

Abstract
Several clinical trials have shown anti-CD3 treatment to be a promising therapy for autoimmune diabetes, but its mechanism of action remains unclear. Foxp3+ regulatory T (Treg) cells are likely to be involved, but through unknown mechanistic pathways. We profiled the transcriptional consequences in CD4+ Treg and conventional T (Tconv) cells in the first hours and days after anti-CD3 treatment of NOD mice. Anti-CD3 treatment led to a transient transcriptional response, terminating faster than most antigen-induced responses. Most transcripts were similarly induced in Treg and Tconv cells, but several were differential, in particular those encoding the IL7 receptor (IL7R) and transcription factors Id2/3 and Gfi1, upregulated in Treg but repressed in Tconv cells. As IL7R was a plausible candidate for driving the homeostatic response of Treg cells to anti-CD3, we tested its relevance by supplementation of anti-CD3 treatment with IL7/anti-IL7 complexes. Although ineffective alone, IL7 significantly improved the rate of remission induced by anti-CD3. Four anti-human CD3 mAbs exhibited the same differential effect on IL7R expression in human as in mouse cells, suggesting that the mechanism also underlie therapeutic effect in human cells, and perhaps a rationale for testing a combination of anti-CD3 and IL7 for the treatment of recent-onset human type-1 diabetes (T1D). Thus, systems level analysis of the response to anti-CD3 in the early phase of the treatment demonstrates different responses in Treg and Tconv cells, and provides new leads to a mechanistic understanding of its mechanism of action in reverting recent-onset diabetes.
INTRODUCTION
Among the treatments being explored for T1D, anti-CD3 is one of the most promising. Based on leads from the NOD mouse model (1-4), anti-CD3 proved to have some efficacy when used in a short-course treatment in patients with recently diagnosed diabetes (5-7). Stabilization of disease and maintenance of endogenous capacity for insulin production were observed in two independent clinical trials with different anti-CD3 reagents (5,6,8); more recent phase 3 trials did not meet their clinical endpoints (9,10), although long-term preservation of C-peptide was still observed in one case (9), and failure in the other case may well be attributable to insufficient dosing (11). The anti-CD3 treatment effect tends to wane after a few years (12).
To improve therapeutic protocols in terms of timing regimen, dose, and potential outcomes, it is essential to understand the mechanisms underlying the effects that have been observed. Unfortunately, there is only a limited understanding of anti-CD3’s mechanism of action. For instance, it is not clear whether the “cytokine storm” induced by anti-CD3, which is not without side-effects, is actually required for therapeutic efficacy. Because mechanistic studies on human patients are of necessity limited to blood cells, which give an incomplete representation of events occurring in lymphoid organs or in the pancreas, most results have been obtained in the NOD model of T1D (or, more recently, in humanized mice (13)). The effects of anti-CD3 on autoimmune disease are typically long-lasting, in NOD mice as well as human patients, persisting long after clearance of the antibody, which implies some resetting of the balance between autoreactive effector cells and regulatory cells, lasting beyond the relatively short timeframe (a few days) during which the T cell receptor (TCR) is blocked or internalized by anti-CD3 engagement. Induction of Tconv anergy, perturbation of the T helper (Th)-1 vs -2 balance, or inactivation of autoreactive T cells have been invoked (14-17). Several investigators have suggested that dominant tolerance may be induced by anti-CD3 therapy (18), in particular via effects on CD4+FoxP3+ Treg cells (13,14,19-22).
Foxp3+ Treg cells play an important part in the control of immunologic self-tolerance, as well as of anti-infectious and anti-tumor responses (23). These diverse regulatory activities involve several specialized subphenotypes and molecular pathways (24,25). Tregs clearly influence the development of T1D: their experimental depletion or genetic deficiency in their numbers or activity promote a more aggressive disease (26,27); while their transfer or therapeutic enhancement are protective (26,28,29).
Studies on anti-CD3-treated mice have shown variable modifications of Treg cells, no or quantitatively modest effects (17,20-22,30), or restricted to particular anatomical locations (14,20). The consensus seems to be, then, that there are no large-scale changes in Treg populations in anti-CD3 treated mice. On the other hand, we have recently shown that anti-CD3 has profound effects on Treg cells whose expansion is constrained by homeostatic limitations, lifting these niche constraints through a striking and selective burst of amplification during the first few days after anti-CD3 administration (22). Using a humanized mouse model, Waldron-Lynch et al reported that anti-hCD3 induced only modest depletion of, but instead the differentiation of a gut-homing population of FoxP3+ and IL-10-producing CD4+ T cells (13). Thus, anti-CD3 may induce, rather than a wholesale expansion of Tregs, specific modifications in the homeostatic control of their repertoire or phenotypic redistribution.
The burst of DNA replication and proliferation in the first few days of anti-CD3 treatment was substantially more extensive in Treg than in Tconv cells (22), implying that the two populations respond differentially to what is otherwise the same activating ligand. In general, little is known about the comparative transcriptional responses of Treg and Tconv cells to TCR engagement. To address these issues, and more generally to obtain a better perspective on the immediate effects of anti-CD3 in vivo, we have dissected the responses of Treg and Tconv cells to anti-CD3 treatment. Our data argue for the potential of anti-CD3/IL7 combination therapy.
MATERIALS AND METHODS
Mice and treatments
NOD/LtJDoi, Foxp3GFPNOD mice (31,32) were obtained from the JDRF Center for Immunological Tolerance at Harvard Transgenic Mouse Core, and maintained in SPF facilities at Harvard Medical School (IACUC 02954). Diabetes was monitored with tests of urine glucose, confirmed by blood glucose measurements when positive and after diagnosis. Mice were considered diabetic with 2 reads > 250 mg/dL on consecutive days. For anti-CD3 treatment, mice were injected i.v. with anti-CD3 mAb (clone KT3, protein G-purified), typically 50 μg daily for 5 consecutive days. For IL7 treatment, IL7/anti-IL7 complexes were generated by mixing 0.75 μg of recombinant human IL7 (Peprotech) with 15 μg anti-hIL7 M25 mAb (BioXcell) in PBS at total volume 30ul for 15min at room temperature, before dilution in 100ul PBS for i.p. injection 3 times at 3 day intervals.
Human samples
For isolation of mononuclear cells from human peripheral blood, freshly collected human peripheral blood were diluted with equal volume of PBS with 2mM EDTA, layered over Ficoll-Hypaque solution and centrifuged at 900 × g, at 25°C for 20min. Remaining red blood cells were lysed by RBC lysis buffer, and the remaining cells were washed twice with PBS/2mM EDTA. Platelets were removed by overlaying the cells with FBS, centrifuged and the mononuclear cells were resuspended in complete RPMI media supplemented with 10% FBS.
In vitro cultures
For in vitro activation assay, mouse spleen cell suspensions were prepared in ice cold PBS. The spleen cell suspension was resuspended at 1 × 106 cells/ml in culture medium consisting of RPMI 1640 supplemented with 50 μM 2-ME (Sigma), 1× nonessential amino acids, 1 mM sodium pyruvate, and 100 U/ml penicillin. Spleen cells were incubated in a 24-well flat-bottom microtiter plate (1 ml/well) with addition of soluble anti-CD3 mAb using. Purified human PBMC were cultured in the same conditions, with stimulation of anti-huCD3 mAbs (HIT3A, OKT3, UCHT1, and TRX4).
Flow cytometry
Intracellular staining for Foxp3 staining was performed with manufacturer’s protocols (clone FJK-16s, eBioscience). For co-staining of Foxp3 and EdU, mice were injected with 1 mg EdU 6hr prior to sacrifice. EdU staining (Click-iT® EdU Flow Cytometry Assay Kits, Invitrogen) was performed after Foxp3 staining. To determine the amount of free and Ab-complexed CD3 molecules at the cell surface, splenocytes were stained with FITC-conjugated goat anti-rat or anti-human IgG, with or without pre-incubation with saturating amounts of the same anti-CD3 antibody as used for treatment (for mouse cells KT3, for human cells HIT3A, OKT3, UCHT1 (Biolegend) or TRX4 (Otelixizumab), yielding measures of total and occupied CD3 (free CD3 being the difference between the two). Results were expressed as a proportion of the mean fluorescence intensity (MFI) relative to that on untreated cells.
