamide (only the dichloro and trichloro species are formed). Hence, the occurrence of a mono-chloro methylene moiety in these two metabolites suggests the functioning of a radical halogenase with differing catalytic properties than any of the currently characterized or partially characterized halogenases. It is still unclear whether lyngbyabellin M (4) is an extraction artifact or a naturally occurring compound. While lyngbyabellin M (4) has an ethyl ester at C-10, previously reported linear lyngbyabellins have a methyl ester at C-16. The position of the ethyl ester in 4 is consistent with the proposed biosynthetic pathway of the lyngbyabellins as predicted from our knowledge of hectochlorin biosynthesis.[26] It has been shown that methyl esters of hybrid polyketides/non-ribosomal peptides are produced by an unusual S-adenosyl-l-methionine (SAM)-dependent methyltransferase in myxobacteria.[27] Methyl and ethyl esters have been found as naturally occurring compounds in cyanobacterial extracts,[28] and thus, may be formed by pathway termination reactions similar to those in myxobacteria.
Conclusions
A chemical investigation of anti-cancer active extracts of Moorea bouillonii collected in Palmyra Atoll led to the isolation of five new lyngbyabellins 1-5. Their planar structures and absolute configurations were determined by the combination of various techniques in spectroscopy, chromatography and synthetic chemistry. While compounds 1-4 were inactive in the H-460 cytotoxicity assay, 5 exhibited strong yet variable cytotoxicity (IC50 0.0048-1.8 μM). We do not understand the basis for this variable level of activity, however, it most likely relates to solubility issues in the assay buffer. These new lyngbyabellin metabolites possess several unique structural features, a monochloro methylene group and two conceptual points of chain initiation, which reflect unique biosynthetic reactions not yet characterized nor understood.
Experimental Section
General Experimental Procedures
Optical rotations were measured on a JASCO P-2000 polarimeter, CD spectra were taken in EtOH using a JASCO J-810 spectropolarimeter. UV spectra and IR spectra were recorded on a Beckman Coulter DU800 spectrophotometer and a Nicolet ThermoElectron Nicolet IR100 FT-IR spectrometer using KBr plates, respectively. NMR spectra were recorded with chloroform as internal standards (δC 77.2, δH 7.26) on a Varian 500 MHz spectrometer (500 and 125 MHz for 1H and 13C NMR, respectively) or a Bruker 600 MHz spectrometer (600 and 150 MHz for 1H and 13C NMR, respectively) equipped with 1.7 mm MicroCryoProbe. For the HETLOC experiments, 128 scans (4K) and 512 experiments were obtained with zero-filing at F1 to 4 K. A mixing time of 60–80 ms was used. HR ESIMS spectra were obtained on an Agilent 6230 ESI-TOF mass spectrometer. X-ray diffraction data is obtained by a state-of-the-art Bruker Single-Crystal Diffractometers with CCD detectors. HPLC was carried out using Waters 515 pumps system with a Waters 996 PDA detector. Acid hydrolysis was performed using a Biotage (Initiator) microwave reactor equipped with high-pressure vessels.
Cyanobacterial Collections and Taxonomic Identification
The lyngbyabellins K-M producing cyanobacterium (collection code: PAL 8/3/09-1) was collected by SCUBA in the north of lagoon at Strawn Island, Palmyra Atoll, USA, in August 2008 (05°53.783’N, 162°07.’W). The lyngbyabellin N producing cyanobacterium (collection code: PAL 8/16/08-3) was collected by SCUBA on reefs 9-15 m deep around Palmyra Atoll, USA. The environmental samples were stored in EtOH/H2O (1:1) at −20 °C, while the genetic materials were preserved in RNA stabilization solution at −20 °C (RNAlater, Ambion Inc.). Morphological characterization was performed using an Olympus IX51 epifluorescent microscope (100×) equipped with an Olympus U-CMAD3 camera. Taxonomic identification of cyanobacterial specimens was performed in accordance with current phycological systems.[29, 30]
Isolation of lyngbyabellins K, L, M, and 7-epi-lyngbyabellin L (1-4)
The cyanobacterial tissue (PAL 8/3/09-1, morphologically identified as Moorea bouillonii) was repetitively extracted with 2:1 CH2Cl2/CH3OH to afford 3.9 g of crude extract. A portion of the extract (3.2 g) was fractionated by silica gel VLC with a stepwise gradient solvent system of increasing polarity starting from 10% EtOAc in hexanes to 100% CH3OH, to produce nine fractions (A–I).The fraction eluting with 80% EtOAc in Hex (fraction F) was subsequently separated using RP HPLC (Phenomenex Fusion RP 4 μ, 250 × 10 mm, 65% CH3CN/H2O at 3 mL/min) to give pure lyngbyabellin K (1, 10 mg, 0.26%), lyngbyabellin M (4, 2 mg, 0.05%), and a mixture of lyngbyabellin L and 7-epi-lyngbyabellin L. The mixtures were subjected to chiral HPLC (Chiral-AGP, 150 × 4.0 mm, 5 μm, UV = 210 nm) and lyngbyabellin L (2, 0.7 mg, 0.018%) and 7-epi-lyngbyabellin L (3, 0.6 mg, 0.015%) were eluted with 13% CH3CN in H2O.
