

Abstract
Triglyceride-lipase (TGL) is a major fat body lipase in Manduca sexta. The knowledge of how TGL activity is regulated is very limited. A WWE domain, presumably involved in protein-protein interactions, has been previously identified in the N-terminal region of TGL. In this study, we searched for proteins partners that interact with the N-terminal region of TGL. Thirteen proteins were identified by mass spectrometry, and the interaction with four of these proteins was confirmed by immunoblot. The oxidoreductase lipoamide-dehydrogenase (LipDH) and the apolipoprotein components of the lipid transporter, HDLp, were among these proteins. LipDH is the common component of the mitochondrial α-keto acid dehydrogenase complexes whereas HDLp occurs in the hemolymph. However, subcellular fractionation demonstrated that these two proteins are relatively abundant in the soluble fraction of fat body adipocytes. The cofactor lipoate found in typical LipDH substrates was not detected in TGL. However, TGL proved to have critical thiol groups. Additional studies with inhibitors are consistent with the notion that LipDH acting as a diaphorase could preserve the activity of TGL by controlling the redox state of thiol groups. On the other hand, when TG hydrolase activity of TGL was assayed in the presence of HDLp, the production of diacylglycerol (DG) increased. TGL-HDLp interaction could drive the intracellular transport of DG. TGL may be directly involved in the lipoprotein assembly and loading with DG, a process that occurs in the fat body and is essential for insects to mobilize fatty acids. Overall the study suggests that TGL occurs as a multi-protein complex supported by interactions through the WWE domain.
1. INTRODUCTION
Lipid is always the major component of the fat body and the main source of metabolic fuel (Arrese and Soulages, 2010). Triglycerides (TG), the major lipid form, are stored in the core of the lipid droplets surrounded by phospholipids and a coat of proteins (Brasaemle, 2007). Lipid droplets are dynamic organelles whose metabolic role is dependent on the protein coat. Several of these proteins are involved in the regulation of triglyceride storage and mobilization (Brasaemle, 2010). Mobilization of TG stores from LDs is catalyzed by lipases. TGL is a major fat body lipase of Manduca sexta that is conserved among insects (Arrese et al., 2010). The most conserved regions of TGL coincide with the presence of functional domains: the WWE domain is located in the amino (N)-terminal part (residues 46-128), the lipase motif is in the central region (GHS367LG), and the DDHD domain is situated between residues 411 to 609 towards the C-terminal (Arrese et al 2010). The DDHD is a long domain commonly present in the proteins of the intracellular phospholipase A1 (iPLA1) family. A recent study showed that DDHD domain is essential for the enzymatic activity of mammalian iPLA1 (Inoue et al., 2012). Moreover, this domain also participates in the formation of homo-oligomers through multiple interactions between the carboxyl (C)-terminal region containing the DDHD domain and the N-terminal half of mammalian iPLA1 (Inoue et al., 2012).
The WWE domain, which is named after three of its conserved residues, has been identified in diverse cytosolic proteins with predicted ubiquitin- and ADP-ribosylation-related functions, and, in general, it is predicted to mediate protein-protein interactions (Aravind, 2001). WWE domain is most commonly found as a single copy motif but proteins of the Deltex family exhibit a tandem pair of WWE domains. Deltex proteins are cytosolic proteins of the Notch pathway that is involved in cell fate determination during several developmental processes (Zweifel et al., 2005). In Drosophila, the WWE tandem modules localize at the N-terminal region of Deltex. Each module forms an autonomous fold in which the N and C termini within the module are in close proximity (Zweifel et al., 2005). Interactions between these two WWE modules create a compact and functional N-term domain by which Deltex interacts with the Notch receptor displaying its activity as modulator of the Notch pathway (Zweifel et al., 2005). The function of WWE domain in TGL or any other lipase remains to be elucidated.
TGL localizes in the cytosol and is able to hydrolyze TG, DG, and phospholipids (Arrese et al., 2006; Patel et al., 2005). Our understanding of how TGL activity is regulated is very limited. In vivo, TGL is constitutively phosphorylated, and the level of phosphorylation remains constant when lipolysis is stimulated by adipokinetic hormone (AKH) (Patel et al., 2006). The cAMP-dependent kinase, PKA, is involved in the activation of the lipolytic response induced by AKH (Arrese et al., 1999; Bednarova et al., 2013). PKA can phosphorylate TGL in vitro but the lipase activity remains unchanged after phosphorylation (Patel et al., 2004; Patel et al., 2005). On the other hand, AKH provokes a rapid phosphorylation of Lsd1, a lipid droplet-associated protein, and this event results in the activation of TGL (Arrese et al., 2008; Patel et al., 2005). Phosphorylation of the lipid droplet accounts for about 70% of the AKH-induced lipolytic response (Patel et al., 2006; Patel et al., 2005).
In addition to the effect on the lipid droplets, AKH also induces lipase activation in the cytosol (Auerswald and Gade, 2006; Auerswald et al., 2005; Patel et al., 2006). In Manduca this effect accounts for the remaining 30% of the lipolytic response to AKH (Patel et al., 2006). The mechanism of this component of the lipase activation that, as mentioned above, is independent of changes in the phosphorylation state of TGL, remains unknown.
To better understand the mechanisms of regulation of TGL, we are interested in defining the protein network involved in the lipolytic process. This study focused on the proteins that interact with TGL. We hypothesized that the WWE domain could be mediating those protein-protein interactions. This hypothesis was tested investigating whether fat body soluble proteins would interact with the lipase region that contains the WWE domain (N-term) by using recombinant protein in an affinity based assay combined with mass spectrometry. Thirteen WWE interacting proteins were identified including the disulfide reductase lipoamide-dehydrogenase and the apolipoprotein components of the lipid transporter, HDLp. Immunoblot analyses confirmed the enrichment of these proteins in the affinity assay. Further studies were undertaken to investigate the possible functional link between TGL and LipDH or HDLp. The identification of proteins that interact with the WWE domain suggests a leading role of this domain in a number of TGL-protein interactions.
2. MATERIALS
pET 32 Ek/LIC vector, E. coli strains Nova Blue and Rosetta 2, were obtained from Novagen (Billerica, MA). Ni-sepharose resin, PD-10 columns, and ECL chemiluminescence reagents were obtained from GE-Healthcare (Pittsburgh, PA). Protein A-Agarose (pre-blocked with albumin) was obtained from Santa Cruz Biotechnology (Dallas, TX). Glutathione (GSH), glutathione disulfide (GSSG), N-ethlymaleimide (NEM), Triton X-100, benzamidine, carmustine and auranofin were obtained from Sigma-Aldrich (St. Louis, MO). Dithiothreitol (DTT) and liquid scintillation counting cocktail were obtained from RPI (Mount Prospect, IL). M. sexta adipokinetic hormone (AKH) was obtained from Peninsula Laboratories (Belmont, CA). [Tri-9,10-3H]-oleoylglycerol was purchased from Perkin Elmer Life Sciences (Boston, MA). Precast 4–20% acrylamide gradient gels and BenchMark™ Protein Ladder containing proteins with molecular masses of 220, 160, 120, 100, 90, 80, 70, 60, 50, 40, 30, 25, 20, 15, and 10 kDa were purchased from Invitrogen (Carlsbad, CA). Pre-cast 4–15% acrylamide gels were purchased from Bio-Rad (Hercules, CA). DNA sequencing was performed by the Core Facility of our department using an ABI Model 3700 DNA Analyzer. All other chemicals were of analytical grade.
2.1. Insects
Manduca sexta eggs were purchased from Carolina Biological (Burlington, NC), and larvae were reared at 25°C on an artificial diet. Adult insects were maintained at room temperature without food. Fat bodies from adult male insects (second day after emergence) were placed in liquid nitrogen immediately after dissection and stored at −80 °C.
2.2. Cloning, Expression and Purification of N terminus region containing WWE domain
The N-terminal region of the Manduca sexta TGL gene (encoding amino acids 1–140) was amplified by polymerase chain reaction (PCR) using the following forward and reverse primers: 5′-GACGACGACAAGATGAACGATAGTACGGAAAGGA-3′ and 5′-GAGGAGAAGCCCGGTCTATCTGGCGTCAGTGGGACCT-TTG-3′, respectively, and a plasmid containing full-length Manduca TGL cDNA (pGEM-TGL) that was previously prepared (Arrese et al., 2010). The amplified product was ligated into pET32 Ek/LIC vector as previously reported (Arrese et al., 2008), and the sequence of the recombinant plasmid was confirmed by DNA sequencing. The N-term region of TGL was expressed as a recombinant fusion protein containing thioredoxin, poly-His and S-tag (Trx-[His]6-Stag-WWE). Expression of the fusion protein was induced with 1mM IPTG for 3 h. For the preparation of cell extracts, the bacterial pellet was resuspended in buffer (50mM Tris pH8, 1mM EDTA, 100mM NaCl, 1mM PMSF) containing 0.3 mg/ml of lysozyme. After 30 min incubation, the preparation was centrifuged at 20,000g for 1h. The fusion protein was found in the pellet, which was resuspended in 20mM Tris buffer pH 7.9 containing 6M urea. The supernatant was collected after centrifugation (20,000g for 30 min) and loaded into a Ni-affinity column. Proteins were eluted by increasing concentrations of imidazole (20–200mM) in the same buffer. The fusion protein (Trx-WWE) was eluted with 200mM imidazole. The purified protein was dialyzed against 10mM Tris buffer pH 7.9 containing 250 mM NaCl, and subsequently treated with enterokinase to remove Trx and the poly-His/S-tag. After incubation, the sample was passed through a small column of Ni-Sepharose and the N-terminal region containing the WWE domain (amino acids 1–140) was recovered in the flow through. The WWE domain was dialyzed against 50mM phosphate buffer, pH 7.4 and analyzed by SDS-PAGE and circular dichroism.