Gene expression profiling
Tconv (CD3+CD4+GFP-) and Treg (CD3+CD4+GFP+) splenocytes were double-sorted from 8 weeks old male Foxp3-iGFP NOD mice treated with anti-CD3 (KT3) for different lengths of time (each sample from a single mouse). Cells were collected directly into Trizol. RNA was purified, and used for probe synthesis for hybridization to Affymetrix Mouse Gene M1.0 ST microarrays. Raw data were background-corrected and normalized with the RMA algorithm in the GenePattern software package. Two independent duplicate experiments were performed. Because there was slight contamination with B cells and erythrocytes in one set, Hemoglobin genes and genes over-expressed in B cells (> 5-fold ratio in ImmGen CD19+ B vs. CD4+ T cell ImmGen data) were filtered out, as were genes with discordant Fold-Change (>2.5 & <1.3). Microarray data have been deposited at NCBI Gene Expression Omnibus, under accession # GSE48210. (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE48210)
Statistical analyses
Student’s t.test was used for comparisons of cell frequencies, and the incidence of diabetes was compared by Kaplan-Meier log-rank test. Findings were considered significant at p ≤ 0.05.
RESULTS
Treg and Tconv cells respond differently to injection of anti-CD3
To study the earliest transcriptional effects of anti-CD3 treatment, we used knockin mice in which Treg cells can be distinguished by expression of a fluorescent reporter, i.e. a mouse line carrying a bicistronic IRES-spaced Foxp3-GFP reporter gene (31) backcrossed onto the NOD genetic background (32) (hereafter referred to as NOD.Foxp3igfp). Adult NOD.FoxP3-iGFP mice were injected at 8-wks of age with the usual 50 μg dose of the anti-CD3 mAb, KT3, used in our published diabetes reversal studies (22). Under these conditions, KT3 induces a limited cytokine storm (33), and in previous studies this protocol reverted the hyperglycemia in approximately 50% of recently diabetic mice. This dose leads to a slight (2-fold or less) reduction in Tconv cells in the spleen and pancreas in the few days after treatment (not shown). CD4+GFP+ Treg and CD4+GFP- Tconv cells were sorted from spleens of treated mice at 2, 8, 24 and 72 hrs after initiation of treatment, and genome-wide gene-expression profiling was performed.
To assess the relative engagement of the TCR in Treg and Tconv cells after such treatment, we first analyzed the response of CD69, an early response marker directly downstream of TCR signaling. CD69 induction was early and comparable in both cell types (Fig. 1A, left). On the other hand, the CD44 response, whose induction is more complex, was higher in Treg cells. Overall results are depicted in Fig. 1B, which compares the induction ratio (relative to the same populations from untreated mice) for Treg and Tconv cells at various times. Several conclusions can be drawn from these plots. First, anti-CD3 elicited an abortive response in both T cell populations: while a robust response was evident 2 hours after administration (at an arbitrary Fold Change cutoff of 2.5, 416 and 550 genes being induced in Tconv and Treg cells, respectively), it had faded out by 72 hrs (only 70 and 64 changes apparent). This transient activation differs from gene-expression profiles observed when T cells were activated in vivo by persistent cognate antigen or strong costimulation, where responses continued to amplify at 72 hrs (e.g. (34) or T cell activation data at www.immgen.org).
Figure 1. Tconv and Treg cell respond differently to anti-CD3.

Transcriptional profiles were generated from Tconv and Treg CD4+ splenocytes from pooled mice (n=3) at 2 to 72 hrs after treatment with anti-CD3. (A) Changes in activation markers that reflect TCR signalling. (B) Comparison of the changes elicited in Treg and Tconv cells (visualized as the ratio of expression at indicated times relative to untreated (t0)); numbers indicate the number of transcrips that change by 2.5-fold or more (B) A Tconv-Treg differential index was computed for each gene to reflect these differential changes (genes towards the top of the graph preferentially respond in Tconv, those at the bottom in Treg), and is plotted against the maximum change observed
Second, the changes induced in Treg and Tconv cells were very comparable for the majority of genes, as indicated by the diagonal disposition of the transcripts in Fig. 1B. However, the slightly off-diagonal placement indicates that the immediate response was more intense in Treg cells at 2 hrs (above the diagonal) (also indicated by the number of responding transcripts), while the response in Tconv cells dominated at later times (below the diagonal). This faster start in Treg cells may be linked to their pre-activated state.
Third, the behavior of a number of transcripts did differ in the two cell types, being uniquely or preferentially induced in one or the other. This observation was true at both early (e.g. Il2, strikingly unique to Tconvs; or Scin, found only in Tregs) and late times (Tnfsf4 in Tconvs). To better visualize these differences, we computed a Tconv/Treg differential response index (by subtracting the Fold Change in Treg from that in Treg, and normalizing to the mean change), where a positive value reflects a preferential response in Tconv cells and a negative one indicates a preferential induction in Treg cells). In Fig. 1C, this index was plotted against the maximum change observed in Treg and Tconv cells (see Table S1 for gene details). The general disposition at 2 and 8 hrs after anti-CD3 administration reflected the different intensity of the responses noted above, but at 24 and 72 hrs the profiles showed clear “horns” of genes preferentially induced in one or the other of the two cell-types. At 24 hrs, more of the induction was seen in Tconv cells, but the residual induction at 72 hrs was more marked in Treg cells (Fig. 1C).
These findings raised the question of whether the differential response to anti-CD3 in Treg and Tconv cells reflected differences in baseline expression profiles between the two cell-types (i.e. the “Treg signature” (35)). The Tc/Tr differential response index versus the baseline Tconv/Treg ratio in unstimulated cells is plotted in Fig. 2 (see also Fig. S1 for 2 and 24 hr time points). To a great extent, the differential transcript changes in the two cell types amounted to a “catching up”, where genes under-expressed at the start in one cell-type were preferentially activated there. For example, Il2ra (CD25), Ctla4, Gpr83 and Tigit were under-represented in naïve Tconv cells, and their induction was stronger than in Tregs cells, where they were already expressed; similarly, Pde3b, Vipr, and Igfbp4 responded preferentially in Treg cells. On the other hand, some transcriptional responses were unique to Tconv cells, such as the increase in Il2, Il21 and Tnfsf4 (OX40L) transcripts mentioned above, suggesting that their expression was completely repressed in Tregs (Fig. 2B); the converse was true for Scin induction in Treg vis-à-vis Tconv cells. Genes encoding transcription factors followed these trends, and we noted an interesting pattern in the response of the E-protein-regulating factors Id2 and Id3, which control several aspects of T cell homeostasis: Id2, preferentially expressed in Tconv cells, was temporarily shut-down there, with an inverse pattern in Treg cells; conversely, its homolog Id3 was induced in Tconv cells, but transiently downregulated in Tregs. Most intriguing was the behavior of the Il7r gene, in the context of the breach of homeostatic control elicited by anti-CD3: it was the most strongly differential transcript at both 8 and 72 hrs (Fig. 2A); being strongly repressed in Tconv cells, but much less so, and actually induced at 24 and 72 hrs in Treg cells (Fig. 2B).
Figure 2. Elements of the differential response of Tconv and Treg to anti-CD3.

(A) The Tconv-Treg differential response, computed as in Fig1B, at 8 hrs (top panel) and 72 hrs (bottom) is plotted against the difference in expression between Tconv and Treg cells at baseline (y-axis), and the most extreme transcrips are shown by name. (B) Gene expression values (arbitrary units) for selected transcripts that are distinctively induced by anti-CD3 in Tconv and Treg cells. Values shown are average from two independent experiments.
Similar responses were elicited by a non-FcR-binding form of anti-CD3 (the Fab’2 fragment of the 2C11mAb) (data not shown). We also asked whether these patterns could be reproduced after anti-CD3 treatment in vitro, which would open the possibility of experimental manipulation and of analysis of responses by human cells. To most closely mimic the in vivo conditions, we cultured whole splenocytes with soluble anti-CD3 (1.25 μg/ml) for the same times as above. As illustrated in Fig. S2A-D, the patterns of differential responses in Tconv vs. Treg cells found in vivo were reproduced to a substantial extent in vitro (with perhaps the exception of Il2 and Il21, less responsive in vitro).
