

Abstract
Fluorescent proteins (FPs) are invaluable tools for biomedical research. Useful FPs have desirable fluorescence properties such as brightness and photostability, but a limitation is that many orange, red, and far-red FPs are cytotoxic when expressed in the cytosol. This cytotoxicity stems from aggregation. To reduce aggregation, we engineered the surface of DsRed-Express to generate DsRed-Express2, a highly soluble tetrameric FP that is noncytotoxic in bacterial and mammalian cells. Directed evolution of DsRed-Express2 yielded the color variants E2-Orange, E2-Red/Green, and E2-Crimson. These variants can be used to label whole cells for single- and multi-color experiments employing microscopy or flow cytometry. Methods are described for reducing the higher-order aggregation of oligomeric FPs and for analyzing FP cytotoxicity in E. coli and HeLa cells.
Keywords: Fluorescent protein, DsRed, whole-cell labeling, protein engineering, cytotoxicity
1. Introduction
Since the introduction of the green fluorescent protein (GFP) from Aequoria victoria (1), fluorescent proteins (FPs) have become invaluable research tools for microscopy and flow cytometry. Dozens of FPs have been discovered or engineered to span the visible spectrum (2). In particular, a number of red FPs have been engineered for use alone or in multi-color studies with GFP. A major goal has been to engineer monomeric red FPs from proteins that are dimeric or tetrameric in their wild-type forms. These efforts have produced the widely-used mFruits (3, 4) as well as DsRed-Monomer (5), TagRFP (6), TagRFP-T (7), mKate (8), mKate2 (9), mRuby (10), mKO (11), and mKO2 (12).
Although monomeric red FPs are theoretically useful for any application, they tend to be dimmer and more photolabile than the corresponding oligomers (Table 1) (13). Therefore, researchers have also created improved oligomeric red FPs such as DsRed-Express (14), Kusabira-Orange (KO), TurboRFP (6), Katushka (8), and RFP611, RFP637, and RFP639 (15). These oligomeric FPs cannot be used as fusion tags, but they are suitable for labeling organelles, whole cells, tissues, and entire organisms.
Table 1.
Fluorescence properties of orange, red, and far-red FPsa
| Fluorescent protein | Excitation / Emission maxima | Extinction coefficient | Quantum yield | Relative brightnessb | Maturation half-time (h) | Photobleaching half-time (s)c | pKa |
|---|---|---|---|---|---|---|---|
| E2-Orange | 540 / 561 | 36,500 | 0.54 | 0.54 | 1.3 | 81 ± 3 | 4.5 |
| mOrange2 | 549 / 563 | 56,300 | 0.49 | 0.75 | 4.5 | 40 ± 3 | 7.5 |
| KO | 548 / 560 | 72,800 | 0.55 | 1.1 | 3.8 | 21 ± 2 | 5.0 |
| mKO2 | 549 / 563 | 54,300 | 0.82 | 1.2 | 1.8 | 5 ± 1 | 5.0 |
| WT DsRed | 558 / 583 | 51,500 | 0.71 | 1.0 | 11 | --- | --- |
| DsRed-Express2 | 554 / 591 | 35,600 | 0.42 | 0.41 | 0.7 | 64 ± 4 | 4.5 |
| DsRed-Max | 560 / 589 | 48,000 | 0.41 | 0.54 | 1.2 | 9 ± 1 | |
| E2-Red/Green (green) | 484 / 498 | 100,200 | 0.06 | 0.17 | 0.4 | 236 ± 8d | 4.0 |
| E2-Red/Green (red) | 560 / 585 | 53,800 | 0.67 | 0.98 | 1.2 | 93 ± 3 | 4.5 |
| DsRed-Express | 554 / 586 | 33,800 | 0.44 | 0.41 | 0.6 | 71 ± 3 | --- |
| DsRed-Monomer | 557 / 592 | 27,300 | 0.14 | 0.10 | 1.3 | 15 ± 1 | --- |
| mCherry | 585 / 609 | 66,400 | 0.23 | 0.42 | 0.6 | 18 ± 1 | --- |
| tdTomato | 553 / 581 | 85,700 | 0.69 | 1.6 | 2.0 | 5 ± 1 | --- |
| TagRFP | 554 / 582 | 77,000 | 0.47 | 0.98 | 1.5 | 8 ± 4 | --- |
| TagRFP-T | 554 / 584 | 67,800 | 0.40 | 0.73 | 1.6 | 20 ± 2 | --- |
| TurboRFPe | 550 / 573 | --- | --- | --- | 1.5 | 32 ± 1 | --- |
| RFP611 | 555 / 606 | 109,700 | 0.60 | 1.8 | 2.7 | 7 ± 2 | --- |
| E2-Crimson | 611 / 646 | 126,000 | 0.23 | 0.79 | 0.4 | 26 ± 3 | 4.5 |
| mPlum | 587 / 649 | 29,300 | 0.10 | 0.08 | 1.6 | 31 ± 2 | 5.5 |
| mRaspberry | 594 / 627 | 42,000 | 0.14 | 0.16 | 2.1 | 21 ± 2 | 5 |
| mKate | 584 / 632 | 45,500 | 0.33 | 0.41 | 1.3 | 15 ± 2 | --- |
| mKate2 | 586 / 630 | 56,400 | 0.39 | 0.60 | 0.8 | 23 ± 2 | 6.5 |
| Katushka | 584 / 631 | 76,300 | 0.32 | 0.67 | 0.6 | 15 ± 1 | 7.5 |
| RFP637 | 585 / 637 | 53,800 | 0.22 | 0.32 | 2.4 | 60 ± 4 | 4.5 |
| RFP639 | 587 / 639 | 74,700 | 0.21 | 0.43 | 1.7 | 29 ± 3 | 4.5 |
All values are reproduced with permission from refs. 5 and 17–19. pKa values were determined for only a subset of the FPs. Photobleaching was not measured for WT DsRed because maturation of this protein is too slow for our in vivo assay.