2.3. Circular Dichroism (CD)
CD spectroscopy was carried out in a Jasco-715 (Jasco Corporation, Tokyo, Japan) spectropolarimeter. Quadruplicates of the spectra were averaged, corrected for background, and smoothed and the mean residue ellipticity (deg•cm2•dmol−1) was calculated. The secondary structure of the recombinant WWE domain was estimated with the program DichroWeb (Whitmore and Wallace, 2008) (http://dichroweb.cryst.bbk.ac.uk).
2.4. WWE Interacting Proteins Assay
Immobilization of Fusion Protein in Ni-Sepharose beads: Trx-WWE (840μg) was bound to Ni-sepharose beads (500mg) and washed with PBS (1mM KH2PO4, 3mM Na2HPO4, 155mM NaCl; pH 7.4). The resin (Trx-WWE-Ni) was split into two identical fractions of 250mg, which from now on, were handled in parallel. In one fraction, the lipase domain was removed by proteolytic cleavage with thrombin followed by one step wash with PBS to generate Trx-Ni (“control” resin). The other half of the resin (Trx-WWE-Ni) was subjected to the same procedures as “control” resin but thrombin was omitted in the buffer. All steps were carried out on ice or at 4°C. Fat body soluble proteins: two fat bodies were homogenized in 2 ml of PBS containing 1 mg/l aprotinin and 1mM PMSF. Infranatant was collected after centrifugation (20,000g for 30 min), and protein concentration adjusted to 20mg/ml. Assay: 150μl of fat body soluble proteins were incubated with each type of resin for 2hr and subsequently washed four times with 1 ml PBS. Proteins were eluted in one step by incubation with 150μl of 1M imidazole. Aliquots of the eluted proteins were subjected to SDS-PAGE (4–20% gradient gels) and analyzed further by mass spectrometry and western blotting. Data were collected from two independent experiments.
2.5. Protein Identification by Mass Spectrometry
Each lane of the Coomassie Blue stained gel was sliced in six regions. During all the steps control and test samples were treated in exactly the same way. Each slice was minced and proteins were prepared for trypsin digestion as previously reported (Soulages et al., 2012). Digestion products were analyzed by LC-MS/MS on a hybrid LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific) coupled to a New Objectives PV-550 nanoelectrospray ion source and an Eksigent NanoLC-2D chromatography system. Other technical details and the methods used for peptide and protein identification were done as described previously (Soulages et al., 2012).
2.6. Western blotting
Proteins were separated by SDS-PAGE on 4–20 % (Invitrogen) or 4–15% (Bio-Rad) acrylamide gels. The gels were blotted onto nitrocellulose membranes using a wet system from Invitrogen or the Trans-Blot Turbo system from Bio-Rad and further incubated with rabbit polyclonal antibody specific for TGL that was raised using recombinant N-terminal region (amino acids 1–140) by Cocalico Biologicals (Reamstown, PA), apolipophorin I/II that was raised in rabbits using purified HDLp from the hemolymph of larval insects by Cocalico Biologicals (Reamstown, PA). The following commercial antibodies that were raised in rabbits were also used: glutathione S-transferase I (Abcam, Cambridge, MA), lipoamide-dehydrogenase (Abcam, Cambridge, MA), Stag (Novagen, Billerica, MA) and lipoyted proteins (Calbiochem, Billerica, MA). After incubation of the blots with horseradish peroxidase-conjugated anti-rabbit secondary antibodies (Santa Cruz Biotechnology, Dallas, TX), the immunocomplexes were detected by enhanced chemiluminescence (ECL GE Healthcare).
2.7. Subcellular Fractionation of Fat Body
Fat body tissue from two male adult insects were dissected, thoroughly washed, pooled and immediately homogenized with a Potter-Elvehjem glass homogenizer fitted with a Teflon pestle, using 3 ml of HB buffer (50mM Tris, pH 7.4, 1mM EDTA, 250mM sucrose, 0.1mM benzamidine, 10mM DTT, 1mM PMSF). The homogenate was centrifuged to 1,000g for 10 min. The resulting soluble fraction was centrifuged at 20,000g for 30min. The pellet was collected and the supernatant of 20,000g (SN20) was overlaid with 1ml buffer PBS and centrifuged at 100,000g for 1h. Three fractions were collected: fat cake, infranatant (cytosol) and pellet (P100). Both pellets (P20 and P100) were resuspended in 3ml PBS and re-centrifuged. Both pellets were resuspended 1ml of buffer and centrifuged at 500g for 15min. The resulting supernatant was used as membranes fraction (P20 and P100, respectively). Fat cake containing the lipid droplets were collected from the top and resuspended in 0.5ml PBS. Protein concentration was determined in all fractions and samples were analyzed by western blotting for LipDH and HDLp.
2.8. Lipase Activity
Four fat bodies were combined with 3 ml of HB buffer (50mM Tris, pH 7.4, 1 mM EDTA, 250 mM sucrose, 1mM benzamidine, 10 mg/l leupeptin, 1 mg/l aprotinin) and homogenized. All steps were carried out on ice or at 4°C. The homogenate was overlaid with 1 ml of buffer without sucrose, and subjected to centrifugation at 20,000g for 30min. The infranatant (soluble fraction) was collected and total protein was determined. Aliquots of the soluble fraction of fat body containing 150–200μg total protein were used to measure the lipase activity that was assayed with a micellar TG substrate containing [3H]-TG and Triton-X-100 as previously described (Arrese and Wells, 1994). The assay mixture (0.1 ml) contained 50mM Tris, 500mM NaCl, 0.05%(v/v) Triton X-100, 0.25mM triolein [0.002 Ci/mmol], pH 7.9. To determine the effect of carmustine (75 to 560μM) and auranofin (1 and 75 μM) on lipase activity, samples were pre-incubated with increasing concentrations of inhibitors for 30min on ice. Control assays were carried out in the absence of inhibitors. Lipase activity was measured in the absence and presence of 10 mM DTT in triplicate reactions in a total of three independent experiments. Lipase activity was expressed as fold change over control (no inhibitor).
2.9. Triglyceride-Lipase Thiol Groups
TGL was partially purified from the cytosolic fraction of M. sexta fat body homogenates using anion-exchange, hydroxyl-apatite, and hydrophobic interaction chromatography as reported previously (Arrese et al., 2006). To determine the effect of DTT, GSH, and GSSG on the lipase activity, 15μg of partially purified TGL was pre-incubated for 30min on ice with increasing concentrations (0, 3, 6 and 12mM) of each reagent followed by lipase activity assay that was carried out as described above. To determine the effect of N-ethlymaleimide (NEM), 1 to 6μl (or nothing in the control sample) of NEM (500mM in ethanol) were added to 80μl of lipase preparation (0.75μg/μl) that was previously treated with 10 mM DTT. The final volume was 100 μl, and the amount of ethanol was adjusted to be the same in all samples. After 30 min incubation on ice, lipase activity was determined as indicated above. Lipase activity was expressed as fold change over control (no addition).
2.10. Effect of AKH on Lipase Activity
Adult insects were decapitated for 24hr and injected with 13mg trehalose. After 2 hr, 100 pmol of AKH were injected and fat body tissue was dissected 20min after the hormonal injection. Control group was injected with PBS. Freshly prepared fat bodies from two insects were pooled, homogenized and centrifuged as described above (section 2.8). Lipase activity in soluble fraction was immediately assayed in the absence and presence of 10 mM DTT (section 2.8). Lipase activity was expressed as nmol TG/mg protein min.
2.10. Co-immunoprecipitation
Fat body soluble proteins (50μl having approximately 500μg total protein) was combined with 2μl of rabbit anti-HDLp antibody for 2h at 4°C, followed by addition of 20μl of Protein-A-Agarose beads and incubated overnight. After centrifugation (1000g for 5 minutes), the pellet was washed four times with 1ml PBS. The resulting immunoprecipitates were resuspended in Laemmli sample buffer, and proteins were subjected to SDS-PAGE (4–15% gradient gels) to be analyzed further by mass spectrometry and western blotting.
2.11. Intracellular Distribution of HDLp, TG and DG
Sucrose density centrifugation: freshly dissected adult fat bodies were homogenized as indicated above using 3mL of HB Buffer. The homogenate was centrifuged at 1000g for 10min and the resulting supernatant was adjusted to 50% sucrose and transferred to a SW40 centrifuge tube to be subsequently overlaid with 1.0 ml each of 40%, 35%, 20%, 10%, 5% in HB buffer plus an additional layer of 1.5 ml of PBS. The tube was centrifuged at 160,000g for 4 h. The gradient was fractionated into seven fractions and each fraction analyzed to determine protein, apoLp I/II, DG and TG. Proteins were separated by 4–15% SDS-PAGE. For the top six fractions the gels were loaded with 0.6% of each fraction. For the cytosolic fraction 0.1% of the fraction was loaded in the gel. The separated proteins were transferred to nitrocellulose membranes and subjected to western blotting using anti-HDLp antibody. The proportions of DG and TG were determined by thin layer chromatography (TLC) of lipid extracts. To this end, lipids from aliquots of the gradient fractions were extracted with chloroform: methanol (2:1 v/v) and separated by TLC in hexane/ethyl ether/formic acid (70:30:3 v/v/v) (Arrese et al., 2001). MG, DG, FA and TG spots were visualized using iodine vapor, scanned and quantified with AlphaEaseFCTM software. TG and DG contents were expressed as percentage of total neutral lipid.