Differential IL7R protein expression in response to anti-CD3 treatment
The transcriptional response of the Il7r gene immediately caught our attention given the role of IL7 as a key trophic cytokine throughout lymphocyte differentiation and memory formation (36), its differential expression in Treg and Tconv cells at baseline (37), and its role in the early resetting of Treg homeostasis after anti-CD3 administration (22). Changes in the expression of IL2R subunits followed a different course (Fig. S2E). The effect of anti-CD3 on the Il7r gene is consistent with the described down-regulation of IL7R protein during the activation of Tconv cells (38-40), and with its up-regulation in Treg cells activated in vivo and in vitro (41). We verified that the transcriptional changes we observed were also reflected in the amount of protein. Indeed, anti-CD3 treatment induced comparable changes in surface levels of IL7R protein, with a reciprocal shift-up in Treg and shift-down in Tconv cells from spleen and LN (72 hr post-treatment in Fig. 3A). These effects were dose-dependent (Fig. 3B), and were relatively transient, with a return to pre-treatment levels by day 16 in both the spleen and pancreatic lymph nodes (PLNs).
Figure 3. IL7R expression on Tconv and Treg cells in response to anti-CD3.
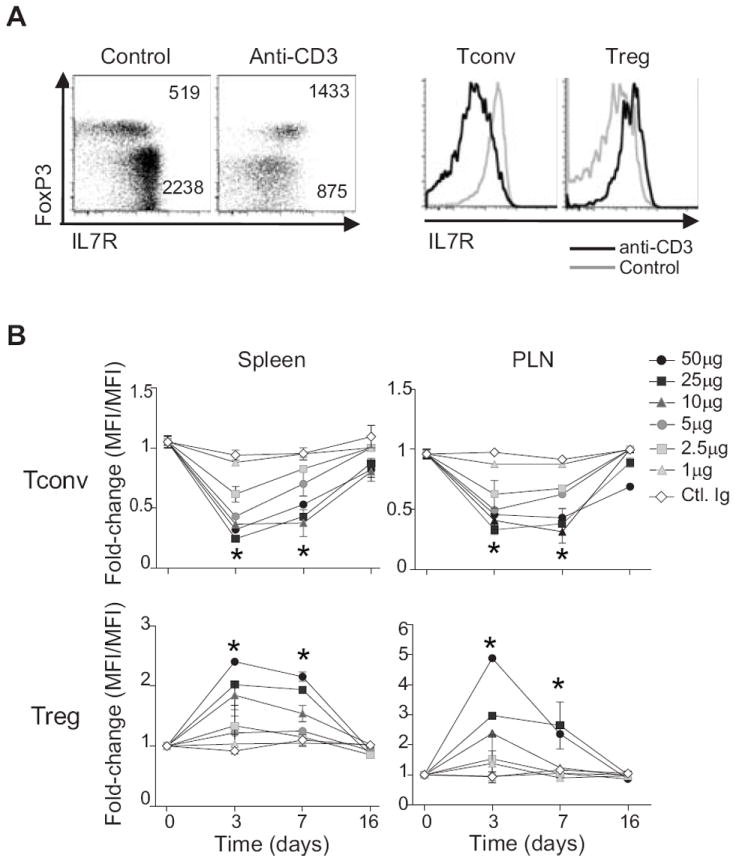
(A) Representative flow cytometric profile of IL7R and FoxP3 expression on TCRβ+CD4+ splenocytes of NOD mice, 72 hrs after treatment with anti-CD3 mAb (profiles representative of more than 5 different experiments. (B) Temporal analysis of IL7R expression on Tconv and Treg CD4+ splenocytes at different times after treatment with graded doses of anti-CD3 mAb i.p. *P ≤ 0.01 (Student’s t-test). Data are combined from 3 independent experiments, with 6 mice per dose.
We then asked how the reciprocal changes in IL7R levels reflected the extent of anti-CD3 engagement the TCR complex, and its ensuing clearance by internalization or shedding. The levels of free or engaged CD3 were measured by indirect staining with anti-CD3 (KT3), detected with anti-rat IgG (Fig. 4A displays data at 72 hrs post-treatment). At the dose used in most of the experiments described above (50 μg intraperitoneally (i.p.)), the majority of the CD3 molecules were lost from the cell surface, and the remainder were mainly occupied by the mAb; free complexes were more abundant at lower doses of injected KT3. As expected, surface levels of the CD3ε molecule reflected levels of the whole TCR complex, which was also cleared from the cell surface following similar kinetics (detected with anti-TCR-Vβ reagent) (Fig. S2F-G). The extent of TCR clearance was identical on Treg and Tconv cells (Fig. 4B), indicating that the differential transcriptional responses were not due to unequal TCR engagement or internalization. Importantly, the post-treatment levels of IL7R correlated strongly with the amount of remaining TCR, both for the disappearance of IL7R in Tconv cells and its increase in Treg cells (Fig. 4C). Similar responses were observed for CD4+ T cells when splenocytes were cultured in vitro with graded concentrations of anti-CD3 (Fig.4D): a dose-dependent drop in surface IL7R on Tconv cells, which correlated with levels of remaining CD3; an increase on Treg cells, most apparent after 72 hrs (after a transient decline at early time points, but mainly at high concentrations). Thus, the transcriptional changes at the Il7r result in alterations in the IL7R receptor, and these alterations are directly related to TCR engagement by anti-CD3.
Figure 4. Inverse relationship between IL7R expression and remaining surface CD3 after anti-CD3 mAb treatment.
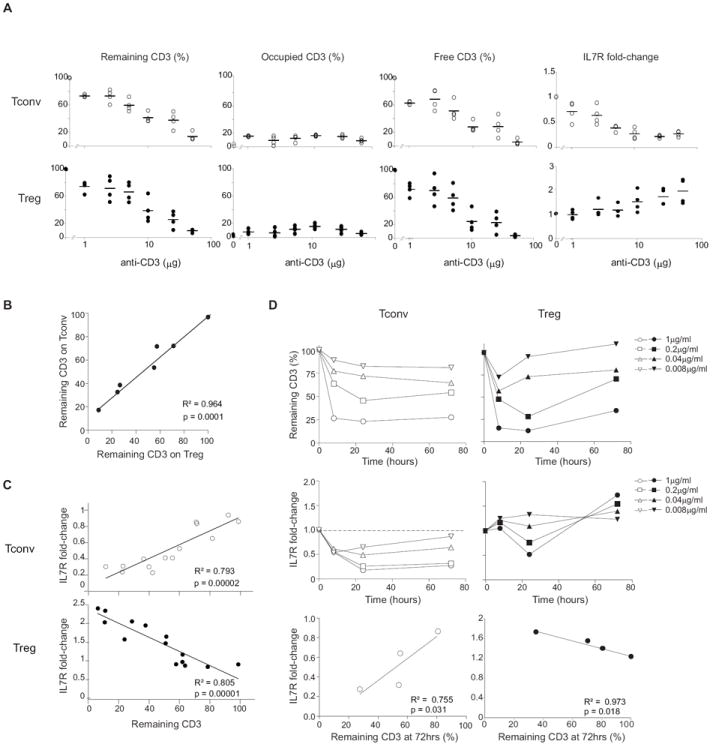
(A) Occupancy and clearance of CD3 molecules, and changes in IL7R expression, over a range of doses of anti-CD3. The amount of total, antibody-bound and free CD3 molecules, and of IL7R (as a FoldChange from untreated controls), on the surface of CD4+ Treg or Tconv cells was determined by flow cytometry 72 hrs after treatment with a range of doses of i.p. anti-CD3. Each dot is an individual mouse (B) CD3 molecules are similarly cleared in Treg and Tconv cells. Percent of remaining surface CD3 on Tconv and Treg cells from the same mouse, over a range of anti-CD3 doses (same experiments as Fig. 4A). (C) IL7R surface expression correlates with CD3 clearance in vivo. Changes in IL7R expression (as a FoldChange from untreated controls) plotted versus the percent of remaining surface CD3 on Tconv and Treg cells 72 hrs after treatment with a range of doses (same experiments as Fig. 4A). (D) In vitro effects of anti-CD3. Whole splenocytes were cultured with soluble anti-CD3 mAb at the concentrations shown, and remaining CD3 and IL7R expression on Tconv and Treg cells were determined at different times of culture. (E) IL7R surface expression correlates with CD3 clearance in culture. Change of IL7R expression plotted against the remaining CD3 expression on Tconv and Treg cell surface analyzed at 72 hrs of culture.