Isolation of lyngbyabellin N (5)
One liter of cyanobacterial tissue (previously identified as Moorea bouillonii)[3] was repetitively extracted with 2:1 CH2Cl2/CH3OH to afford 4.2 g of crude extract. The extract was fractionated by silica gel VLC with a stepwise gradient solvent system of increasing polarity starting from 10% EtOAc in hexanes to 100% CH3OH, to produce nine fractions (A–I). The fraction eluting with 75% EtOAc in CH3OH (fraction H) was subsequently separated using a 5 g RP SPE with a stepwise gradient solvent system decreasing in polarity starting from 55% CH3CN in H2O to 100% CH2Cl2, to produce five fractions (1-5). The fraction eluting with 70% CH3CN in H2O (fraction 3) was further separated using prepTLC, with an isocratic solvent system of 100% EtOAc, to yield pure lyngbyabellin N (5, 5.4 mg, 0.12 %).
Lyngbyabellin K (1)
pale yellow oil; [α]D25-28.0 (c 0.5, MeOH); UV (MeOH)λmax 236 nm (log ε 3.76); CD (MeOH) λmax (Δε), 211 nm (+1.11), 227 (−1.45), 238 (−0.58), 245 (−0.69), 265 (+0.28); IR (KBr) γmax3421, 3127, 2965, 2935, 2880, 1737, 1616, 1481, 1379, 1320, 1211, 1163, 1096 cm−1; 1H, 13C and 2D NMR data, see Table 1; HR ESIMS m/z [M+Na]+ 601.0609 (calcd for C23H28N2O7Cl2S2Na 601.0607, Δ +0.2 mmu).
Lyngbyabellin L (2)
pale yellow oil; [α]D25-33.0 (c 0.5, MeOH); UV (MeOH) λmax 236 nm (log ε 3.69); CD (MeOH) λmax (Δε), 210 nm (+0.19), 225 (−1.98), 239 (−0.70), 245 (−0.74), 265 (+0.17); IR (KBr) γmax 3440, 3116, 2962, 2921, 1738, 1480, 1367, 1320, 1211, 1100 cm−1; 1H, 13C and 2D NMR data, see Table 1 and Supporting Information; HR ESIMS m/z [M+Na]+ 567.1000 (calcd for C23H29N2O7ClS2Na 567.0997, Δ +0.3 mmu).
7-epi-Lyngbyabellin L (3)
pale yellow oil; [α]D25-34.8 (c 0.3, MeOH); UV (MeOH) λmax 236 nm (log ε 3.59); CD (MeOH) λmax (Δε), 210 nm (+0.25), 225 (−1.42), 239 (−0.51), 245 (−0.52), 265 (+0.12); IR (KBr) γmax 3731, 3623, 2962, 2920, 2846, 1737, 1458, 1368, 1319, 1211, 1101 cm−1; 1H, 13C and 2D NMR data, see Table 1 and Supporting Information; HR ESIMS m/z [M+Na]+ 567.1000 (calcd for C23H29N2O7ClS2Na 567.0997, Δ +0.3 mmu).
Lyngbyabellin M (4)
pale yellow oil; [α]D25-4.5 (c 0.5, MeOH); UV (MeOH) λmax 234 nm (log ε 3.71); IR (KBr) γmax 3377, 2963, 2926, 2853, 1730, 1644, 1465, 1334, 1224, 1175 cm−1; 1H, 13C and 2D NMR data, see Table 1 and Supporting Information; HR ESIMS m/z [M+Na]+ 647.1029 (calcd for C25H34N2O8Cl2S2Na 647.1026, Δ +0.3 mmu).