2.12. Lipophorin Purification
Hemolymph from adult insects was collected in the presence of 30mM KH2PO4, 2mM EDTA and 10mM DTT. Hemolymph was centrifuged at 4°C for 15min at 4,500g. The supernatant was adjusted to 0.5g/ml KBr and overlaid with PBS containing 0.1g/ml KBr. The sample was centrifuged at 340,000g in the Beckman rotor VTi65.2 at 10°C for 1 h. The density gradient was fractionated from top to bottom, and the fractions containing HDLp were collected (fractions 3 and 4). After desalting the fractions by gel filtration, the purity of HDLp was assessed by SDS-PAGE, and the protein concentration was determined.
2.13. Effect of HDLp on TGL activity
The activity of partially purified TGL was assayed in the presence of HDLp or albumin (BSA) as control. The assay mixture (0.1 ml) contained 50mM Tris, pH 7.9, 25nomles of [9,10-3H]-triolein, 0.002Ci/mmol, 0.05%(v/v) Triton X-100, 500mM NaCl, 10 mM DTT and the corresponding amount of HDLp or BSA (0–5μg). The reaction was started by adding TGL (15μg) and incubated at 37°C with constant shaking for 30min. Blank reactions did not contain lipase. The reaction was stopped by adding 500μl of CHCl3/methanol/benzene (2:2.4:1 vol) and 40μl 1N HCl. The organic phase was obtained, dried, and separated by TLC. Spots corresponding to each lipid form were scraped. Silica was transferred to a vial containing 4 ml of scintillation cocktail and the radioactivity determined by liquid scintillation counting to determine the hydrolysis products as previously described (Arrese and Wells, 1994). Results were expressed as nmoles of lipid produced (DG, MG) and consumed (TG) per reaction. Values represent the mean ± SEM of three determinations.
2.14. Other Methods
SDS–PAGE was performed according to Laemmli and proteins were visualized by Coomassie Brilliant Blue R staining. Protein concentrations were determined by the Bradford dye-binding assay using bovine serum albumin (BSA) as standard. Data were presented as the mean ± SEM. Statistical comparisons were made by one way ANOVA with post test.
3. RESULTS AND DISCUSSION
3.1. Identification of TGL interacting proteins
The WWE domain is a ~90-amino acids fragment situated between amino acids 40 to 128 of Manduca TGL (Arrese et al., 2010). The function of this domain in TGL is unknown. A significant sequence alignment between amino acid 40 and 140 of TGL and the second WWE domain of Drosophila Deltex, for which the structural information is available, was found. Fig 1A shows the alignment of Manduca and Drosophila WWE sequence motif, and the three conserved residues–two tryptophans and glutamic acid-characteristic of WWE domain are indicated. We cloned the cDNA encoding amino acids 1–140 of TGL, and the recombinant protein was overexpressed in E. coli as a fusion protein with thioredoxin, polyHis and Stag (Trx-[His]6-Stag-WWE). The expression of the fusion protein was confirmed by western blot using Stag antibodies (not shown). Purified fusion protein was cleaved with enterokinase to remove the fusion tags (Fig 1B), and the secondary structure of resulting protein (WWE) was analyzed by circular dichroism (CD) (Fig 1C). Deconvolution of the CD spectrum indicated that the N-term region was folded. The estimated proportions of α–helix, β-strands, turns, and unordered structures were 22.5%, 22.4%, 21.7% and 33.4%, respectively. This structural composition is compatible with the fold of Deltex WWE domains deduced from the crystal structure (Zweifel et al., 2005).
Figure 1. Sequence, Expression and Circular Dichroism of N-terminal region of TGL.

A) The amino acid sequence alignment of the N-terminal region of MsTGL with Drosophila WWE-Deltex sequence is shown. The sequence alignment was generated using Alignment Explorer/Muscle (Edgar 2004). Identical residues in the sequences are denoted by asterisks; conservative substitutions are denoted by dots. The locations of α-helices (rectangles) and β-strands (arrows) identified from the crystal structure of WWE-Deltex were adapted from Zweifel et al (Zweifel et al. 2005); B) Coomassie Blue stained SDS-PAGE of the purified 140-residue polypeptide, which was expressed as a fusion protein with thioredoxin, cleaved with enterokinase and then purified; C) Far-UV CD spectrum of the purified N-terminal domain of TGL [amino acids 1–140].
Recombinant TGL N-term proved to be a stable polypeptide having the predicted fold, and we utilized this protein to search for TGL interacting proteins. Fusion protein, Trx-[His]6-Stag-WWE, was immobilized to Ni-sepharose beads and incubated with the soluble fraction of fat body homogenate. As a control, an identical assay was carried out in parallel using resin in which only the tag portion of the fusion protein (Trx-[His]6) was bound. After multiple washes, proteins were eluted with imidazole and analyzed by SDS-PAGE coupled to mass spectrometry (MS/MS). Fig 2A shows a representative acrylamide gel with the protein profiles for test (lane 1) and control (lane 2) resin. The fusion protein (MW=33.3kDa) and control protein (MW=13.9kDa) can be seen as major bands of lanes 1 and 2, respectively, together with co-eluted proteins. Each gel line was sliced in sections that were analyzed by MS/MS as indicated in Methods. A comparison of the levels of spectral counts was used to distinguish the proteins that were more abundant in the test samples. A threshold of 1.5-fold increase in the test sample over control was chosen to minimize potential artifacts. Table 1 shows the list of proteins that matched this criterion in two independent experiments. A total of thirteen proteins were identified. Antibodies were available for four of these proteins (apolipophorin precursor - apoLp-I/II-, LipDH, glutathione S-transferase and TGL). Western blotting analyses of the eluate from control and test resins confirmed the enrichment detected by MS/MS (Fig 2B). In addition, to confirm the interaction of TGL and apoLp-I/II we carried out immunoprecipitation using anti-HDLp antibodies. The immunoprecipitates were analyzed by MS/MS and western blotting using anti-TGL antibodies. TGL was detected in the immunocomplex confirming that TGL binds HDLp (Fig 3). In addition to TGL, four other proteins were also identified in the immunocomplex by MS/MS. These proteins are indicated in Table 1.
Figure 2. WWE Interacting Proteins Assay.
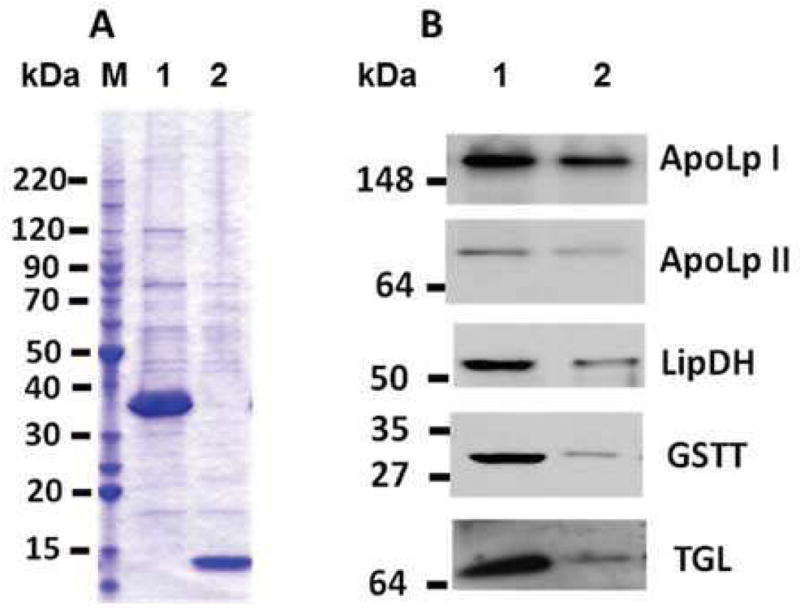
A) Coomassie blue stained gel of the soluble proteins eluted from by Ni-sepharose beads containing Trx-WWE fusion protein (lane 1) or Trx (lane 2). Fat body soluble proteins were incubated with 12.5nmol of Trx-WWE bound to Ni-sepharose beads for 2h and the resin was subsequently washed four times with buffer. Proteins were eluted in one step by incubation with 1M imidazole. Co-eluted proteins were analyzed by SDS-PAGE in 4–20% acrylamide gradient gels (lane 1). Control experiment was carried out using 12.5nmol of Trx bound to Ni-sepharose beads (lane 2). Protein markers (BenchMark protein ladder) are shown in lane M (see Materials). B) Western blotting: aliquots of eluted proteins from test (lane 1) and control (lane 2) resins were separated by SDS-PAGE in 4–20% acrylamide gel and transferred to nitrocellulose. Blots were analyzed by immunodetection for the following proteins: ApoLp-I and II, LipDH, GST and TGL as indicated in Materials and Methods.
Table 1. WWE interacting proteins identified by MS/MS.
The table shows the proteins that interact with the WWE domain. The list includes proteins whose spectral counts (SC) were at least 1.5-fold greater in the test (Trx-WWE) resin than in the control resin (Trx), in two experiments. The SC and the SC ratio between the samples eluted from test and control resins observed in one of the experiments are shown. In bold are the proteins in which antibody was available and MS/MS results were confirmed by Western blot.
| Identified Protein | Accession Number | Spectral Counts(SC) | ||
|---|---|---|---|---|
| Trx-WWE | Trx | SC Ratio | ||
| 1- Apolipophorin Precursor^ | 2498144 | 88 | 19 | 4.6 |
| 2- Arylphorin subunit beta^ | 1168527 | 77 | 3 | 25 |
| 3- Trehalose 6-phosphate synthase^ | 281372519 | 71 | 48 | 1.5 |
| 4- Dihydrolipoamide dehydrogenase | 6014978 | 67 | 39 | 1.7 |
| 5- Pyruvate-kinase | 259450896 | 53 | 29 | 1.8 |
| 6- V-type proton ATPse subunit H | 12585497 | 53 | 32 | 1.7 |
| 7- Transketolase^ | 114050833 | 43 | 21 | 2.0 |
| 8- Glutamate dehydrogenase | 114052462 | 31 | 15 | 2.0 |
| 9- Kinesin-like protein 1 | 309378082 | 27 | 5 | 5 |
| 10- Glutathione S-transferase theta | 170779021 | 22 | 15 | 1.5 |
| 11-V-type proton ATPase subunit D | 12585494 | 20 | 6 | 3.3 |
| 12- Tubulin beta-1 chain^ | 121987496 | 18 | 5 | 3.6 |
| 13- Triglyceride-lipase^ | 238846408 | 70 | 11 | 6.4 |
Proteins also identified by MS/MS in immunocomplexes using HDLp antibody.