IL7 synergizes with anti-CD3 in vivo
The differential expression of Il7r mRNA and protein was an attractive explanation for the differences in population dynamics between Treg and Tconv cells after anti-CD3 treatment. Thus we asked whether supplementation with IL7 could enhance the effects of anti-CD3 treatment. We chose to add IL7/anti-IL7 complexes, in which the non-blocking antibody increases the stability and availability of the cytokine (42). The dose of 0.75 μg IL7 with 15 μg of anti-IL7 mAb M25 (hereafter simplified as “IL7”), administered daily at the same time as anti-CD3, resulted in only modest increases in the number of B or CD8+ T lymphocytes (Fig. S2H). Our protocol led to a slight increase in the proportion of FoxP3+ cells among CD4+ T cells in spleen, PLN and pancreas over what was achieved by anti-CD3 alone, an increase which was not statistically significant (Fig. 5A). IL7 induced an expansion of Treg cells in the spleen (Fig. 5A, lower panel), which was also seen for Tconv cells, explaining the limited effect on Treg cell proportions. These increases corresponded to a boost in cell cycling, as might be expected, evidenced by incorporation of EdU (Fig. 5B).
Figure 5. IL7 synergizes with anti-CD3 to induce expansion of Treg cells in vivo.
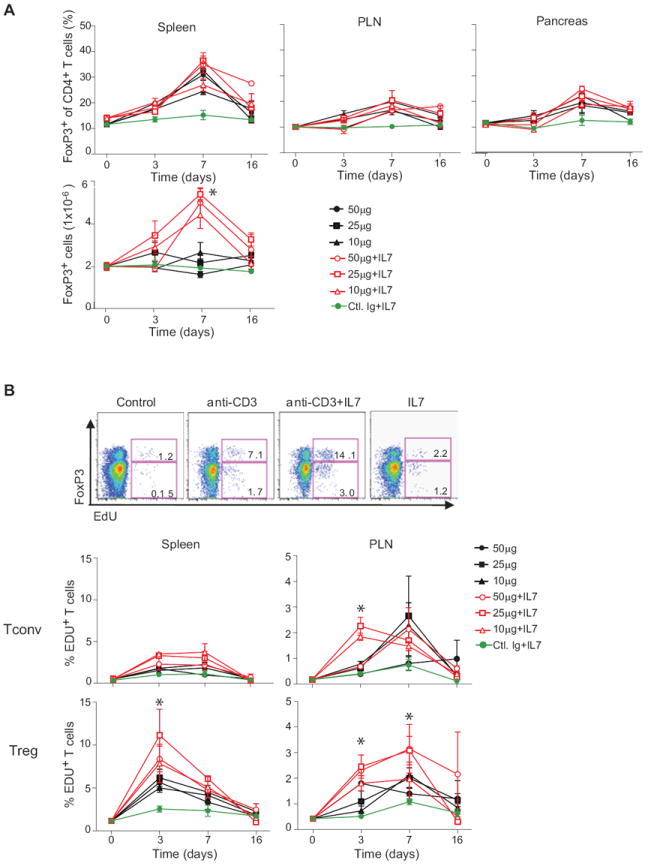
(A) Percentage and absolute numbers (bottom panel) of FoxP3+CD4+ cells in the spleen, PLN, and pancreas of anti-CD3 treated (solid symbols, doses as shown) or anti-CD3 plus IL7/anti-IL7 (0.75μg/15μg) complexes treated (open symbols); grey symbol: IL7/anti-IL7 complexes alone. *P ≤ 0.0001. Data are combined from two independent experiments with 3 mice for each dose. (B) Proliferative response. CD4+ splenocytes from mice treated as indicated untreated, anti-CD3, anti-CD3 plus IL7, or control IgG and IL7 treated were analyzed by flow cytometry, 6 hrs after administration of with. EdU; top: profiles representative of 3 independent experiments. Bottom: time course analysis in response to graded doses of anti-CD3 alone or in combination with IL7/anti-IL7 complexes. *P ≤ 0.01. Data are combined with two independent experiments with 3 mice for each dose.
We then asked whether the changes in population dynamics observed after supplementation of anti-CD3 with IL7 might also translate to an improved therapeutic outcome. NOD mice with recent-onset diabetes were treated within one week of diagnosis with the usual daily dose of anti-CD3 (50 μg), alone or supplemented with IL7/anti-IL7 complexes during the 5 day treatment period. As illustrated in Fig. 6, the addition of IL7 significantly improved the outcome (Kaplan-Meier p=0.02), with 13/16 mice showing long-term diabetes-free survival, compared to 4/9 with anti-CD3 alone. IL7 alone had no protective effect.
Figure 6. Synergistic effect of combined anti-CD3 and IL7 treatment in newly diabetic NOD mice.
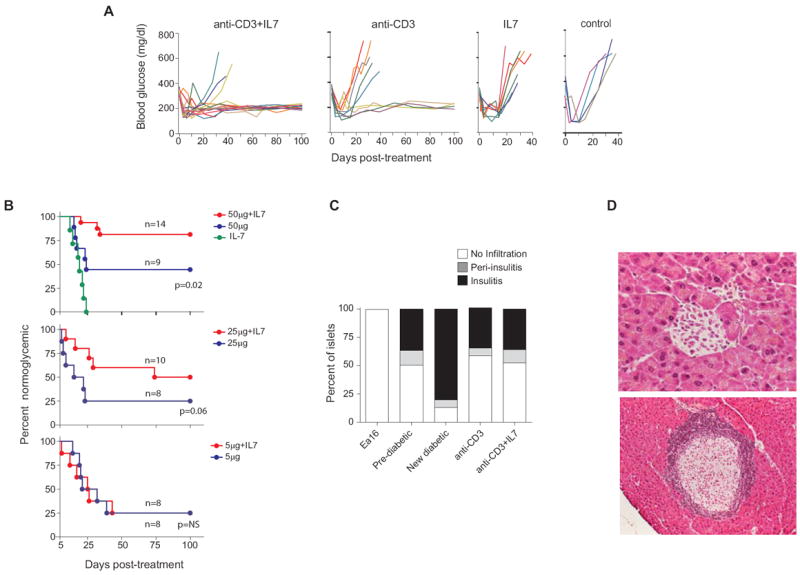
(A) Monitoring of blood glucose in recent-onset diabetic NOD females treated with anti-CD3 alone (50 μg daily, n=9), anti-CD3 together with IL7/anti-IL7 complexes (n=14), Il7 complexes alone, or none for five consecutive days; all mice received an insulin pellet to maintain glycemic control for 15-20 days. (B) Compilation of disease incidence in experiments as in A for several doses of anti-CD3 (IL7/anti-IL7 complexes at a constant dose of 0.75μg/15μg). (C) Insulitis in the pancreata of previously diabetic NOD female mice treated as in A (anti-CD3 n =5; anti-CD3 + IL7 complexes n =5), evaluated histologically after 100 days. Non-insulitic Ea16/NOD mice were included as a negative control. A minimum of 30 islets was examined for each mouse. (D) Representative islets from mice in long-term remission after treatment with anti-CD3 + IL7 complexes.
It would be of interest for the practical application of anti-CD3 therapy, to reduce the dose of antibody in order to limit its undesirable effects (i.e. the cytokine storm or the EBV reactivation observed in some patients). So we tested the effect of IL7 in recent-onset diabetic NOD mice treated under the same regimen, but with reduced dosing of anti-CD3. At 25 μg anti-CD3, IL7 was still effective (although with only 50% long-term remission), but little protection was seen with the less effective dose of 5 μg anti-CD3 (Fig. 6B). Thus, supplementation with IL7 does not seem to allow a substantial reduction in anti-CD3 dosage.
We also investigated the residual autoimmune lesion after treatment with anti-CD3 + IL7. We and others have previously reported that anti-CD3 does not clear insulitis, which persists but in a milder and histological more organized form (similar to the “respectful insulitis” observed in the BDC2.5 TCR transgenic model on non-progressor genetic backgrounds (43)). The same result was obtained with mice protected by anti-CD3 plus IL7 (Fig. 6C). Thus, while IL7 reproducibly improves the efficacy of anti-CD3, it does not appear to qualitatively alter the outcome.
Effect of anti-CD3 treatment of human cells
Are the above results relevant for anti-CD3 therapy in humans? The similarities between the transcriptional changes induced by anti-CD3 in mouse cells in vivo and in vitro encouraged us to search for an equivalent response in human cells challenged with anti-CD3 in culture. Peripheral blood mononuclear cells (PBMCs) were obtained from healthy volunteers. To best approximate in vivo conditions of mixed cell populations, we used whole PBMCs rather than purified CD4+ T cells. PBMCs were cultured in the presence of anti-huCD3 mAb for 72 hrs and were analyzed by flow cytometry for expression of IL7R on Treg and Tconv cells. In order to evaluate possible variation between mAbs of different affinity and ability to bind Fc receptors, we used several anti-CD3 mAb preparations, two of which have been used in clinical trials: three mouse anti-huCD3 (HIT3A, OKT3 (Teplizumab) and UCHT1) and an aglycosyl form of chimeric anti-huCD3 mAb (TRX4, Otelixizumab). The same reciprocal effects of anti-CD3 treatment on IL7R expression by Treg vs. Tconv cells were observed with human cells, although not as clearly as with mouse Treg cells in vivo: a clear shift-down for Tconv cells, a slight upregulation of IL7R in Treg cells, particularly marked on cells expressing high levels of FoxP3 (Fig. 7A, top; titration in Fig. S3). The effect was essentially identical with the four different mAbs examined.