Lyngbyabellin N (5)
pale yellow oil; [α]D27-24.0 (c 1.05, MeOH); UV (MeOH) λmax 202 nm (log ε 4.36), 235 (log ε 3.99); CD (MeOH) λmax (Δε), 212 nm (−0.64), 224 (−1.60), 237 (−0.27), 248 (−1.36), 268 (+0.03); IR (KBr) γmax 3436, 2961, 2933, 1742, 1677, 1467, 1372, 1321, 1233, 1166, 1097, 1037cm−1; 1H, 13C and 2D NMR data, see Table 2; HRESIMS m/z [M+H]+ 905.2997 (calcd for C40H59Cl2N4O11S2 905.2993, Δ +0.4 mmu).
Table 2.
NMR Spectral Data for Lyngbyabellin N (5) in d6-DMSO at 500 MHz (1H) and 125 MHz (13C).
| 5 | ||||
|---|---|---|---|---|
| Position | δ C | δH multi (J in Hz) | COSY | HMBC |
| 1 | 173.2 | |||
| 2 | 42.9 | 2.84 dd (9.4, 6.8) | 3, 9 | 1, 3, 9 |
| 3 | 74.5 | 5.13 m | 2, 4a, 4b | |
| 4a | 30.0 | 1.80 m | 3 | 3. 6 |
| 4b | 1.69 m | 3 | 3, 6 | |
| 5a | 29.0 | 1.24 m | 6 | 4 |
| 6a | 48.3 | 2.27 m | 4a, 4b, 5 | 7, 8 |
| 6b | 2.20 m | 4a, 4b, 5 | 7, 8 | |
| 7 | 92.1 | |||
| 8 | 37.0 | 2.09 s | 6, 7 | |
| 9 | 14.6 | 1.16 d (7.1) | 2 | 1, 2, 4 |
| 10 | 159.4 | |||
| 11 | 145.4 | |||
| 12 | 129.7 | 8.44 s | 10, 11, 13 | |
| 13 | 165.5 | |||
| 14 | 70.2 | 6.27 t (6.1) | 15 | 15 |
| 15a | 63.6 | 4.81 dd (11.2, 5.2) | 14 | 14 |
| 15b | 4.57 dd (11.7, 6.8) | 14 | 14 | |
| 16 | 160.2 | |||
| 17 | 145.0 | |||
| 18 | 130.2 | 8.46 s | 17, 19 | |
| 19 | 167.2 | |||
| 20 | 76.2 | 5.55 d (8.1) | 21 | 1, 19, 21, 22, 23 |
| 21 | 31.9 | 2.24 m | 20, 22, 23 | 20, 23 |
| 22 | 18.4 | 0.80 d (6.5) | 21 | 20, 21, 23 |
| 23 | 17.9 | 1.00 d (6.5) | 21 | 20, 21, 22 |
| 24 | 168.9 | |||
| 25a | 34.0 | 2.91 m | 26 | 24 |
| 25b | 2.70 m | 26 | 24, 26 | |
| 26 | 71.6 | 5.12 m | 25a, 25b, 27 | |
| 27 | 47.9 | 4.35 t (4.4) | 26, 28a, 28b, NH-1 | |
| 28a | 37.5 | 1.41 m | 29 | 29, 31 |
| 28b | 1.23 m | 29 | 29, 31 | |
| 29 | 24.4 | 1.55 m | 28, 30, 31 | |
| 30 | 24.0 | 0.90 d (6.4) | 29 | 28, 29, 31 |
| 31 | 20.8 | 0.81 d (6.2) | 29 | 28, 29, 31 |
| NH-1 | 8.81 d (8.7) | 27 | ||
| 32 | 165.8 | |||
| 33 | 71.8 | 3.63 t (6.7) | 34 | 32, 34, 35, 36 |
| 34 | 26.3 | 2.31 m | 33, 35, 36 | 32, 33, 35, 36 |
| 35 | 19.5 | 1.07 d (6.8) | 34 | 33, 34, 36 |
| 36 | 16.5 | 0.94 d (6.6) | 34 | 33, 34, 35 |
| 37 | 41.5 | 2.72 d (4.0) | 33, 38 | |
| 38 | 41.0 | 2.77 d (4.2) | 33, 37 | |
| NH-2 | 9.38 bs | |||
| 39 | 169.4 | |||
| 40 | 20.8 | 1.91 s | 39 | |
X-ray Crystallography
Eight milligrams of 1, dissolved in 400 μL of CH3CN, was transferred into a tube and the tube was placed in a vial. The vial was sealsed and monitored for crystal growth over a month, then colorless needle shaped crystals were observed.