Figure 3. Co-immunoprecipitation of TGL with anti-HDLp antibodies.
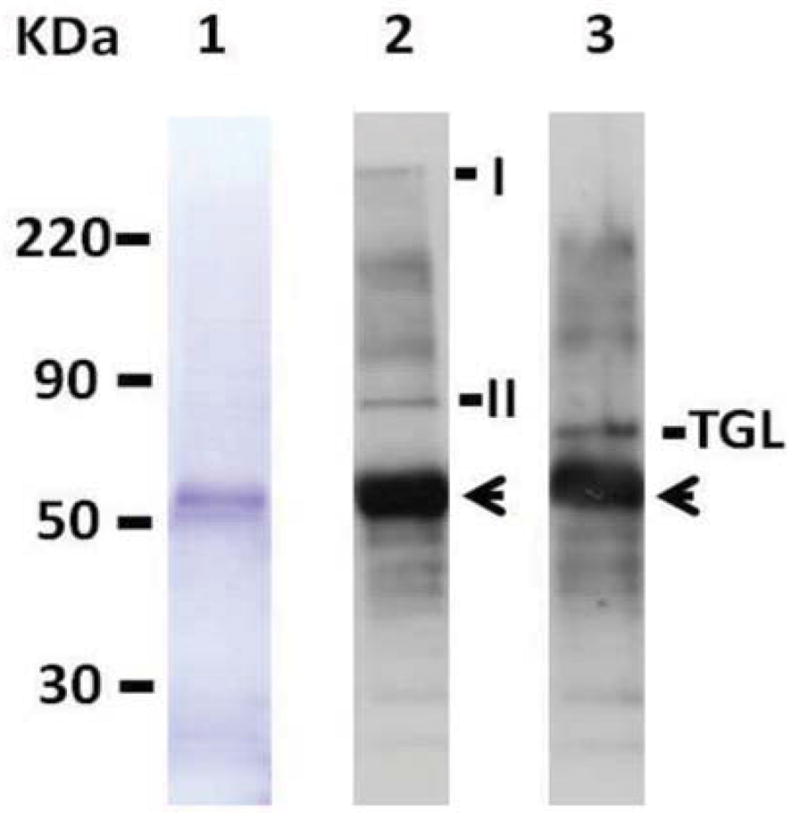
Fat body soluble proteins were incubated with anti-HDLp antibody for 2h and immunoprecipitated using protein-A-agarose beads preblocked with BSA. After washing the beads four times with PBS the associated-proteins were separated by SDS-PAGE gel in 4–15% acrylamide gel (lane 1), and analyzed by western blotting using anti-HDLp (lane 2) and anti-TGL antibodies (lane 3). The arrow indicates IgG. ApoLp I/II and TGL bands are also shown in lane 2 and 3, respectively.
Several of the proteins shown in Table 1 are enzymes including TGL. The presence of TGL indicates that the lipase is able to form homodimers. Dimers could be maintained through WWE-WWE interactions such as occurs in proteins of the Deltex family (Zweifel et al., 2005). However, the large DDHD domain present in the C-term of TGL could also interact with the N-term region of a second TGL molecule, as recently reported in the homodimer formation of vertebrate phospholipase iPLA1 (Inoue et al., 2012).
Three of the TGL interacting proteins–transketolase, trehalose synthase and pyruvate kinase- are central enzymes of carbohydrate metabolism. Transketolase connects the pentose pathway and glycolysis whereas trehalose synthase catalyzes the synthesis of trehalose. Pyruvate kinase catalyzes the last step of glycolysis where pyruvate and ATP are produced. Pyruvate kinase and trehalose synthase are linked in the sense that both compete for glucose. The glycolytic pathway has to be inhibited for trehalose synthesis to occur. Interestingly, ATP is an inhibitor of both TGL (Arrese and Wells, 1994) and vertebrates’ pyruvate kinase (Voet, 2011). Moreover, glutamate dehydrogenase (GDH), a TGL interacting protein (Table 1), is also inhibited by ATP in mammals (Voet, 2011). GDH catalyzes the oxidative deamination of glutamate raising the cellular content of NAD(P)H (Voet, 2011). Lipoamide dehydrogenase (LipDH) was also identified as TGL interacting protein (Table 1). This enzyme catalyzes a reversible reaction in which NADH is a substrate (Voet, 2011). Despite the fact that all the proteins identified in Table 1 belong to different metabolic pathways, yet a certain connection can be recognized. One would think that the regulation of the activity of these enzymes that have substrates or products in common would be enhanced if they are in close proximity as these results are suggesting. Likewise, the transfer of NADH from one enzyme to another would be facilitated if the enzymes are in a complex.
Glutathione S-transferase (GST) was also identified in Table 1. GSTs are cytosolic enzymes that catalyze the conjugation of glutathione to a wide range of substrates including the product of lipid peroxidation that are toxic for the cell (Ålin et al., 1985; Sharma et al., 1991). Two subunits of the V-type proton ATPase, a multimeric enzyme involved in ATP-dependent acidification processes related to vesicular transport including endocytosis and exocytosis (Stevens and Forgac, 1997), in addition to the structural components of high density lipophorin (HDLp), and some elements of cytoskeletal structure (kinesin-like protein and tubulin b) were also identified as TGL interacting proteins.
We have demonstrated that the N-terminal region of TGL forms a protein fold compatible with that of the WWE domain. Furthermore, this region was able to mediate protein-protein interactions corroborating the predicted functionality of the WWE domain. The data suggest that TGL occurs as a multi-protein complex mediated in part by interactions through the WWE domain. In addition to the possibility of forming homodimers, TGL interacts with other enzymes of intermediary metabolism including redox-active proteins, lipid transport proteins (HDLp lipophorin), and proteins of the cytoskeleton. The physiological connection between TGL and these proteins is unknown. In this study we focused on two of these proteins: LipDH, and apoLp I/II, the apolipoprotein component of HDLp, the main carrier of diacylglycerol in the hemolymph. Further studies were undertaken to try to understand the implications of the interaction between TGL and each of these proteins.
3.2. TGL-LipDH interaction
LipDH (EC 1.8.1.4) is a conserved protein best known as an integral component of mitochondrial α-keto acid dehydrogenase complexes. In rat liver 90% of the enzyme localizes in the mitochondria (Matuda and Saheki, 1982). It catalyzes the oxidation of the dihydrolipoyl cofactor of the acyltransferase (E2) component of the multienzymatic complexes with NAD+ as the electron acceptor (Williams, 1992). M. sexta LipDH has been cloned and sequenced (Pullikuth and Gill, 1997). The predicted protein has an estimated mass of 51 kDa and is a relatively well conserved protein (Pullikuth and Gill, 1997). Preliminary immunoblot studies showed that the protein can be detected in the fat body using antibodies raised against LipDH isolated from porcine heart. First we studied the subcellular distribution of LipDH in the fat body of M. sexta adult insects by differential centrifugation and western blotting. We discovered the presence of LipDH in all subcellular fractions including cytosol, fat cake and membranes (Fig 4). Taking into account the distribution of total protein among subcellular fractions, it was estimated that 65 ± 14 % of total cellular LipDH localizes to the cytosol whereas 34 ± 14 % localized to the fractions containing membranes (mitochondria, plasma membrane, microsomes, and lipid droplets). To our knowledge the high levels of cytosolic LipDH represents a new finding in animals. The presence of LipDH outside the mitochondrial complexes has been reported in trypanosomes and archaebacteria so far. In these species LipDH is found associated to the plasma membrane (Danson, 1988; Danson et al., 1987; Else et al., 1994) and the cytosolic fraction (Portela and Stopopani, 1991). No physiological substrate for these forms of the enzyme in those organisms has been reported.
Figure 4. Subcellular Localization of LipDH in M.sexta Fat Body.

Subcellular fractions of adult fat body homogenates were separated by SDS-PAGE and analyzed by western blotting using anti-LipDH antibody. Approximately 10 μg (lanes “a” and “d”) and 30 μg (lanes “b” and “c”) of total proteins were loaded in the corresponding lane. Lane a: lipid droplets; lane b: cytosol; lane c: 100,000g pellet (P100); lane d: 20,000g pellet (P20). A representative western blotting result is shown.
The function of LipDH is to catalyze the oxidation of dihydrolipoyl covalently attached to protein. Next we examined whether recombinant fusion protein has lipoate as a cofactor. Western blotting using antibody specific for lipoylated proteins revealed a very strong response suggesting the presence of a lipoyl residue in the fusion protein (data not shown). Next we analyzed the protein by MS/MS to identify lipoylated peptides. However, MS/MS analyses failed to identify any lipoylation site in the fusion protein despite that all peptides were accounted (100% sequence coverage). No peptide showing a lipoyl modification was detected by MS/MS after triplicate analyses using protein samples obtained from independent preparations including a sample in which recombinant protein was expressed in a culture supplemented with lipoic acid. The robust response in the western analyses using antibodies specific for protein-bound lipoic acid suggested a high level of lipoylation in the N-term region of TGL. Although MS/MS result cannot exclude lipoylation in a small fraction of the protein, it indicates that the vast majority of protein did not contain lipoic acid as cofactor. Lipoylation is a post-translational modification in which lipoate is attached via amide linkage to the amino group of the side chain of a specific lysine residue of the target protein. Several lysine residues are present in the N-term region of the lipase but the conserved lipoyl attachment domain (PF00364) that is identified in lipoylated proteins is not found in TGL.