Figure 7. Differential IL7R expression on Tconv and Treg cells from human blood in response to anti-CD3.
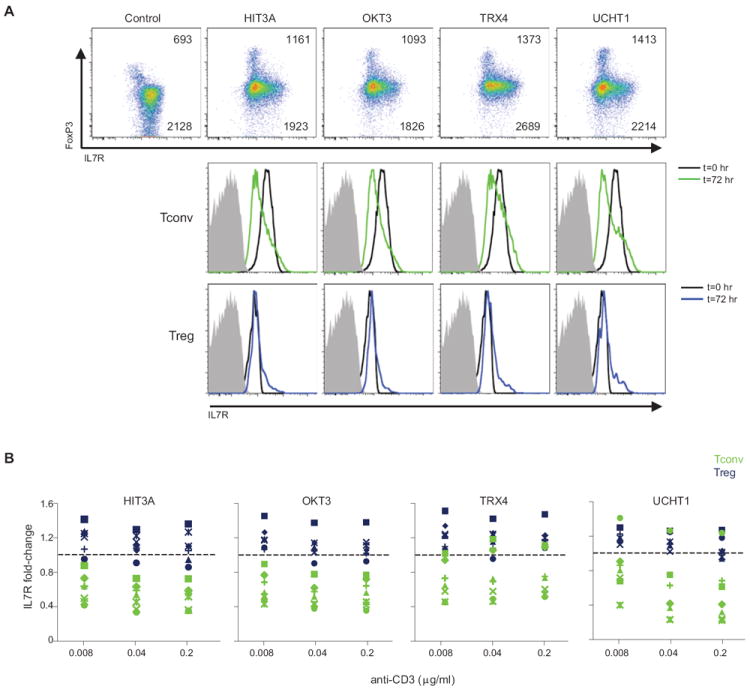
PBMCs from healthy volunteers were treated in culture with soluble anti-CD3 (4 different mAbs), and IL7R expression assessed in Treg and Tconv cells after 72 hrs by flow cytometry (A) Top: Scatter plots of FoxP3 and IL7R staining, values are MFI in FoxP3hi and FoxP3- cells; bottom: corresponding overlaid histograms. (B) Similar responses in Treg and Tconv cells from several independent donors. Changes in IL7R expression at 72 hrs (as FoldChange relative to t=0 control) induced by the different anti-CD3 mAbs on blood Tconv (green) and Treg (blue) from seven Caucasian donors.
Finally, we asked whether the changes in IL7R expression might be variable between unrelated individuals. There has been variability in responses to anti-CD3 treatment in clinical trials, which could reflect differences in their diabetes status or conditions of treatment, but also perhaps pharmacogenetic variability in their response to the drug. Blood PBMCs were obtained from seven healthy donors of European ancestry (4 females, 3 males, ages 21-44), and were treated as described above each of the four anti-CD3 mAbs. Cells from all donors showed largely similar IL7R responses at 72 hrs, with the usual increase in Treg and drop in Tconv cells (Fig. 7B). There was one exception, in that one donor failed to respond to UCHT1, but this observation is most likely explained by the known polymorphism in FcgRII, which conditions the ability to bind mouse IgG1 (which UCHT1 contains), and which had actually been discovered on the basis of differential responses to UCHT1 (44).
Thus, these results show that anti-CD3 elicits the same differential response by human Treg vs. Tconv cells as it does in the mouse counterparts, suggesting that the underlying differential signaling pathways are the same in both species.
DISCUSSION
The re-establishment of self-tolerance induced by anti-CD3 treatment of autoimmune diabetes likely involves the resetting of active modes of peripheral tolerance. Here, we have analyzed potential mechanisms of action of anti-CD3, following leads that pointed to differential effects on the homeostasis of CD4+ Tconv and Treg cells. We found that administration of anti-CD3 induced transient and clearly different responses in Treg and Tconv cells. In both human and mouse cells, one of the most distinctive divergences concerned IL7R, a meaningful finding given the pleiotropic role of IL7 in lymphocyte homeostasis and, most relevant, in the proliferation of Treg cells unleashed by anti-CD3 (22). This result encouraged us to hypothesize, and ultimately find, a synergy between anti-CD3 and IL7 treatments in vivo in the reversion of recent-onset diabetes in NOD mice.
The results highlight a clear difference in the transcriptional response of CD4+ Treg and Tconv cells to engagement by anti-CD3. Although most alterations in gene expression were similar in the two cell types, including the activation of cell-cycle and metabolic genes, several loci showed clearly differential changes. Importantly, many of the differences observed in vivo were reproduced upon anti-CD3 challenge in vitro. Some of these divergences were observed several days after treatment, reflecting indirect consequences, but others were visible at the 2-hr time point, and thus reflected more immediate consequences of different signaling pathways in Treg and Tconv cells. Several reports have indicated that such differences exist: lower activation of Akt and S6 kinase in Treg cells (45), a reduced recruitment of PKCΘ at the TCR synapses of Treg cells, with lower Carma-1 relative to Dlgh1 activation, which might lead to relatively higher NFATc but lower NF-κB and Akt activation (46,47); a kinase-independent signaling pathway seeming to operate, uniquely in Treg cells, between ZAP-70 and the GTPase Rap1 (48). Alone or in combination, these could easily result in the quantitative differences in transcriptional activation observed here.
Most intriguing were the differential alterations in IL7R expression. IL7 is a key trophic cytokine across the immune system, affecting all lymphoid lineages. IL7 is of demonstrated importance in the homeostasis of Treg cells and in the maintenance of FoxP3 expression, particularly when IL2 is limiting or absent (49-52) although thymic stromal lymphopoietin (TSLP) can partially compensate for IL7 deficiency in the maintenance of Treg cells (53). The results of Di Caro et al (54), showing that IL7 helps mature Treg cells and enhance FoxP3 expression, are also consistent with a scenario in which Treg cells gain a relative advantage over Tconv cells from the these reciprocal changes in the Il7R express. As might be expected, IL7R is tightly regulated by many extracellular stimuli, such as TCR activation signals or IL7 and other pro-survival cytokines (36). Low expression of IL7R is a hallmark of Treg cells (37), or at least some of them (41), perhaps via direct down-regulation by FoxP3 (37). Several transcription factors (TF) regulate expression of Il7r. Ets family transcription factors, Ets-1 and GABPa, bind the Il7r promoter region at the same location as PU.1 in B cells, and promote its expression in immature thymocytes and activated CD8+ T cells (55,56). Foxo-1, a member of the forkhead family of TFs, binds an intronic Il7r enhancer, and is required for the expression of Il7r in naive T cells (57). In contrast, Gfi-1 and FoxP1 suppress Il7r expression in T cells, the latter by antagonizing Foxo1 (56,58). Finally, the E-protein inhibitor Id3 also influences the differential expression of IL7R in effector vs. memory-precursor cells during CD8+ T cell responses (59). Thus, there appears to be a convoluted interplay of TFs responsible for Il7r regulation, and one can assume that different TFs affect the sequence of events that unfolds (a precipitous drop by 8hrs, followed by a stagnant recovery). Interestingly, we found that anti-CD3 elicited an early decrease in Foxo1 expression, together with an induction of Gfi1 that predominated in Tconv cells, which might account for Il7r transcription, as well as reciprocal changes in Id2 and Id3 (Fig. 2B, Supplementary Table 2). One or the combination of these rapid changes in TF representation might be responsible for the unique behavior of the Il7r locus after anti-CD3 treatment, which may be grounds for future interesting exploration.