Crystal data for lyngbyabellin K (1)
monoclinic, space group P21/c, unit cell dimensions a = 10.6419(8) Å, α = 90°, b = 5.8776(5) Å, β = 101.928(6)°, c = 23.859(2) Å, γ = 90°, V = 1460.1(2) Å3, Z = 2, ρcalcd. = 1.411 gcm−3, crystal dimensions 0.21 × 0.11 × 0.05 mm. A total of 7218 reflections were collected covering the indices, −12 ≤ h ≤ 12, −6 ≤ k ≤ 6, −27 ≤ l ≤ 10. 4362 reflections were found to be symmetry independent, with an Rint of 0.0358. Final R(F2) = −0.02(2). Relevant data collection parameters and results of structural refinement for this structural determination are given in the Supporting Information. Crystallographic data for lyngbyabellin K (1) have been deposited with the Cambridge Crystallographic Data Centre (deposition number CCDC 882823). Copies of the data can be obtained, free of charge, via www.ccdc.cam.ac.uk/data_request/cif or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44(0)1223-336033.
Ozonolysis, acid hydrolysis, and chiral GC-MS
A portion (100μg) of 4 was dissolved in 500 μL of CH2Cl2 at – 78 °C and O3 was bubbled through the sample for 5 min. The pale blue solution was dried under N2 (g). The products were resuspended in 200 μL of 6 N HCl and reacted at 110 °C for 2 h. The acid hydrolysates were dried under N2 (g), treated with excessive CH2N2 for 30 min at room temperature, and dried under N2 (g). The products were dissolved in CH2Cl2 and injected over chiral GC-MS (Chirasil-Val, Agilent Technologies J&W Scinetific, 30 m × 0.25 mm) under the following conditions: the initial oven temperature was 32 °C, held for 15 min, followed by a ramp from 32 °C to 60 °C at a rate of 10 °C/min, followed by another ramp to 200 °C , at a rate of 15 °C/min and held at 200 °C for 5 min. The retention time of products resulting from the acid hydrolysate of 4 matched the synthetic 2S-HIVA standard (9.7 min; 2RHIVA, 10.2 min).. Synthetic standards of 2S-hydroxy isovaleric acid (HIVA) and 2R-HIVA were methylated with the excessive CH2N2, dried under N2 (g), resuspended in CH2Cl2, then analyzed with chiral GC-MS. The retention time of products resulting from the acid hydrolysate of 4 matched the synthetic 2S-HIVA standard (9.7 min; 2R-HIVA, 10.2 min).
Ozonolysis, and Acid Hydrolysis of Lyngbyabellin N (5)
A portion (1 mg) of 5 was dissolved in 1 mL of CH2Cl2 at – 78 °C and O3 was bubbled through the sample for 10 min. The pale blue solution was dried under N2 (g). The products were re-suspended in 500 μL of 6 n HCl and reacted at 160 °C for 5 min in a microwave reactor.
Modified Marfey's Analysis to Determine Configuration of Statine Units
An aliquot (~500 μg) of the acid hydrolysate was dried under N2 (g), and dissolve in 1 mL of 1 m sodium bicarbonate, and 12 μL of 1% d-FDAA (1-fluoro-2,4-dinitrophenyl-5-d-alanine amide) was added in acetone. The solution was maintained at 40 °C for 90 min, at which time the reaction was quenched by the addition of CH3CN, and 10 μL of the solution was analyzed by LC-ESIMS. The Marfey's derivatives of the hydrolysate and standards were analyzed by RP HPLC using Phenomenex Luna 5 μ C18 column (4.6 × 250 mm). The HPLC conditions began with 10% CH3CN/H2O acidified with 0.1% formic acid (FA) followed by a gradient profile to 50% CH3CN/H2O acidified with 0.1% FA over 85 min at a flow of 0.4 mL/min, monitoring from 200 to 600 nm. The retention times of the authentic acid d-FDAA derivatives were (3R,4R)-Sta-d-FDAA (78.2 min), (3S,4R)-Sta-d-FDAA (80.9 min), (3S,4S)-Sta-d-FDAA (92.2 min), and (3R,4S)-Sta-d-FDAA (93.1 min); the hydrolysate product gave a peak with retention time of 93.3 min, indicating an absolute configuration of 3R,4S.