LipDH is a member of the pyridine nucleotide-disulfide oxidoreductase family that includes glutathione reductase (GR) and thioredoxin reductase, TrxR (Williams, 1992). The last two enzymes catalyze the NADPH-dependent reduction of oxidized glutathione (disulfide glutathione) and thioredoxin, respectively. In vertebrates, GR and TrxR have a prevalent role in the balance of cellular redox homoeostasis (Schafer and Buettner, 2001). However, Drosophila lacks GR and its function is substituted by TrxR (Kanzok et al., 2001). Inspection of the available genomes suggests that this could be a general case in insects. LipDH catalyzes the electron transfer between pyridine nucleotides and disulphide compounds such as lipoic acid and the reaction is reversible (Igamberdiev et al., 2004). The presence of LipDH in the cytosol could be needed to substitute for the absence of GR and maintain the redox state of cytosol. The disulfide-oxidoreductase activity of cytosolic LipDH could also serve to keep the thiol groups of TGL in a reduced state. TGL exhibits four strictly conserved Cys (Arrese et al., 2010). Cys 130 and Cys 234 are flanking the predicted lid region of TGL (Arrese et al., 2010),. The lid, which is present in lipases of the α/β hydrolase family, covers the active site, and rotates allowing the interaction with the substrate. The flanking Cys are thought to be involved in the mechanism of lipase activation (Derewenda, 1994). To verify the occurrence of thiol groups in TGL that are essential for its activity, we measured the activity of purified TGL in the presence of increasing concentrations of reduced (GSH) and oxidized glutathione (GSSG) and DTT. As shown in Fig 5A, the activity gradually increased in the presence of reducing agents-DTT and GSH-doubling its value at 12mM. In contrast, the incubation with 12mM GSSG decreased the lipase activity to ~50 % of the activity control. The inhibitory effect of GSSG could be explained by the formation of mixed disulfide or glutatyonylation between the GSSG and reactive lipase thiol groups as being described for other proteins such as isocitrate dehydrogenase (Hansen et al., 2009, Kil and Park, 2005). Furthermore, alkylation of thiol groups with NEM also showed a deleterious effect on the activity of TGL (Fig 5B). These results indicate that TGL activity is sensitive to the redox status of the environment. The enzyme activity is preserved when the redox-sensitive thiol groups of the lipase are in the reduced-state.
Figure 5. Effect of DTT, GSH, GSSG and NEM on TGL Activity.
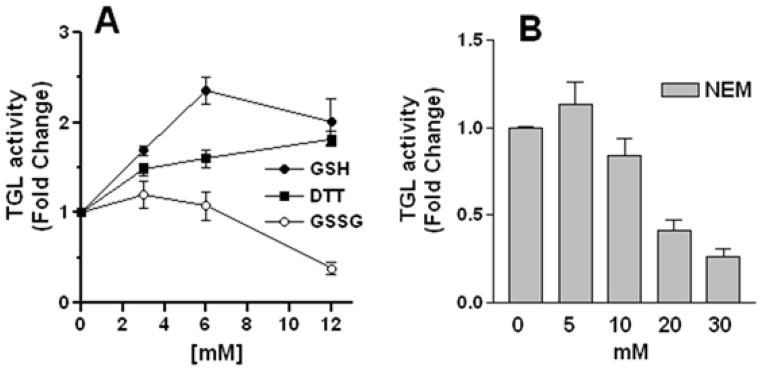
Partially purified TGL was pre-incubated with the indicated concentrations of DTT, GSH, GSSG (A), NEM (B) for 30 min on ice prior to measure the lipase activity. Lipase activity was expressed as a fold change relative to control (no addition). Values are the mean ± SEM of three independents experiments.
3.2.1. Effect of carmustine and auranofin on lipase activity
Carmustine (Becker and Schirmer, 1995; Lohrer and Krauth-Siegel, 1990) and Cd+2 (Searls et al., 1961) are LipDH inhibitors. Next we tested the effect of carmustine on the lipase activity of fat body homogenates. Carmustine was assayed at concentrations ranging between 75 to 560μM and showed a significant inhibitory effect on the lipase activity starting at 75μM (Fig 6A). In contrast, carmustine did not affect the activity of purified TGL even at 1.5mM (not shown). Therefore the inhibitory effect of carmustine on the lipase activity of fat body homogenates could be interpreted as an indirect effect due to the inhibition of LipDH. Carmustine has been originally described as a LipDH and GR inhibitor (Becker and Schirmer, 1995). However, carmustine can also act on TrxR at concentrations ranging from 0.1 to 1mM (Arscott et al., 1997). To investigate whether the effect of carmustine on the lipase activity was due TrxR inhibition rather than LipDH, we tested the effect of auranofin, which is a very potent inhibitor of TrxR that is effective at nanomolar range, whereas 1000-fold higher concentrations inhibit GR (Angelucci et al., 2009). The effect of auranofin was tested in a range of concentrations between 1 and 75 μM. We chose this range because it was shown that 1μM auranofin efficiently inhibited (90%) Drosophila TrxR (Gromer et al., 2003). However, no significant inhibition on the lipase activity was observed at concentrations ranging from 1 to 10 μM. The inhibitory effect of auranofin on the lipase activity was observed at the concentration range in which auranofin inhibits GR and LipDH (25 μM) (Fig 6B). The inhibition in both cases was reversed by 10 mM DTT indicating that the action of these inhibitors indirectly impacted the redox state of thiol groups of lipase (Fig 5A–B). Furthermore, LipDH inhibition by carmustine caused a greater lipase inhibition than the inhibition of thioredoxin reductase (1–10 μM range) by auranofin. Taken together these results are consistent with the idea of LipDH playing a role in the control of the redox state of TGL critical thiol groups. In this way, LipDH would catalyze the reverse reaction acting as a disulfide reductase.
Figure 6. Effect of Carmustine (A) and Auranofin (B) on lipase activity.
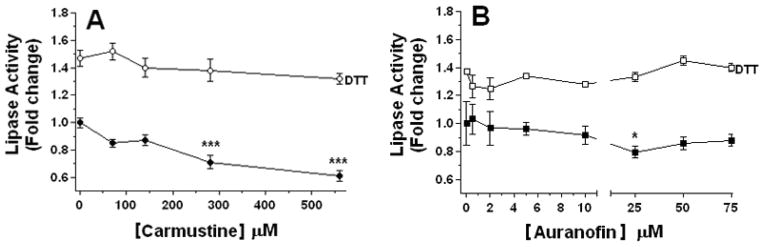
Fat body soluble fractions were pre-incubated with the indicated concentrations of inhibitor for 30 min on ice prior to measure lipase activity. Experiments were conducted in the presence and absence of 10 mM DTT. Lipase activity was expressed as a fold change relative to control (no inhibitor). Values are the mean ± SEM of three independent experiments. Significant differences determined by one way ANOVA are shown with asterisks. [***] indicates p<0.001 and [*] indicates p <0.05.
As mentioned above, the physiological function of LipDH is to catalyze the oxidation of the dihydrolipoyl cofactor of the acyltransferase component of the multienzymatic complexes with NAD+ as the electrom acceptor (Williams, 1992). However, in vitro, the enzyme can also act as a diaphorase and catalyzes the reverse reaction, in which the oxidation of NADH occurs. Some diaphorase substrates of LipDH are: NO (Igamberdiev et al., 2004), O2 (Argyrou and Blanchard, 2001), lipoamide (Igamberdiev et al., 2004), cytochrome c, quinones (Argyrou and Blanchard, 2001), and methylene blue (Corran et al., 1939).
Manduca LipDH is more related to GR than TrxR. The mechanisms of LipDH and GR have been found to be very similar to each other in several organisms (Arscott et al., 1997). LipDH could also compensate for the lack of GR and be involved in the metabolism of glutathione catalyzing the recycling of GSSG to GSH. Alternatively, cytosolic LipDH could catalyze the reduction of protein-dithiol-disulfide using reducing equivalents from NADH. Likewise, the enzyme could act on mixed disulphide bonds, such as those formed between GSH and protein-thiols under oxidative stress conditions to prevent further oxidation of the protein-thiols (Dalle-Donne et al., 2009; Schafer and Buettner, 2001). Unlike the mammalian system in which only 10% of LipDH is extramitochondrial, in the fat body more than 50% of the enzyme localizes in the soluble fraction. A study on Bombyx mori LipDH showed that recombinant protein was a soluble enzyme able to catalyze the lipoamide-dependent oxidation of NADH (Huo et al., 2010). Altogether this information suggests that LipDH plays an additional function in insects. According to this study, LipDH seems to be part of the antioxidative system of the cell in control of the redox status of protein-thiols, and its activity prevents lipase inactivation.
3.2.2. Effect of AKH on lipase activity in the presence and absence of reducing agents
Previous work in our laboratory has shown that TGL is a major cytosolic lipase, and AKH induces an increase in the lipase activity of cytosol (Arrese et al., 1996). In addition, the activation is independent of the lipase phosphorylation state (Patel et al., 2004; Patel et al., 2006). We asked whether the AKH-induced lipase activation could be due to a change in the redox state of critical thiols. In that case, AKH could shift the ratio between the oxidized (inactive) and reduced (active) thiol groups of TGL. In this scenario, it could be possible to observe a smaller change in lipase activity of AKH-treated cytosols when assayed in the absence of DTT compared to the activity of control cytosols measured in the presence of DTT. This possibility was examined by measuring the lipase activity of freshly prepared cytosols from control and AKH-treated insects in the presence and absence of 10mM DTT. Results confirmed the increase in lipase activity induced by AKH as previously observed (pbd <0.001) (Arrese et al., 1996; Patel et al., 2006). However, the extent of the lipase activation observed in the absence of DTT (Fig 7, bar c) was lower than in the presence of DTT (Fig 7, bar d, pcd <0.001) and higher than the activity of control samples measured in the presence of DTT (Fig 7, bar b, pbc <0.05). Thus, these results imply that changes in the redox state of lipase thiol groups cannot account for the lipase activation that was observed in AKH-treated insects. The above results suggest that some other undefined mechanism is involved in the AKH-induced lipase activation.