The differential transcriptional effects elicited by anti-CD3 injection were largely reproduced by treatment of mouse splenocytes in culture, and this similarity also extended to cultured human PBMCs, for which very similar effects on IL7R expression were detected in response to four different anti-CD3 preparations, two of which are in clinical development. Because the differential shift in IL7R expression occurs rapidly, the likeliest explanation is that changes in mRNA/protein content are indeed taking place in each cell, but we cannot rule out that preferential survival of IL-7R cells contributes to the result. It was not possible to generate gene-expression profiles from human Treg and Tconv cells after treatment, as the activation obscures the CD25 marker normally used for sorting. Attempts were made to perform restricted signature profiling after sorting of permeabilized and anti-FoxP3-stained human cells: although too variable for robust exploitation, the results did point to differential changes in Treg vs Tconv cells for some of the genes that responded differentially in mouse cells (e.g. Il7r, Il2, TRAT1, and Il12rb2 – LL, unpublished). However, because the effects were generally less obvious in vitro than in vivo, it would be desirable to analyze human subjects at early times after anti-CD3 treatment. Analysis of a small group of individual donors showed no evidence of inter-individual variability of the response, except for one outlier likely due to the FcgRII polymorphism that affects binding of mouse IgG1 (44), since the low response was seen only with UCHT1 and not the IgG2a mAbs (Fig. 7C).
These differential effects led us to hypothesize that a combined anti-CD3/IL7 therapy might have enhanced efficacy in restoring glycemic control to recont-onset diabetic NOD mice. This proved to be the case, validating our interpretation, but also opening the door to potential clinical application of this new combination therapy to human T1D patients. Unfortunately, although IL7 co-administration proved effective at increasing the penetrance of optimal doses of anti-CD3, it did not allow a reduction in dosing of anti-CD3, which would be a therapeutically desirable goal. As a side note, while our results mirrored those of Mehta et al in terms of dose-dependent TCR occupancy and clearing (~90% or greater reduction in TCR at the 50 or 25 μg therapeutic doses, only ~50% reduction at the 5 μg dose, Fig. 4A) (60), we did not reproduce the observation of good clinical efficacy in the 5 μg dose range. There was a clear linear relationship between the persistence of TCR and magnitude of changes affecting IL7R, whether in mouse or human cells. Optimal effects were seen with the highest level of TCR downregulation, those at which we also fund best clinical efficacy. If our interpretation is correct, this relationship may suggest that a rapidly maximal treatment would be most effective, rather than a gradual ramp-up from repeated low doses.
Whether anti-CD3 induces an alteration in dominant tolerance, or a attenuation of pathogenic cells has been a long open question. Evidence has been provided for the latter (14-17), most recently with the suggestion that anti-CD3 redirects IL17-producing pathogenic cells to the small intestine (13,61). While our results provide a plausible mechanism for a resetting of Treg homeostasis, they certainly do not rule out complementary effects on pathogenic T cells themselves. Indeed, the transient response observed in Tconv cells, which mimics the differential response elicited with or without costimulatory signals (E. Wakamatsu, DM and CB, unpublished data), might underlie the anergic phenotypes reported previously (14-16).
IL7 therapy is being attempted in several settings where it might be desirable to restore lymphoid homeostasis, such as primary or acquired immunodeficiencies or after cancer chemotherapy (62). It might seem paradoxical, then, to propose IL7 supplementation in a context where autoreactive Tconv cells are the root of the problem. IL7 blockade by anti-IL7 mAbs has been used as a therapy in preclinical models of autoimmune disease. It can reduce pathogenic Th17 cells in Experimental Allergic Encephalomyelitis (63), and recent reports show it is effective in reversing recent-onset NOD diabetes (64,65). As an adjunct to anti-CD3 therapy, however, the concept is that IL7 serves to transiently enhance homeostatic changes that favor Treg over autoreactive efector cells.
In conclusion, we have demonstrated divergent responses of CD4+ Treg and Tconv cells to anti-CD3 engagement, suggesting a molecular underpinning for the drug’s mechanism of action and prompting us to propose a combination approach to resetting Treg homeostasis for therapy of autoimmune diabetes.
Supplementary Material
Acknowledgments
We thank K. Hattori for help with mice, J. LaVecchio and G. Buruzala for flow cytometry, and Jeff Ericson and Scott Davis for help with the microarray analyses and reference datasets. This work benefitted from data assembled by the ImmGen consortium. It was supported by a Sponsored Research Agreement from GlaxoSmithKline, by a grant from the NIH (RC2-GM093080), and by the core facilities of Joslin Diabetes Center’s National Institutes of Diabetes and Digestive and Kidney Diseases funded Diabetes and Endocrinology Research Center and of the JDRF Center on Immunological Tolerance in Type-1 Diabetes at Harvard Medical School. LL and JN were supported by mentor-based fellowship grants from the American Diabetes Association.
It was supported in part by a Sponsored Research Agreement with GlaxoSmithKline (GSK), one of the authors is from GSK as acknowledgement of materials and contributions to the experimental design and interpretation of the data, and GSK provided one of the reagents used. GSK has an ongoing interest in anti-CD3 therapy.
Footnotes
Competing interests: This work was performed at Harvard Medical School, and the Harvard authors have no personal conflicts of interest.
References
- 1.Herold KC, Bluestone JA, Montag AG, Parihar A, Wiegner A, Gress RE, Hirsch R. Prevention of autoimmune diabetes with nonactivating anti-CD3 monoclonal antibody. Diabetes. 1992;41:385–391. doi: 10.2337/diab.41.3.385. [DOI] [PubMed] [Google Scholar]
- 2.Vallera DA, Carroll SF, Brief S, Blazar BR. Anti-CD3 immunotoxin prevents low-dose STZ/interferon-induced autoimmune diabetes in mouse. Diabetes. 1992;41:457–464. doi: 10.2337/diab.41.4.457. [DOI] [PubMed] [Google Scholar]
- 3.Hayward AR, Shriber M. Reduced incidence of insulitis in NOD mice following anti-CD3 injection: requirement for neonatal injection. J Autoimmun. 1992;5:59–67. doi: 10.1016/s0896-8411(05)80051-4. [DOI] [PubMed] [Google Scholar]
- 4.Chatenoud L, Thervet E, Primo J, Bach JF. Anti-CD3 antibody induces long-term remission of overt autoimmunity in nonobese diabetic mice. Proc Natl Acad Sci U S A. 1994;91:123–127. doi: 10.1073/pnas.91.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keymeulen B, Vandemeulebroucke E, Ziegler AG, Mathieu C, Kaufman L, Hale G, Gorus F, Goldman M, Walter M, Candon S, Schandene L, Crenier L, De Block C, Seigneurin JM, De Pauw P, Pierard D, Weets I, Rebello P, Bird P, Berrie E, Frewin M, Waldmann H, Bach JF, Pipeleers D, Chatenoud L. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med. 2005;352:2598–2608. doi: 10.1056/NEJMoa043980. [DOI] [PubMed] [Google Scholar]
- 6.Herold KC, Hagopian W, Auger JA, Poumian-Ruiz E, Taylor L, Donaldson D, Gitelman SE, Harlan DM, Xu D, Zivin RA, Bluestone JA. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med. 2002;346:1692–1698. doi: 10.1056/NEJMoa012864. [DOI] [PubMed] [Google Scholar]
- 7.Keymeulen B, Walter M, Mathieu C, Kaufman L, Gorus F, Hilbrands R, Vandemeulebroucke E, d Van V, Crenier L, De BC, Candon S, Waldmann H, Ziegler AG, Chatenoud L, Pipeleers D. Four-year metabolic outcome of a randomised controlled CD3-antibody trial in recent-onset type 1 diabetic patients depends on their age and baseline residual beta cell mass. Diabetologia. 2010;53:614–623. doi: 10.1007/s00125-009-1644-9. [DOI] [PubMed] [Google Scholar]
- 8.Herold KC, Gitelman S, Greenbaum C, Puck J, Hagopian W, Gottlieb P, Sayre P, Bianchine P, Wong E, Seyfert-Margolis V, Bourcier K, Bluestone JA. Treatment of patients with new onset Type 1 diabetes with a single course of anti-CD3 mAb Teplizumab preserves insulin production for up to 5 years. Clin Immunol. 2009;132:166–173. doi: 10.1016/j.clim.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sherry N, Hagopian W, Ludvigsson J, Jain SM, Wahlen J, Ferry RJ, Jr, Bode B, Aronoff S, Holland C, Carlin D, King KL, Wilder RL, Pillemer S, Bonvini E, Johnson S, Stein KE, Koenig S, Herold KC, Daifotis AG. Teplizumab for treatment of type 1 diabetes (Protege study): 1-year results from a randomised, placebo-controlled trial. Lancet. 2011;378:487–497. doi: 10.1016/S0140-6736(11)60931-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.GlaxoSmithKline: Press release. GlaxoSmithKline and Tolerx announce phase III DEFEND-1 study of otelixizumab in type 1 diabetes did not meet its primary endpoint. 2011 http://www.gsk.com/media/pressreleases/2011/2011_pressrelease_10039.htm.