Correspondingly, (3S,4S)-statine, (3S,4R)-statine, (3R,4R)-statine and (3R,4S)-statine were synthesized from l-Leucine (1 g) and d-Leucine (1 g), respectively, following Reetz et al. and to yield the benzyl protected aldehyde.[20] An aliquot (3.38 mmol) of the benzyl protected aldehydes dissolved in 15 mL were then added to a solution of with 3.72 mmol tertbutyl acetate and freshly prepared lithium disoproplyamine (5.06 mmol). The reaction was warmed to – 40 °C for 1 h and then quenched with 85 mL NaHCO3, filtered and the aqueous layer was separated and washed 2× with 20 mL of Et2O. The organic layer was then washed with brine and dried with NaSO4 and filtered to yield diastereomeric tert-butyl 4-(dibenzylamino)-3-hydroxy-6-methylheptanoate. The mixture of diasteromers were inseparable, thus the method outlined by Andrés et al. was used, where the benzyl protected amine was converted to the Boc protection allowing for purification of a small amount of each of the diastereomers.[21] Each of the diastereomers were identified by the comparison of 1H NMR each of the pure standards to the known compounds in the above paper. An aliquot (2 mg) of each protected statins were treated with 1 mL of 6 N HCl and heated to 160 °C for 5 min in a microwave reactor. Each of the hydrolysate products were dried down by N2 (g) and then reacted with the Marfey's reagent as mentioned above, to yield the four Marfey's derived statine standards.
Preparation and GC-MS Anaylsis of 2-Hydroxyisovaleric Acid (HIVA)
An aliquot (~500 μg) of the hyrdolysate was treated with excessive CH2N2 for 30 min at room temperature, and dried under N2 (g). Correspondingly, l-HIVA and d-HIVA were synthesized as described above. The products were dissolved in CH2Cl2 and injected over chiral GC-MS using a Chiralsil-Val column (Agilent Technologies J&W Scinetific, 30 m × 0.25 mm) under the following conditions: the initial oven temperature was 32 °C, held for 15 min, followed by a ramp from 32 °C to 60 °C at a rate of 10 °C/min, followed by another ramp to 200 °C , at a rate of 15 °C/min and held at 200 °C for 5 min. The retention time of products resulting from the acid hydrolysate of 4 matched the synthetic 2S-HIVA standard (9.7 min; 2R-HIVA, 10.2 min).
Preparation and GC-MS Analysis of N’,N’-Dimethylvaline
The methylated hydrolysate product of 5 was analyzed by Chiral GC-MS using a Cyclosil B column (Agilent Technologies J&W Scientific, 30 m × 0.25 mm) under the following conditions: the initial oven temperature was 34 °C and was held for 68 min, followed by a ramp from 34 °C to 100 °C at a rate of 30 °C/min and held at 100 °C for 5 min. Synthetic standards of 2S-N’N’-dimehtylvaline and 2R-N’N’-dimehtylvaline were first methylated by dissolving 10 mg of each starting material into 433 μL of H2O, followed by the addition of 27 μL of formaldehyde and 10.4 mg of 10% Pd/C. The system was then treated with H2 (g) for 16 h. After 16 h the reactions were brought to a boil and then concentrated via rotorvap. Each of the synthetic standards were then treated with CH2N2 for 5 min and then dried down under N2 (g), re-suspended in CH2Cl2, then analyzed with chiral GC-MS. The retention time of products resulting from the acid hydrolysate of 5 matched to that of the authentic 2S-N’N’-dimethylvaline standard (63.7 min; 2R-N’N’-dimethylvaline, 64.2 min).
Preparation of MTPA ester of lyngbyabellin M
Duplicate samples of compound 4 (0.5 mg) were dried and dissolved in 1 mL of anhydrous pyridine and a catalytic amount of dimethyl amino pyridine was added. Separately and into each vial, 15 μL of R-MTPA chloride and 15 μL of S-MTPA chloride were added. The reaction vials were stored at 40°C for 48 h with stirring. The acylation products were purified using RP HPLC (Phenomenex Jupiter 5 μ C18, 4.6× 250 mm, 85% CH3OH/H2O with at 1 mL/min). The m/z values of the two diastereomeric MTPA derivatives of compound 4 were observed by ESIMS, and the 1H NMR chemical shift was assigned by COSY.