Brightness was calculated as the product of extinction coefficient and quantum yield, and was normalized to a value of 1 for wild-type DsRed.
Photobleaching half-times during widefield illumination are listed as mean ± s.e.m. for three independent replicates.
The photobleaching half-time reported for the green chromophore of E2-Red/Green cannot be compared directly to the photobleaching half-times of red chromophores because a different filter set was used.
Because TurboRFP showed very poor solubility during extraction from bacteria, we were unable to perform brightness measurements for this protein.
A serious problem with both monomeric and oligomeric FPs is cytotoxicity (16). Most of the available red FPs show pronounced cytotoxicity that presumably stems from aggregation (17–19). For this reason, we modified the surface of DsRed-Express to create a tetrameric derivative that shows minimal higher-order aggregation. The resulting FP, DsRed-Express2, combines low cytotoxicity with favorable photophysical properties such as brightness, fast maturation, photostability, and pH stability (17). As a result, the tetrameric DsRed-Express2 is an ideal red FP for whole-cell labeling.
Noncytotoxic color variants were then engineered by modifying the interior of DsRed-Express2. This approach yielded three tetrameric derivatives termed E2-Orange (18), E2-Red/Green (18), and E2-Crimson (19). Like the parental DsRed-Express2, these new variants substantially outperform other FPs with regard to cytotoxicity in bacterial and mammalian systems (17–19). E2-Orange is ideal for multi-color experiments involving green and far-red FPs (Fig. 1A), while E2-Red/Green is useful as a “third color” for flow cytometry in combination with red and green FPs (Fig. 1B). E2-Crimson is particularly noteworthy because it is the fastest-maturing red or far-red FP, the brightest far-red FP, and the only known member of the GFP family that is efficiently excited with standard 633-nm lasers. As a result, E2-Crimson is useful for multi-color microscopy and flow cytometry (Fig. 1C). The methods used to engineer and evaluate DsRed-Express2 and its derivatives are detailed below, and can be used as a guide for the development of new noncytotoxic FPs.
Fig 1.

Novel applications of DsRed-Express2 derivatives. (A) E2-Orange and E2-Crimson are useful for three-color imaging with GFP. S. cerevisiae cells with the endoplasmic reticulum labeled with E2-Crimson, Golgi cisternae labeled with enhanced GFP (EGFP), and the cytosol labeled with E2-Orange were imaged using widefield microscopy with standard filter sets. (Reproduced with permission from ref. 19.)(B) E2-Red/Green is useful for three-color flow cytometry together with red and green FPs. S. cerevisiae cells expressing either EGFP (green box), DsRed-Express2 (red box), E2-Red/Green (blue box), or no FP were pooled and analyzed by flow cytometry with a 488-nm excitation laser. These four populations were cleanly resolvable. (Reproduced with permission from ref. 18.) (C) E2-Crimson is uniquely suited to flow cytometry with 633-nm excitation lasers. E. coli cells expressing various FPs or no FP (“Control”) were analyzed by flow cytometry with 633-nm excitation. Only the population expressing E2-Crimson was fully resolvable from nonfluorescent control cells. (Reproduced with permission from ref. 19.)
2. Materials
2.1. Screening E. coli colonies for brightness
Luria Broth (LB) plates supplemented with 100 μg/mL ampicillin.
Chemically competent Escherichia coli (E. coli) DH5α cells.