- 11.Bach JF. Anti-CD3 antibodies for type 1 diabetes: beyond expectations. Lancet. 2011;378:459–460. doi: 10.1016/S0140-6736(11)60980-X. [DOI] [PubMed] [Google Scholar]
- 12.Herold KC, Gitelman SE, Masharani U, Hagopian W, Bisikirska B, Donaldson D, Rother K, Diamond B, Harlan DM, Bluestone JA. A single course of anti-CD3 monoclonal antibody hOKT3gamma1(Ala-Ala) results in improvement in C-peptide responses and clinical parameters for at least 2 years after onset of type 1 diabetes. Diabetes. 2005;54:1763–1769. doi: 10.2337/diabetes.54.6.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waldron-Lynch F, Henegariu O, Deng S, Preston-Hurlburt P, Tooley J, Flavell R, Herold KC. Teplizumab induces human gut-tropic regulatory cells in humanized mice and patients. Sci Transl Med. 2012;4:118ra12. doi: 10.1126/scitranslmed.3003401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kohm AP, Williams JS, Bickford AL, McMahon JS, Chatenoud L, Bach JF, Bluestone JA, Miller SD. Treatment with nonmitogenic anti-CD3 monoclonal antibody induces CD4+ T cell unresponsiveness and functional reversal of established experimental autoimmune encephalomyelitis. J Immunol. 2005;174:4525–4534. doi: 10.4049/jimmunol.174.8.4525. [DOI] [PubMed] [Google Scholar]
- 15.Alegre ML, Tso JY, Sattar HA, Smith J, Desalle F, Cole M, Bluestone JA. An anti-murine CD3 monoclonal antibody with a low affinity for Fc gamma receptors suppresses transplantation responses while minimizing acute toxicity and immunogenicity. J Immunol. 1995;155:1544–1555. [Google Scholar]
- 16.Smith JA, Tso JY, Clark MR, Cole MS, Bluestone JA. Nonmitogenic anti-CD3 monoclonal antibodies deliver a partial T cell receptor signal and induce clonal anergy. J Exp Med. 1997;185:1413–1422. doi: 10.1084/jem.185.8.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Penaranda C, Tang Q, Bluestone JA. Anti-CD3 therapy promotes tolerance by selectively depleting pathogenic cells while preserving regulatory T cells. J Immunol. 2011;187:2015–2022. doi: 10.4049/jimmunol.1100713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chatenoud L, Primo J, Bach JF. CD3 antibody-induced dominant self tolerance in overtly diabetic NOD mice. J Immunol. 1997;158:2947–2954. [PubMed] [Google Scholar]
- 19.You S, Leforban B, Garcia C, Bach JF, Bluestone JA, Chatenoud L. Adaptive TGF-beta-dependent regulatory T cells control autoimmune diabetes and are a privileged target of anti-CD3 antibody treatment. Proc Natl Acad Sci U S A. 2007;104:6335–6340. doi: 10.1073/pnas.0701171104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Belghith M, Bluestone JA, Barriot S, Megret J, Bach JF, Chatenoud L. TGF-beta-dependent mechanisms mediate restoration of self-tolerance induced by antibodies to CD3 in overt autoimmune diabetes. Nat Med. 2003;9:1202–1208. doi: 10.1038/nm924. [DOI] [PubMed] [Google Scholar]
- 21.Bresson D, Togher L, Rodrigo E, Chen Y, Bluestone JA, Herold KC, Von Herrath M. Anti-CD3 and nasal proinsulin combination therapy enhances remission from recent-onset autoimmune diabetes by inducing Tregs. J Clin Invest. 2006;116:1371–1381. doi: 10.1172/JCI27191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishio J, Feuerer M, Wong J, Mathis D, Benoist C. Anti-CD3 therapy permits regulatory T cells to surmount T cell receptor-specified peripheral niche constraints. J Exp Med. 2010;207:1879–1889. doi: 10.1084/jem.20100205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T Cells: Mechanisms of Differentiation and Function. Annu Rev Immunol. 2012;30:531–564. doi: 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8:523–532. doi: 10.1038/nri2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feuerer M, Hill JA, Mathis D, Benoist C. Foxp3+ regulatory T cells: differentiation, specification, subphenotypes. Nat Immunol. 2009;10:689–695. doi: 10.1038/ni.1760. [DOI] [PubMed] [Google Scholar]
- 26.Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- 27.Feuerer M, Shen Y, Littman DR, Benoist C, Mathis D. How punctual ablation of regulatory T cells unleashes an autoimmune lesion within the pancreatic islets. Immunity. 2009;31:654–664. doi: 10.1016/j.immuni.2009.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Z, Herman AE, Matos M, Mathis D, Benoist C. Where CD4+CD25+ T reg cells impinge on autoimmune diabetes. J Exp Med. 2005;202:1387–1397. doi: 10.1084/jem.20051409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tarbell KV, Yamazaki S, Olson K, Toy P, Steinman RM. CD25+ CD4+ T Cells, Expanded with Dendritic Cells Presenting a Single Autoantigenic Peptide, Suppress Autoimmune Diabetes. J Exp Med. 2004;199:1467–1477. doi: 10.1084/jem.20040180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen G, Han G, Wang J, Wang R, Xu R, Shen B, Qian J, Li Y. Essential roles of TGF-beta in anti-CD3 antibody therapy: reversal of diabetes in nonobese diabetic mice independent of Foxp3+CD4+ regulatory T cells. J Leukoc Biol. 2008;83:280–287. doi: 10.1189/jlb.0707498. [DOI] [PubMed] [Google Scholar]
- 31.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 32.Darce J, Rudra D, Li L, Nishio J, Cipolletta D, Rudensky AY, Mathis D, Benoist C. An N-terminal mutation of the Foxp3 transcription factor alleviates arthritis but exacerbates diabetes. Immunity. 2012;36:731–741. doi: 10.1016/j.immuni.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mottram PL, Murray-Segal LJ, Han W, Maguire J, Stein-Oakley AN. Remission and pancreas isograft survival in recent onset diabetic NOD mice after treatment with low-dose anti-CD3 monoclonal antibodies. Transpl Immunol. 2002;10:63–72. doi: 10.1016/s0966-3274(02)00050-3. [DOI] [PubMed] [Google Scholar]
- 34.Obst R, Van Santen HM, Melamed R, Kamphorst AO, Benoist C, Mathis D. Sustained antigen presentation can promote an immunogenic T cell response, like dendritic cell activation. Proc Natl Acad Sci U S A. 2007 doi: 10.1073/pnas.0707331104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hill JA, Feuerer M, Tash K, Haxhinasto S, Perez J, Melamed R, Mathis D, Benoist C. Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity. 2007;27:786–800. doi: 10.1016/j.immuni.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 36.Ma A, Koka R, Burkett P. Diverse functions of IL-2, IL-15, and IL-7 in lymphoid homeostasis. Annu Rev Immunol. 2006;24:657–679. doi: 10.1146/annurev.immunol.24.021605.090727. [DOI] [PubMed] [Google Scholar]
- 37.Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, Gottlieb PA, Kapranov P, Gingeras TR, Fazekas de St GB, Clayberger C, Soper DM, Ziegler SF, Bluestone JA. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med. 2006;203:1701–1711. doi: 10.1084/jem.20060772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xue HH, Kovanen PE, Pise-Masison CA, Berg M, Radovich MF, Brady JN, Leonard WJ. IL-2 negatively regulates IL-7 receptor alpha chain expression in activated T lymphocytes. Proc Natl Acad Sci U S A. 2002;99:13759–13764. doi: 10.1073/pnas.212214999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Swainson L, Verhoeyen E, Cosset FL, Taylor N. IL-7R alpha gene expression is inversely correlated with cell cycle progression in IL-7-stimulated T lymphocytes. J Immunol. 2006;176:6702–6708. doi: 10.4049/jimmunol.176.11.6702. [DOI] [PubMed] [Google Scholar]
- 40.Alves NL, van Leeuwen EM, Derks IA, van Lier RA. Differential regulation of human IL-7 receptor alpha expression by IL-7 and TCR signaling. J Immunol. 2008;180:5201–5210. doi: 10.4049/jimmunol.180.8.5201. [DOI] [PubMed] [Google Scholar]