Figure 7. Effect of AKH on lipase activity in the presence and absence of reducing agents.
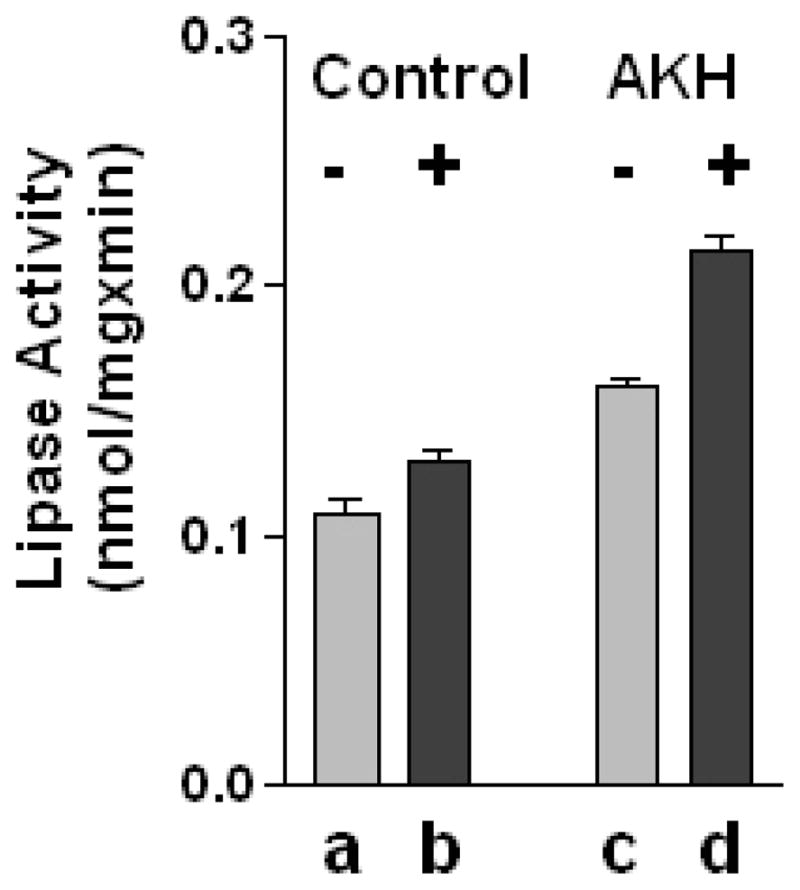
Freshly dissected fat bodies from two insects that were injected with PBS (Control, bars a–b) or AKH (AKH, bars c–d) were pooled and homogenized. Fat body soluble fractions were tested for lipase activity in the absence (−) and presence (+) of 10 mM DTT. Lipase activity was expressed in nmol TG hydrolyzed/min.mg of total protein. Values are the mean ± SEM of three independents experiments. P values are: pab >0.05; pac<0.001; pad<0.001; pbc <0.05; pbd <0.001; pcd <0.001.
3.4. TGL-HDLp interaction
Apolipoprotein I and II (apoLp I/II) - the structural apolipoproteins of lipophorin (HDLp) - were identified as TGL interacting proteins (Table 1, Fig 2B and Fig 3). ApoLp I/II are synthesized in the fat body from a common precursor which transforms into apoLp-I and –II after post-translational cleavage (Weers et al., 1993). In Manduca, apoLp I/II were also found associated to LD, the cytosolic organelles housing cellular TG stores (Soulages et al., 2012). Since apoLp I/II interact with both TGL and LD suggests that HDLp could play a role in the intracellular traffic of DG. The mechanism by which DGs are transported inside the cell from the lipid droplets to the plasma membrane remains unknown. First we examined the intracellular localization of HDLp, DG and TG. Fat body homogenate was subjected to ultracentrifugation in a discontinuous sucrose gradient and the distribution of HDLp (apoLps) along the density gradient was obtained by Western blot (Fig 8A). The distribution of DG (Fig 8B), TG (Fig 8C) and total protein among the fractions was also determined. As expected, a small but significant amount of lipophorin was found in the lipid droplet fraction (fraction 1, 0% sucrose), which contains most of the TG and DG of the fat body. Another small fraction of lipophorin was found floating at ~20 % and ~40% sucrose where plasma membrane and other membranes are expected (fractions 4 and 6). Most of the lipophorin was detected at the bottom of the gradient in the cytosolic fraction (fraction 7) where most of the cellular proteins were also present (not shown). The distribution of intracellular DG was similar to the distribution of lipophorin (Fig 8 B and C).
Figure 8. Subcellular Distribution of Lipophorin, DG and TG.

Fat body homogenate from adult male insects was subjected to ultracentrifugation in a sucrose gradient. The gradient was fractionated into seven fractions. The distribution of ApoLp-I/II, DG and TG is shown in panel A, B and C, respectively. Top panel shows a representative western blot depicting the distribution of ApoLp-II among fractions. For this purpose, 0.6% of fraction 1 to 6, and 0.1% of fraction 7 were loaded in 4–20 % acrylamide gel, transferred to nitrocellulose and analyzed by western blotting. The concentration of sucrose of the fractions is given in g/100ml. Panel A graph comes from the densitometry of the blot; panel B and C were obtained after separation of lipids by TLC as described under Materials and Methods. After visualizing lipids spots with iodine, plates were scanned and lipids quantified with AlphaEaseFCTM software. TG and DG contents were expressed as percentage of total neutral lipid. Two independent experiments were carried and Fig 8 shows a representative result.
Previous studies showed that TGL is always detected in the cytosol. The lipase hydrolyzes TG contained in the lipid droplets but the interaction between TGL and LD is transient (Arrese et al., 2006; Patel et al., 2005). Results shown in Fig 8 confirmed that HDLp is found in the cytosol. On the other hand, the studies in which we observed the interaction of TGL and HDLp were conducted with the soluble fraction of the fat body indicating that the cytosolic fraction of HDLp interacts with TGL (Fig 2–3).
Because lipophorin is synthesized in the fat body (van Heusden et al., 1998; Weers et al., 1993), finding intracellular lipoprotein in fat body adipocytes is somehow expected. The pathway of Lp synthesis and secretion in insects has been proposed to be similar to that in mammals (Van der Horst et al., 2009). Thus, newly formed lipoprotein is expected to undergo vesicular transport from the ER through Golgi followed by the release of the secretory vesicles to the hemolymph. However, a very significant amount of Lp was found in the cytosol. The origin of cytosolic Lp is unclear and may be different from the origin of the nascent particle that is secreted to the hemolymph as a particle with low lipid content (Prasad et al., 1986). Perhaps cytosolic HDLp could come from the hemolymph for reloading of the lipid cargo. It could be transporting intracellular DG from the lipid droplet–the site in which TGL acts- to the plasma membrane for secretion into circulation.
Circulating Lp unloads lipids into the different tissues and the remaining particle returns to the fat body where it is re-loaded with lipids and starts a new cycle of DG transport (Van Der Host, 2010). Because the turnover of DG occurs at a much higher rate than that of the protein component of lipophorin, a characteristic feature of Lp is that it acts as a reusable lipid shuttle (Downer and Chino, 1985; van der Horst et al., 2002). However, the precise mechanism by which lipophorin reloads the lipid cargo is not completely understood. The original idea is that DG is transferred at the surface of the adipocytes without internalization of the particle in a process facilitated by lipid transfer particle (LTP) (Canavoso et al., 2004; Van Heusden and Law, 1989). However, receptor mediated endocytic uptake of HDLp also occurs in the fat body of insects (Van Hoof et al., 2002, 2003, 2005). This process is mediated by the lipophorin receptor (LpR). So far the intracellular route of endocytosed Lp is unknown but it appears that the particle is recycled avoiding degradation in lysosomes (Van der Horst et al., 2009). The Lp taken up by adipocytes perhaps re-loads lipids at the surface of the lipid droplet in a process mediated by lipases such as TGL. Consistent with this notion, the fat body DG pools that are secreted to the hemolymph seem to localize mainly in the lipid droplets and cytosol (Arrese et al., 2001).
3.4.1. Effect of HDLp on lipase activity
To gain insight into the possible role of cytosolic Lp on lipid mobilization we investigated whether Lp affects the catalytic properties of TGL. We previously showed that TGL is able to hydrolyze TG to 2-monoacylglycerol (MG) (Arrese and Wells, 1994), and we asked if the interaction between TGL and HDLp would impact the hydrolysis products of TG. The activity of partially purified TGL was assayed against [3H]-triolein (25nmoles) in the presence of increasing amounts of lipophorin (0, 1, 3 and 5μg of HDLp) that was isolated and quantified as described under Materials and Methods. Following the lipase reaction, lipids from the reaction mixture were extracted by organic solvent and analyzed by TLC to quantify the products of hydrolysis. The amount in nmoles of DG and MG produced during the reaction as well as the amount of TG consumed is shown Fig 9A. The addition of HDLp altered the properties of TGL as judged by the ratio of [3H]-MG to [3H]-DG produced. In the absence of HDLp this molar ratio was about ~20 but gradually decreased to 2 when the lipase assay was carried out in the presence of 5μg of HDLp. Control experiments were carried out using bovine albumin (BSA). Addition of increasing amounts of BSA into the assay mixture did not change the lipase activity or the product of hydrolysis (not shown). However, the addition of lipophorin to the assay mixture had an inhibitory effect on the lipase activity. Lipophorin inhibits mostly the DG-lipase activity of TGL, as seen by the decrease in MG production as the concentration of lipophorin is increased. This effect changes the proportion of products formed favoring the production of DG. It seems possible that once DG is formed is somehow shielded in the particle preventing further lipase action.