Mutagenic library encoded in pQE-60NA (17), which is a modified version of the pQE-60 vector (Qiagen, Valencia, CA) lacking the hexahistidine tag. pQE-60NA is a low-copy vector that constitutively expresses genes from a medium-strength T5 promoter flanked by two lac operator sequences.
Carousel 4200 slide projector (Eastman Kodak Co., Rochester, NY) with a 300-W bulb (General Electric, Fairfield, CT).
Glass bandpass filters for excitation (Chroma, Rockingham, VT). The nm ranges are 540/20, 560/20, and 620/20 for orange, red, and far-red FPs, respectively. Excitation filters are hung in front of the slide projector light source using a small hook.
Emission filter goggles. Red FP emission was observed using 580-nm longpass goggles (CE-EN207 with Krypton/Copper Vapor Filter; NoIR Laser Co., South Lyon, MI). Goggles for viewing orange and far-red FPs were made in-house by covering standard laboratory goggles with Kodak Wratten 560-nm or 650-nm longpass filters, respectively.
Sterile toothpicks.
2.2. Screening for aggregation using the bacterial lysis assay
Chemically competent E. coli DH5α cells (see Note 1).
Liquid LB medium supplemented with 100 μg/mL ampicillin.
Sterile, transparent, round-bottom 96-well plates with 300-μL well volume (Costar).
Black round-bottom 96-well plates with 300-μL well volume (Costar).
Sterile toothpicks.
BPER II lysis reagent (ThermoFisher Scientific, Waltham, MA).
Microplate fluorometer, such as a Tecan Safire2 plate reader.
2.3. Assaying cytotoxicity in bacteria
Chemically competent E. coli DH10B cells harboring the pREP4 repressor plasmid (Qiagen).
LB plates supplemented with 100 μg/mL ampicillin and 30 μg/mL kanamycin, either with or without 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG).
8–16% precast SDS polyacrylamide gels (Pierce).
BupH Tris-HEPES-SDS Running Buffer (Pierce) dissolved in deionized H2O.
PageRuler prestained protein ladder (Fermentas Inc., Glen Burnie, MD).
5X SDS-PAGE sample loading buffer: 280 mM Tris-HCl, pH 6.8, 10% (w/v) sodium dodecyl sulfate (SDS), 22.5 % (v/v) glycerol, 0.04% (w/v) bromophenol blue, 10% (v/v) β-mercaptoethanol (see Note 2).
2.4. HeLa cell transient transfection assay for cytotoxicity
HeLa cells (catalog number CCL-2; ATCC, Manassas, VA).
Culture medium consisting of Dulbecco’s Modified Eagle’s Medium (DMEM) with high glucose and L-glutamine (DMEM High Glucose; HyClone) supplemented with 10% fetal bovine serum (FBS; HyClone) and 1X MEM Non-Essential Amino Acids (Invitrogen, Carlsbad, CA).
OptiMEM (Invitrogen).
0.05% Trypsin/EDTA (Invitrogen) for cell dissociation.
1X PBS (Gibco).
Lipofectamine 2000 (Invitrogen) for transient transfections.
FPs encoded by a mammalian transient transfection vector with the SV40 origin of replication. The FPs are expressed under control of the CMV promoter with a Kozak sequence immediately upstream of the start codon.
Polybrene (Sigma-Aldrich Corp., St. Louis, MO) diluted to 10 mg/mL in sterile deionized H2O for lentiviral transduction.
Hemocytometer.
Flow cytometer and analysis software.
2.5. Generation of lentiviral vectors and HeLa cell lentiviral transduction assay for cytotoxicity
293-T/17 cells (catalog number CRL-11268; ATCC).
Culture medium (see Subheading 2.4.).
Lenti-X Expression Kit (Clontech, Mountain View, CA), including the pLVX-Puro plasmid.
QuickTiter Lentivirus Quantitation Kit (Cell Biolabs, Inc., San Diego, CA).
3. Methods
The engineering of useful FPs for whole-cell labeling typically involves multiple rounds of mutagenesis. To generate DsRed-Express2, we employed targeted or random mutagenesis followed by screening for improved variants. Each screen was conducted in two stages, in which a preliminary selection for bright clones was followed by a bacterial lysis assay to measure aggregation. This general approach can be used to create noncytotoxic derivatives of FPs that have desirable photophysical properties. Oligomeric FPs are best suited to optimization by this strategy, because the multivalent nature of an oligomeric FP enhances the functional affinity that can be detected using the bacterial lysis assay.