3,14-di-S-MTPA ester of lyngbyabellin M (6)
pale yellow amorphous solid; 1H NMR (CDCl3, 600MHz): δ = 8.16 (s, 1 H,12-H), 7.96 (s, 1 H, 13-H), 6.78 (dd,J = 7.6, 2.9 Hz, 1 H, 14-H), 5.98 (d,J = 5.4 Hz, 1 H,20-H), 5.48 (td, J = 7.6, 3.4 Hz, 1 H, 3-H), 5.07 (dd, J = 12.3, 2.9 Hz, 1 H, 15-Ha), 4.79 (dd, J = 12.3, 7.6 Hz, 1 H, 15-Hb), 4.43 (q, J = 7.1 Hz, 2 H, 24-H2), 3.05 (dq, J = 7.6, 7.2 Hz, 1 H, 2-H), 2.41 (m, 1 H, 21-H), 2.12 (m, 1 H, 6-Ha), 2.07 (m, 1 H, 6-Hb), 2.06 (s, 3 H, 8-H3), 1.73 (m, 1 H, 5-Ha), 1.68 (m, 1 H, 5-Hb), 1.59 (m, 2 H, 4-H2), 1.41 (t, J = 7.1 Hz, 3 H, 25-H3), 1.28 (d, J = 7.2 Hz, 3 H, 9-H3), 0.96 (d, J = 6.8 Hz, 3 H, 22-H3), 0.95 (d, J = 6.8 Hz, 3 H, 23-H3); LR ESIMS m/z 1057.02 [M+H]+, 1079.12 [M+Na]+.
3,14-di-R-MTPA ester of lyngbyabellin M (7)
pale yellow amorphous solid; 1H NMR (CDCl3, 600MHz): δ = 8.22 (s, 1 H,12-H), 7.88 (s, 1 H, 18-H), 6.72 (dd,J = 7.1, 3.1 Hz, 1 H, 14-H), 5.95 (d,J = 5.4 Hz, 1 H,20-H), 5.47 (m, 1 H, H-3), 4.98 (dd, J = 12.3, 3.1 Hz, 1 H, 15-Ha), 4.78 (dd, J = 12.3, 7.1 Hz, 1 H, 15-Hb), 4.44 (q, J = 7.1 Hz, 2 H, 24-H2), 3.05 (m, 1 H, 2-H), 2.42 (m, 1 H, 21-H), 2.19 (m, 1 H, 6-Ha), 2.14 (m, 1 H, 6-Hb), 2.11 (s, 3 H, 8-H3), 1.78 (m, 1 H, 5-Ha), 1.68 (m, 1 H, 5-Hb), 1.76 (m, 2 H, 4-H2), 1.42 (t, J = 7.1 Hz, 3 H, 25-H3), 1.16 (d, J = 7.2 Hz, 1 H, 9-H), 0.95 (d, J = 6.8 Hz, 6 H, 22-H3, 23-H3); LR ESIMS m/z 1056.99 [M+H]+, 1079.07.58 [M+Na]+.
Cytotoxicity Assay
H-460 cells were added to 96-well plates at 3.33 × 104 cells/mL of Roswell Park Memorial Institute (RPMI) 1640 medium with 10% fetal bovine serum (FBS) and 1% Penicillin/Streptomycin. The cells, in a volume of 180 μL per well, were incubated overnight (37°C, 5% CO2) to allow recovery before treatment with test compounds. Compounds were dissolved in DMSO to a stock concentration of 10 mg/mL. Working solutions of the compounds were made in RPMI 1640 medium without FBS, with a volume of 20 μL added to each well to give a final compound concentration of either 30 or 3 μg/mL. An equal volume of RPMI 1640 medium without FBS was added to wells designated as negative controls for each plate. Plates were incubated for approximately 48 h before staining with MTT. Using a ThermoElectron Multiskan Ascent plate reader, plates were read at 570 and 630 nm. Concentration response graphs were generated using GraphPad Prism (GraphPad Software Inc., San Diego, CA).
Supplementary Material
Acknowledgments
We thank A. C. Jones, A. R. Pereira and R. C. Coates for assistance with collection of the lyngbyabellin-producing cyanobacterial samples from Palmyra Atoll, C. E. Moore for assistance with crystallographic data analysis of lyngbyabellin K, as well as the UCSD mass spectrometry facilities for their analytical services.We further acknowledge support of this research from UC San Diego and NIH CA092143. We also acknowledge The Growth Regulation & Oncogenesis Training Grant NIH/NCI (T32A009523-24) for a fellowship to E.M.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/ejoc.xxxxxxxxx.
Dedicated to Professor Ernesto Fattorusso on the occasion of his 75th birthday.