Figure 9. Effect of Lipophorin on the Lipase Activity of TGL.

A) The lipase activity of partially purified lipase was determined by incubating 25 nmoles of [3H]-triolein-Triton X-100 with 15μg of TGL in the presence of increasing amounts of lipophorin (HDLp; δ=1.15g/cm3) for 30 min. The formation of individual products (DG, MG) was determined after analyzing the reaction products by TLC coupled to scintillation counting. A) Data representative of one experiment are shown in panel A. Data were expressed in nmoles and are represented as the mean ± SEM of three determinations ; B) The effect of lipophorin concentration on the molar ratio DG to MG produced were calculated from the data shown in panel A.
When the organism needs to utilize fatty acids stored in the fat body as TG, the fat body secretes DG into the hemolymph where they are transported by lipophorin to the utilization sites, e.g. flight muscle. The loading of lipids in the fat body cell is not completely understood. The interaction of TGL with HDLp is significant for two reasons: 1) HDLp modifies the catalytic properties of TGL leading to a higher production of DG; 2) the interaction may be involved in the process of lipoprotein assembly and loading with DG. HDLp could participate in the transport of DG from the lipid droplet–the site in which TGL acts- to the plasma membrane to be secreted into the hemolymph. Finding HDLp in the cytosol, and most importantly, knowing that HDLp interacts with TGL, suggests that lipophorin could take a role in the intracellular transport and/or loading of DG. Future work will be aimed to define the mechanism of intracellular transport and secretion of DG.
Acknowledgments
We are grateful to Janet Rogers for providing excellent technical assistance for the MS/MS work. This research was supported by the National Institute of Health (GM64677) and the Oklahoma Experiment Station. Zengying Wu is recipient of the Sitlington Enriched Graduate Scholarship from the Division of Agricultural Sciences and Natural Resources at Oklahoma State University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ålin P, Danielson UH, Mannervik B. 4-Hydroxyalk-2-enals are substrates for glutathione transferase. FEBS Lett. 1985;179:267–270. doi: 10.1016/0014-5793(85)80532-9. [DOI] [PubMed] [Google Scholar]
- Angelucci F, Sayed AA, Williams DL, Boumis G, Brunori M, Dimastrogiovanni D, Miele AE, Pauly F, Bellelli A. Inhibition of Schistosoma mansoni thioredoxin-glutathione reductase by auranofin: structural and kinetic aspects. J Biol Chem. 2009;284:28977–28985. doi: 10.1074/jbc.M109.020701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L. The WWE domain: a common interaction module in protein ubiquitination and ADP ribosylation. Trends Biochem Sci. 2001;26:273–275. doi: 10.1016/s0968-0004(01)01787-x. [DOI] [PubMed] [Google Scholar]
- Argyrou A, Blanchard JS. Mycobacterium tuberculosis lipoamide dehydrogenase is encoded by Rv0462 and not by the lpdA or lpdB genes. Biochemistry. 2001;40:11353–11363. doi: 10.1021/bi010575o. [DOI] [PubMed] [Google Scholar]
- Arrese EL, Flowers MT, Gazard JL, Wells MA. Calcium and cAMP are second messengers in the adipokinetic hormone-induced lipolysis of triacylglycerols in Manduca sexta fat body. J Lipid Res. 1999;40:556–564. [PubMed] [Google Scholar]
- Arrese EL, Gazard JL, Flowers MT, Soulages JL, Wells MA. Diacylglycerol transport in the insect fat body: evidence of involvement of lipid droplets and the cytosolic fraction. J Lipid Res. 2001;42:225–234. [PubMed] [Google Scholar]
- Arrese EL, Howard AD, Patel RT, Rimoldi OJ, Soulages JL. Mobilization of lipid stores in Manduca sexta: cDNA cloning and developmental expression of fat body triglyceride lipase, TGL. Insect Biochem Mol Biol. 2010;40:91–99. doi: 10.1016/j.ibmb.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrese EL, Patel RT, Soulages JL. The main triglyceride-lipase from the insect fat body is an active phospholipase A(1): identification and characterization. Journal of lipid research. 2006;47:2656–2667. doi: 10.1194/jlr.M600161-JLR200. [DOI] [PubMed] [Google Scholar]
- Arrese EL, Rivera L, Hamada M, Mirza S, Hartson SD, Weintraub S, Soulages JL. Function and structure of lipid storage droplet protein 1 studied in lipoprotein complexes. Arch Biochem Biophys. 2008;473:42–47. doi: 10.1016/j.abb.2008.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrese EL, Rojas-Rivas BI, Wells MA. The use of decapitated insects to study lipid mobilization in adult Manduca sexta: effects of adipokinetic hormone and trehalose on fat body lipase activity. Insect Biochem Mol Biol. 1996;26:775–782. doi: 10.1016/s0965-1748(96)00024-0. [DOI] [PubMed] [Google Scholar]
- Arrese EL, Soulages JL. Insect fat body: energy, metabolism, and regulation. Annu Rev Entomol. 2010;55:207–225. doi: 10.1146/annurev-ento-112408-085356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrese EL, Wells MA. Purification and properties of a phosphorylatable triacylglycerol lipase from the fat body of an insect, Manduca sexta. J Lipid Res. 1994;35:1652–1660. [PubMed] [Google Scholar]
- Arscott LD, Gromer S, Schirmer RH, Becker K, Williams CH., Jr The mechanism of thioredoxin reductase from human placenta is similar to the mechanisms of lipoamide dehydrogenase and glutathione reductase and is distinct from the mechanism of thioredoxin reductase from Escherichia coli. Proc Natl Acad Sci U S A. 1997;94:3621–3626. doi: 10.1073/pnas.94.8.3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerswald L, Gade G. Endocrine control of TAG lipase in the fat body of the migratory locust, Locusta migratoria. Insect Biochem Mol Biol. 2006;36:759–768. doi: 10.1016/j.ibmb.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Auerswald L, Siegert KJ, Gade G. Activation of triacylglycerol lipase in the fat body of a beetle by adipokinetic hormone. Insect Biochemistry And Molecular Biology. 2005;35:461–470. doi: 10.1016/j.ibmb.2005.01.010. [DOI] [PubMed] [Google Scholar]
- Becker K, Schirmer RH. 1,3-Bis(2-chloroethyl)-1-nitrosourea as thiol-carbamoylating agent in biological systems. Methods in enzymology. 1995;251:173–188. doi: 10.1016/0076-6879(95)51120-2. [DOI] [PubMed] [Google Scholar]
- Bednarova A, Kodrik D, Krishnan N. Adipokinetic hormone exerts its anti-oxidative effects using a conserved signal-transduction mechanism involving both PKC and cAMP by mobilizing extra- and intracellular Ca(2+) stores. Comparative biochemistry and physiology. Toxicology & pharmacology : CBP. 2013;158:142–149. doi: 10.1016/j.cbpc.2013.07.002. [DOI] [PubMed] [Google Scholar]
- Brasaemle DL. Thematic review series: adipocyte biology. The perilipin family of structural lipid droplet proteins: stabilization of lipid droplets and control of lipolysis. J Lipid Res. 2007;48:2547–2559. doi: 10.1194/jlr.R700014-JLR200. [DOI] [PubMed] [Google Scholar]
- Brasaemle DL. Lipolysis control: the plot thickens. Cell Metab. 2010;11:173–174. doi: 10.1016/j.cmet.2010.02.008. [DOI] [PubMed] [Google Scholar]
- Canavoso LE, Yun HK, Jouni ZE, Wells MA. Lipid transfer particle mediates the delivery of diacylglycerol from lipophorin to fat body in larval Manduca sexta. J Lipid Res. 2004;45:456–465. doi: 10.1194/jlr.M300242-JLR200. [DOI] [PubMed] [Google Scholar]
- Corran HS, Green DE, Straub FB. On the catalytic function of heart flavoprotein. Biochem J. 1939;33:793–801. doi: 10.1042/bj0330793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalle-Donne I, Rossi R, Colombo G, Giustarini D, Milzani A. Protein S-glutathionylation: a regulatory device from bacteria to humans. Trends Biochem Sci. 2009;34:85–96. doi: 10.1016/j.tibs.2008.11.002. [DOI] [PubMed] [Google Scholar]
- Danson MJ. Dihydrolipoamide dehydrogenase: a ‘new’ function for an old enzyme? Biochemical Society transactions. 1988;16:87–89. doi: 10.1042/bst0160087. [DOI] [PubMed] [Google Scholar]
- Danson MJ, Conroy K, McQuattie A, Stevenson KJ. Dihydrolipoamide dehydrogenase from Trypanosoma brucei. Characterization and cellular location. Biochem J. 1987;243:661–665. doi: 10.1042/bj2430661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derewenda ZS. Structure and function of lipases. Advances in protein chemistry. 1994;45:1–52. doi: 10.1016/s0065-3233(08)60637-3. [DOI] [PubMed] [Google Scholar]