- 41.Simonetta F, Chiali A, Cordier C, Urrutia A, Girault I, Bloquet S, Tanchot C, Bourgeois C. Increased CD127 expression on activated FOXP3+CD4+ regulatory T cells. Eur J Immunol. 2010;40:2528–2538. doi: 10.1002/eji.201040531. [DOI] [PubMed] [Google Scholar]
- 42.Boyman O, Ramsey C, Kim DM, Sprent J, Surh CD. IL-7/anti-IL-7 mAb complexes restore T cell development and induce homeostatic T Cell expansion without lymphopenia. J Immunol. 2008;180:7265–7275. doi: 10.4049/jimmunol.180.11.7265. [DOI] [PubMed] [Google Scholar]
- 43.Andre I, Gonzalez A, Wang B, Katz J, Benoist C, Mathis D. Checkpoints in the progression of autoimmune disease: lessons from diabetes models. Proc Natl Acad Sci U S A. 1996;93:2260–2263. doi: 10.1073/pnas.93.6.2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tax WJ, Willems HW, Reekers PP, Capel PJ, Koene RA. Polymorphism in mitogenic effect of IgG1 monoclonal antibodies against T3 antigen on human T cells. Nature. 1983;304:445–447. doi: 10.1038/304445a0. [DOI] [PubMed] [Google Scholar]
- 45.Crellin NK, Garcia RV, Levings MK. Altered activation of AKT is required for the suppressive function of human CD4+CD25+ T regulatory cells. Blood. 2007;109:2014–2022. doi: 10.1182/blood-2006-07-035279. [DOI] [PubMed] [Google Scholar]
- 46.Zanin-Zhorov A, Ding Y, Kumari S, Attur M, Hippen KL, Brown M, Blazar BR, Abramson SB, Lafaille JJ, Dustin ML. Protein kinase C-theta mediates negative feedback on regulatory T cell function. Science. 2010;328:372–376. doi: 10.1126/science.1186068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zanin-Zhorov A, Lin J, Scher J, Kumari S, Blair D, Hippen KL, Blazar BR, Abramson SB, Lafaille JJ, Dustin ML. Scaffold protein Disc large homolog 1 is required for T-cell receptor-induced activation of regulatory T-cell function. Proc Natl Acad Sci U S A. 2012;109:1625–1630. doi: 10.1073/pnas.1110120109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Au-Yeung BB, Levin SE, Zhang C, Hsu LY, Cheng DA, Killeen N, Shokat KM, Weiss A. A genetically selective inhibitor demonstrates a function for the kinase Zap70 in regulatory T cells independent of its catalytic activity. Nat Immunol. 2010;11:1085–1092. doi: 10.1038/ni.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bayer AL, Lee JY, de la Barrera A, Surh CD, Malek TR. A function for IL-7R for CD4+CD25+Foxp3+ T regulatory cells. J Immunol. 2008;181:225–234. doi: 10.4049/jimmunol.181.1.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harnaha J, Machen J, Wright M, Lakomy R, Styche A, Trucco M, Makaroun S, Giannoukakis N. Interleukin-7 is a survival factor for CD4+ CD25+ T-cells and is expressed by diabetes-suppressive dendritic cells. Diabetes. 2006;55:158–170. [PubMed] [Google Scholar]
- 51.Gratz IK, Truong HA, Yang SH, Maurano MM, Lee K, Abbas AK, Rosenblum MD. Cutting Edge: memory regulatory t cells require IL-7 and not IL-2 for their maintenance in peripheral tissues. J Immunol. 2013;190:4483–4487. doi: 10.4049/jimmunol.1300212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim GY, Ligons DL, Hong C, Luckey MA, Keller HR, Tai X, Lucas PJ, Gress RE, Park JH. An in vivo IL-7 requirement for peripheral Foxp3+ regulatory T cell homeostasis. J Immunol. 2012;188:5859–5866. doi: 10.4049/jimmunol.1102328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mazzucchelli R, Hixon JA, Spolski R, Chen X, Li WQ, Hall VL, Willette-Brown J, Hurwitz AA, Leonard WJ, Durum SK. Development of regulatory T cells requires IL-7Ralpha stimulation by IL-7 or TSLP. Blood. 2008;112:3283–3292. doi: 10.1182/blood-2008-02-137414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Di C, V, D’Anneo A, Phillips B, Engman C, Harnaha J, Lakomy R, Styche A, Trucco M, Giannoukakis N. Interleukin-7 matures suppressive CD127(+) forkhead box P3 (FoxP3)(+) T cells into CD127(-) CD25(high) FoxP3(+) regulatory T cells. Clin Exp Immunol. 2011;165:60–76. doi: 10.1111/j.1365-2249.2011.04334.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xue HH, Bollenbacher J, Rovella V, Tripuraneni R, Du YB, Liu CY, Williams A, McCoy JP, Leonard WJ. GA binding protein regulates interleukin 7 receptor alpha-chain gene expression in T cells. Nat Immunol. 2004;5:1036–1044. doi: 10.1038/ni1117. [DOI] [PubMed] [Google Scholar]
- 56.Chandele A, Joshi NS, Zhu J, Paul WE, Leonard WJ, Kaech SM. Formation of IL-7Ralphahigh and IL-7Ralphalow CD8 T cells during infection is regulated by the opposing functions of GABPalpha and Gfi-1. J Immunol. 2008;180:5309–5319. doi: 10.4049/jimmunol.180.8.5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kerdiles YM, Beisner DR, Tinoco R, Dejean AS, Castrillon DH, DePinho RA, Hedrick SM. Foxo1 links homing and survival of naive T cells by regulating L-selectin, CCR7 and interleukin 7 receptor. Nat Immunol. 2009;10:176–184. doi: 10.1038/ni.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Feng X, Wang H, Takata H, Day TJ, Willen J, Hu H. Transcription factor Foxp1 exerts essential cell-intrinsic regulation of the quiescence of naive T cells. Nat Immunol. 2011;12:544–550. doi: 10.1038/ni.2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang CY, Best JA, Knell J, Yang E, Sheridan AD, Jesionek AK, Li HS, Rivera RR, Lind KC, D’Cruz LM, Watowich SS, Murre C, Goldrath AW. The transcriptional regulators Id2 and Id3 control the formation of distinct memory CD8+ T cell subsets. Nat Immunol. 2011;12:1221–1229. doi: 10.1038/ni.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mehta DS, Christmas RA, Waldmann H, Rosenzweig M. Partial and transient modulation of the CD3-T-cell receptor complex, elicited by low-dose regimens of monoclonal anti-CD3, is sufficient to induce disease remission in non-obese diabetic mice. Immunology. 2010;130:103–113. doi: 10.1111/j.1365-2567.2009.03217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Esplugues E, Huber S, Gagliani N, Hauser AE, Town T, Wan YY, O’Connor W, Jr, Rongvaux A, van RN, Haberman AM, Iwakura Y, Kuchroo VK, Kolls JK, Bluestone JA, Herold KC, Flavell RA. Control of TH17 cells occurs in the small intestine. Nature. 2011;475:514–518. doi: 10.1038/nature10228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mackall CL, Fry TJ, Gress RE. Harnessing the biology of IL-7 for therapeutic application. Nat Rev Immunol. 2011;11:330–342. doi: 10.1038/nri2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu X, Leung S, Wang C, Tan Z, Wang J, Guo TB, Fang L, Zhao Y, Wan B, Qin X, Lu L, Li R, Pan H, Song M, Liu A, Hong J, Lu H, Zhang JZ. Crucial role of interleukin-7 in T helper type 17 survival and expansion in autoimmune disease. Nat Med. 2010;16:191–197. doi: 10.1038/nm.2077. [DOI] [PubMed] [Google Scholar]
- 64.Lee LF, Logronio K, Tu GH, Zhai W, Ni I, Mei L, Dilley J, Yu J, Rajpal A, Brown C, Appah C, Chin SM, Han B, Affolter T, Lin JC. Anti-IL-7 receptor-alpha reverses established type 1 diabetes in nonobese diabetic mice by modulating effector T-cell function. Proc Natl Acad Sci U S A. 2012;109:12674–12679. doi: 10.1073/pnas.1203795109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Penaranda C, Kuswanto W, Hofmann J, Kenefeck R, Narendran P, Walker LS, Bluestone JA, Abbas AK, Dooms H. IL-7 receptor blockade reverses autoimmune diabetes by promoting inhibition of effector/memory T cells. Proc Natl Acad Sci U S A. 2012;109:12668–12673. doi: 10.1073/pnas.1203692109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.