Supporting Information (see footnote on the first page of this article): 1H NMR, 13C NMR, COSY, gHSQC, and HMBC spectra in CDCl3 for lyngbyabellin K and M (1 and 4). 1H NMR in CDCl3 for the Mosher derivatives of lyngbyabellin M (4). 1H NMR, 13C NMR, gHSQC, COSY, HMBC, DQF-COSY, NOESY, and HETLOC spectra in CDCl3 for lyngbyabellin L (2) and 7-epi-lyngbyabellin L (3). 1H NMR, 13C NMR, gHSQC, COSY, HMBC, and TOCSY spectra in d6-DMSO for lyngbyabellin N (5). 1H NMR and 13C NMR in CDCl3 for lyngbyabellin N (5). ORTEP representation of lyngbyabellin K (1) crystal, and crystal data. CD spectra for lyngbyabellin K, L, N (1, 2, and 5) and 7-epi-lyngbyabellin L (3).
References
- 1.a Nunnery JK, Mevers E, Gerwick WH. Curr. Opin. Biotechnol. 2010;21:787–793. doi: 10.1016/j.copbio.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Tan LT. J. Appl. Phycol. 2010;22:659–676. [Google Scholar]; c Gerwick WH, Coates RC, Engene N, Gerwick L, Grindberg RV, Jones AC, Sorrels CM. Microbe. 2008;3:277–284. [Google Scholar]; d Tidgewell K, Clark BR, Gerwick WH. In: Comprehensive Natural Products Chemistry II. Moore B, Crews P, editors. Vol. 8. Elsevier; Oxford: 2010. pp. 141–188. [Google Scholar]
- 2.Nagle DG, Paul VJ, Phycol J. 1999;35:1412–1421. [Google Scholar]
- 3.a Luesch H, Yoshida WY, Moore RE, Paul VJ, Corbett TH. J. Am. Chem. Soc. 2001;123:5418–5423. doi: 10.1021/ja010453j. [DOI] [PubMed] [Google Scholar]; b Luesch H, Yoshida WY, Moore RE, Paul VJ. Bioorg. Med. Chem. 2002;10:1973–1978. doi: 10.1016/s0968-0896(02)00014-7. [DOI] [PubMed] [Google Scholar]; c Gutiérrez M, Suyama TL, Engene N, Wingerd JS, Matainaho T, Gerwick WH. J. Nat. Prod. 2008;71:1099–1103. doi: 10.1021/np800121a. [DOI] [PubMed] [Google Scholar]; d Matthew S, Schupp PJ, Luesch H. J. Nat. Prod. 2008;71:1113–1116. doi: 10.1021/np700717s. [DOI] [PubMed] [Google Scholar]; e Tidgewell K, Engene N, Byrum T, Media J, Takayuki D, Valeriote FA, Gerwick WH. ChemBioChem. 2010;11:1458–1466. doi: 10.1002/cbic.201000070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orjala J, Nagle DG, Hsu VL, Gerwick WH. J. Am. Chem. Soc. 1995;117:8281–8282. [Google Scholar]
- 5.Cardellina JH, Marner FJ, Moore RE. Science. 1979;204:193–195. doi: 10.1126/science.107586. [DOI] [PubMed] [Google Scholar]
- 6.Gerwick WH, Proteau PJ, Nagle DG, Hamel E, Blokhin A, Slate DL. J. Org. Chem. 1994;59:1243–1245. [Google Scholar]
- 7.Orjala J, Gerwick WH. J. Nat. Prod. 1996;59:427–430. doi: 10.1021/np960085a. [DOI] [PubMed] [Google Scholar]
- 8.Edwards DJ, Marquez BL, Nogle LM, McPhail K, Goeger DE, Roberts MA, Gerwick WH. Chem. Biol. 2004;11:817–833. doi: 10.1016/j.chembiol.2004.03.030. [DOI] [PubMed] [Google Scholar]
- 9.Gerwick WH, Tan LT, Sitachitta N. In: The Alkaloids. Cordell GA, editor. Vol. 57. Academic Press; San Diego: 2001. pp. 75–184. [DOI] [PubMed] [Google Scholar]
- 10.Simmons TL, Andrianasolo E, McPhail K, Flatt P, Gerwick WH. Mol. Cancer Ther. 2005;4:333–342. [PubMed] [Google Scholar]