- Downer RGH, Chino H. Turnover of protein and diacylglycerol components of lipophorin in insect haemolymph. Insect Biochemistry. 1985;15:627–630. [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic acids research. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Else AJ, Clarke JF, Willis A, Jackman SA, Hough DW, Danson MJ. Dihydrolipoamide dehydrogenase in the trypanosoma subgenus, trypanozoon. Molecular and biochemical parasitology. 1994;64:233–239. doi: 10.1016/0166-6851(93)00016-3. [DOI] [PubMed] [Google Scholar]
- Gromer S, Johansson L, Bauer H, Arscott LD, Rauch S, Ballou DP, Williams CH, Jr, Schirmer RH, Arner ES. Active sites of thioredoxin reductases: why selenoproteins? Proc Natl Acad Sci U S A. 2003;100:12618–12623. doi: 10.1073/pnas.2134510100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen RE, Roth D, Winther JR. Quantifying the global cellular thiol-disulfide status. Proc Natl Acad Sci U S A. 2009;106:422–427. doi: 10.1073/pnas.0812149106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo J, Shi H, Yao Q, Chen H, Wang L, Chen K. Cloning and purification of recombinant silkworm dihydrolipoamide dehydrogenase expressed in Escherichia coli. Protein Expression and Purification. 2010;72:95–100. doi: 10.1016/j.pep.2010.01.014. [DOI] [PubMed] [Google Scholar]
- Igamberdiev AU, Bykova NV, Ens W, Hill RD. Dihydrolipoamide dehydrogenase from porcine heart catalyzes NADH-dependent scavenging of nitric oxide. FEBS Lett. 2004;568:146–150. doi: 10.1016/j.febslet.2004.05.024. [DOI] [PubMed] [Google Scholar]
- Inoue H, Baba T, Sato S, Ohtsuki R, Takemori A, Watanabe T, Tagaya M, Tani K. Roles of SAM and DDHD domains in mammalian intracellular phospholipase A1 KIAA0725p. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2012;1823:930–939. doi: 10.1016/j.bbamcr.2012.02.002. [DOI] [PubMed] [Google Scholar]
- Kanzok SM, Fechner A, Bauer H, Ulschmid JK, Muller HM, Botella-Munoz J, Schneuwly S, Schirmer R, Becker K. Substitution of the thioredoxin system for glutathione reductase in Drosophila melanogaster. Science. 2001;291:643–646. doi: 10.1126/science.291.5504.643. [DOI] [PubMed] [Google Scholar]
- Kil IS, Park J-W. Regulation of Mitochondrial NADP+-dependent Isocitrate Dehydrogenase Activity by Glutathionylation. J Biol Chem. 2005;280:10846–10854. doi: 10.1074/jbc.M411306200. [DOI] [PubMed] [Google Scholar]
- Lohrer H, Krauth-Siegel RL. Purification and characterization of lipoamide dehydrogenase from Trypanosoma cruzi. Eur J Biochem. 1990;194:863–869. doi: 10.1111/j.1432-1033.1990.tb19480.x. [DOI] [PubMed] [Google Scholar]
- Matuda S, Saheki T. Intracellular distribution and biosynthesis of lipoamide dehydrogenase in rat liver. Journal of biochemistry. 1982;91:553–561. doi: 10.1093/oxfordjournals.jbchem.a133727. [DOI] [PubMed] [Google Scholar]
- Patel R, Soulages JL, Wells MA, Arrese EL. cAMP-dependent protein kinase of Manduca sexta phosphorylates but does not activate the fat body triglyceride lipase. Insect Biochem Mol Biol. 2004;34:1269–1279. doi: 10.1016/j.ibmb.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Patel RT, Soulages JL, Arrese EL. Adipokinetic hormone-induced mobilization of fat body triglyceride stores in Manduca sexta: role of TG-lipase and lipid droplets. Arch Insect Biochem Physiol. 2006;63:73–81. doi: 10.1002/arch.20143. [DOI] [PubMed] [Google Scholar]
- Patel RT, Soulages JL, Hariharasundaram B, Arrese EL. Activation of the lipid droplet controls the rate of lipolysis of triglycerides in the insect fat body. J Biol Chem. 2005;280:22624–22631. doi: 10.1074/jbc.M413128200. [DOI] [PubMed] [Google Scholar]
- Portela MP, Stopopani AO. Lipoamide dehydrogenase from Trypanosoma cruzi: some properties and cellular localization. Biochemistry international. 1991;24:147–155. [PubMed] [Google Scholar]
- Prasad SV, Ryan RO, Law JH, Wells MA. Changes in lipoprotein composition during larval-pupal metamorphosis of an insect, Manduca sexta. J Biol Chem. 1986;261:558–562. [PubMed] [Google Scholar]
- Pullikuth AK, Gill SS. Primary structure of an invertebrate dihydrolipoamide dehydrogenase with phylogenetic relationship to vertebrate and bacterial disulfide oxidoreductases. Gene. 1997;200:163–172. doi: 10.1016/s0378-1119(97)00413-7. [DOI] [PubMed] [Google Scholar]
- Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free radical biology & medicine. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- Searls RL, Peters JM, Sanadi DR. alpha-Ketoglutaric dehydrogenase. X. On the mechanism of dihydrolipoyl dehydrogenase reaction. J Biol Chem. 1961;236:2317–2322. [PubMed] [Google Scholar]
- Sharma R, Gupta S, Singhal SS, Ansari GA, Awasthi YC. Glutathione S-transferase-catalyzed conjugation of 9,10-epoxystearic acid with glutathione. Journal of biochemical toxicology. 1991;6:147–153. doi: 10.1002/jbt.2570060209. [DOI] [PubMed] [Google Scholar]
- Soulages JL, Brenner RR. Study on the composition-structure relationship of lipophorins. J Lipid Res. 1991;32:407–415. [PubMed] [Google Scholar]
- Soulages JL, Firdaus SJ, Hartson S, Chen X, Howard AD, Arrese EL. Developmental changes in the protein composition of Manduca sexta lipid droplets. Insect Biochem Mol Biol. 2012 doi: 10.1016/j.ibmb.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulages JL, Rivera M, Walker FA, Wells MA. Hydration and localization of diacylglycerol in the insect lipoprotein lipophorin. A 13C-NMR study. Biochemistry. 1994;33:3245–3251. doi: 10.1021/bi00177a015. [DOI] [PubMed] [Google Scholar]
- Stevens TH, Forgac M. Structure, function and regulation of the vacuolar (H+)-ATPase. Annual review of cell and developmental biology. 1997;13:779–808. doi: 10.1146/annurev.cellbio.13.1.779. [DOI] [PubMed] [Google Scholar]
- Van der Horst DJ, van Hoof D, van Marrewijk WJ, Rodenburg KW. Alternative lipid mobilization: the insect shuttle system. Mol Cell Biochem. 2002;239:113–119. [PubMed] [Google Scholar]
- Van der Horst DJ, Roosendaal SD, Rodenburg KW. Circulatory lipid transport: lipoprotein assembly and function from an evolutionary perspective. Mol Cell Biochem. 2009;326:105–119. doi: 10.1007/s11010-008-0011-3. [DOI] [PubMed] [Google Scholar]
- Van der Horst DJ, Rodenburg KW. Lipoprotein assembly and function in an evolutionary perspective. Biomolecular NMR Assignments. 2010;1:165–183. doi: 10.1515/bmc.2010.012. [DOI] [PubMed] [Google Scholar]
- Voet D, Voet JG. Biochemistry. 4. John Wiley & Sons; Hoboken, NJ: 2011. [Google Scholar]
- Van Heusden MC, Law JH. An insect lipid transfer particle promotes lipid loading from fat body to lipoprotein. The Journal of biological chemistry. 1989;264:17287–17292. [PubMed] [Google Scholar]
- van Heusden MC, Thompson F, Dennis J. Biosynthesis of Aedes aegypti lipophorin and gene expression of its apolipoproteins. Insect Biochemistry And Molecular Biology. 1998;28:733–738. doi: 10.1016/s0965-1748(98)00068-x. [DOI] [PubMed] [Google Scholar]
- Van Hoof D, Rodenburg KW, Van der Horst DJ. Insect lipoprotein follows a transferrin-like recycling pathway that is mediated by the insect LDL receptor homologue. J Cell Sci. 2002;115:4001–4012. doi: 10.1242/jcs.00113. [DOI] [PubMed] [Google Scholar]
- Van Hoof D, Rodenburg KW, van der Horst DJ. Lipophorin receptor-mediated lipoprotein endocytosis in insect fat body cells. J Lipid Res. 2003;44:1431–1440. doi: 10.1194/jlr.M300022-JLR200. [DOI] [PubMed] [Google Scholar]
- Van Hoof D, Rodenburg KW, Van der Horst DJ. Receptor-mediated endocytosis and intracellular trafficking of lipoproteins and transferrin in insect cells. Insect Biochem Mol Biol. 2005;35:117–128. doi: 10.1016/j.ibmb.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Voet D, Voet JG. Biochemistry. 4. John Wiley & Sons; Hoboken, NJ: 2011. [Google Scholar]
- Weers PM, Van Marrewijk WJ, Beenakkers AM, Van der Horst DJ. Biosynthesis of locust lipophorin. Apolipophorins I and II originate from a common precursor. J Biol Chem. 1993;268:4300–4303. [PubMed] [Google Scholar]
- Whitmore L, Wallace BA. Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers. 2008;89:392–400. doi: 10.1002/bip.20853. [DOI] [PubMed] [Google Scholar]
- Williams CH., Jr . Lipoamide dehydrogenase, glutathione reductase, thioredoxin reductase, and mercuric ion reductase-a family of flavoenzyme transhydrogenases. CRC Press; Boca Raton: 1992. [Google Scholar]
- Zweifel ME, Leahy DJ, Barrick D. Structure and Notch receptor binding of the tandem WWE domain of Deltex. Structure. 2005;13:1599–1611. doi: 10.1016/j.str.2005.07.015. [DOI] [PubMed] [Google Scholar]