- 11.Matthew S, Ratnayake R, Becerro MA, Ritson-Williams R, Paul VJ, Luesch H. Mar. Drugs. 2010;8:1803–1816. doi: 10.3390/md8061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pereira R, Etzbach L, Engene N, Müller R, Gerwick WH. J. Nat. Prod. 2011;74:1175–1181. doi: 10.1021/np200106b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi H, Mascuch SJ, Villa FA, Byrum T, Teasdale ME, Smith JE, Preskitt LB, Rowley DC, Gerwick L, Gerwick WH. Chem. Biol. 2012;19:589–598. doi: 10.1016/j.chembiol.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grindberg RV, Shuman CF, Sorrels CM, Wingerd J, Gerwick WH. In: Modern Alkaloids: Structure, Isolation, Synthesis and Biology. Fattorusso E, Taglialetela-Scafati O, editors. Vol. 1. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim: 2008. pp. 139–170. [Google Scholar]
- 15.a Luesch H, Yoshida WY, Moore RE, Paul VJ, Mooberry SL. J. Nat. Prod. 2000;63:611–615. doi: 10.1021/np990543q. [DOI] [PubMed] [Google Scholar]; b Matthew S, Salvador LA, Schupp PJ, Paul VJ, Luesch H. J. Nat. Prod. 2010;73:1544–1552. doi: 10.1021/np1004032. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Han B, McPhail KL, Gross H, Goeger DE, Mooberry SL, Gerwick WH. Tetrahedron. 2005;61:11723–11729. [Google Scholar]
- 16.Sone H, Kondo T, Kiryu M, Ishiwata H, Ojika M, Yamada K. J. Org. Chem. 1995;60:4774–4781. [Google Scholar]
- 17.Marquez BL, Watts KS, Yokochi A, Roberts MA, Verdier-Pinard P, Jimenex JI, Hamel E, Scheuer PJ, Gerwick WH. J. Nat. Prod. 2002;65:866–871. doi: 10.1021/np0106283. [DOI] [PubMed] [Google Scholar]
- 18.Matsumori N, Kaneno D, Murata M, Nakamura H, Tachibana K. J. Org. Chem. 1999;64:866–876. doi: 10.1021/jo981810k. [DOI] [PubMed] [Google Scholar]
- 19.Miyata Y, Matsunaga S. Tetrahedron. 2008;49:6334–6336. [Google Scholar]
- 20.Reetz MT, Drewes MW, Schwickardi R. Org. Synth. 1999;76:110. [Google Scholar]
- 21.Andrés JM, Pedrosa R, Pérez A, Pérez-Encabo A. Tetrahedron. 2001;57:8521–8530. [Google Scholar]
- 22.Bowman RE, Stroud HH. J. Chem. Soc. 1950:1322–1345. [Google Scholar]
- 23.Subramanian B, Nakeff A, Tenney K, Crews P, Gunatilaka L, Valeriote J FV. Exp. Ther. Oncol. 2006;11:195–204. [PMC free article] [PubMed] [Google Scholar]
- 24.Galonic DP, Vaillancourt FH, Walsh CT. J. Am. Chem. Soc. 2006;128:3900–3901. doi: 10.1021/ja060151n. [DOI] [PubMed] [Google Scholar]
- 25.Flatt PM, O'Connell SJ, L McPhail K, Zeller G, Willis CL, Sherman DH, Gerwick WH. J. Nat. Prod. 2006;69:938–944. doi: 10.1021/np050523q. [DOI] [PubMed] [Google Scholar]
- 26.Ramnaswamy AV, Sorrels C, Gerwick WH. J. Nat. Prod. 2007;70:1977–1986. doi: 10.1021/np0704250. [DOI] [PubMed] [Google Scholar]
- 27.Weinig S, Hecht H-J, Mahmud T, Müller R. Chem. Biol. 2003;10:939–952. doi: 10.1016/j.chembiol.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 28.Engene N, Choi H, Esquenazi E, Byrum T, Villa FA, Cao Z, Murray TF, Dorrestein PC, Gerwick L, Gerwick WH. J. Nat. Prod. 2011;74:1737–1743. doi: 10.1021/np200236c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Castenholz RW, Rippka R, Herdman M. In: Manual of Systematic Bacteriology. Boone DR, Castenholz RW, editors. Vol. 1. Springer; New York: 2001. pp. 473–599. [Google Scholar]
- 30.Komárek J, Anagnostidis K. In: Süsswasserflora von Mitteleuropa. Büdel B, Gártner G, Krienitz L, Schagerl M, editors. 19/2. Gustav Fischer; Jena: 2005. pp. 576–606. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.