In our experience, high solubility of an oligomeric FP in the bacterial lysis assay correlates strongly with low cytotoxicity in bacterial and mammalian cells. However, cytotoxicity should be measured for each FP. To test for cytotoxicity in bacteria, E. coli colonies expressing a given FP are compared to colonies grown under repressing conditions. Smaller colonies indicate greater cytotoxicity. To test for cytotoxicity in mammalian cells, FPs are expressed in HeLa cells either transiently or by lentiviral transduction, and the fluorescence signal is monitored for several days using flow cytometry. With a cytotoxic FP, the average brightness of the cell population decreases over time, whereas with a noncytotoxic FP such as enhanced GFP (EGFP) or DsRed-Express2, the average brightness remains steady over time.
3.1. Screening E. coli colonies for brightness
Transform pQE60-NA-based libraries into DH5α cells using standard protocols. Spread the transformation mixture on LB + ampicillin plates at a density that yields approximately 1000–1500 colonies per 100-mm Petri plate.
After 12–15 h growth at 37°C (see Note 3), examine colony fluorescence using the slide projector, as follows. In a dark room, place an appropriate bandpass filter over the slide projector light source. Wearing appropriate emission filter goggles, illuminate the Petri plate with the lid off and look for the brightest colonies. Rotate the Petri plate to assure that all regions are examined.
Using sterile toothpicks, pick the brightest colonies (approximately 10% of the total) into 96-well plates for screening in the aggregation assay, or restreak promising colonies for maintenance (see Note 4).
3.2. Screening for aggregation using the bacterial lysis assay
Fill each well of a 96-well round-bottom plate with 175 μl of LB + ampicillin. As described in Subheading 3.1., pick individual fluorescent E. coli colonies into the wells. In each plate, set aside four control wells for clones expressing the FP that was the template for mutagenesis (see Note 5). Grow 10–12 h at 37°C (see Note 6), shaking at an angle in an orbital shaker.
To store clones for later recovery, use a multichannel pipette to spot 2 μL each from 48 wells onto an LB + ampicillin plate in a grid pattern. Label the Petri plates according to well number. Incubate the Petri plates at 37°C for 12–15 h, and store them at 4°C.
Meanwhile, for each 96-well plate, centrifuge the remaining culture samples for 5 min at 3,000g. Carefully remove the supernatants using a multichannel pipette.
Resuspend each cell pellet in 100 μL BPER II using a multichannel pipette. Allow the lysis to proceed for 20 min at 37°C in an orbital shaker.
Centrifuge each 96-well plate for 5 min at 3,000g. Using a multichannel pipette, transfer the supernatants to a pair of black 96-well plates, skipping every other column to leave room for the corresponding pellet fractions. Be careful not to create bubbles during the transfer.
Using a multichannel pipette, thoroughly resuspend each pellet of lysed cells in 100 μL BPER II. Transfer the resuspended pellet samples to black 96-well plates, placing each pellet sample next to the corresponding supernatant sample.
Quantify the fluorescence signals using a 96-well plate fluorometer. It is important to use a fixed gain that is high enough to produce strong signals without overloading the detector. For red FPs, we typically use 560 ± 20 nm excitation and 600 ± 20 nm emission.
Determine the aggregation value for each clone by calculating the percentage of the signal found in the pellet. For a given well, divide the pellet signal by the sum of the pellet and supernatant signals. As a positive control, EGFP should have an aggregation value of 2–4%. Aggregation values for various oligomeric red and far-red FPs are shown in Fig. 2.
For each well, compare the aggregation value to the average aggregation value for the four template wells. Calculate the standard error of the mean (s.e.m.) for the four template wells (see Note 7). Multiply the s.e.m. by two and subtract it from the mean aggregation value for the remaining 92 wells, thereby obtaining a threshold for significance. Any well with an aggregation value less than or equal to the threshold should be considered promising for further analysis.
To evaluate promising candidates, restreak from the replica spots (step 2) on fresh LB + ampicillin plates. For each candidate, pick 4–8 restreaked colonies into 96-well plates, and repeat the aggregation assay with a template control. Candidates that are reproducibly more soluble than the template should be characterized further by preparing miniprep DNA for sequencing.
After obtaining the sequences of the improved variants, build a combinatorial library that includes the solubility-enhancing mutations as well as the original codons. In some cases, it may be relevant to test additional mutations at key positions (see Note 8).
Screen this library as described above (see Note 9) to identify the optimal combination of mutations.
Fig 2.

DsRed-Express2 and its derivatives are as soluble as EGFP and much more soluble than other oligomeric FPs. Oligomeric FPs were expressed in E. coli. For each FP, a bacterial lysis assay was used to determine the aggregation value, which is defined as the percentage of the total fluorescence signal in the pellet fraction. A high aggregation value indicates the formation of higher-order aggregates. As a reference, EGFP has an aggregation value of only 3%. In these assays, green fluorescence was measured with 470 ± 10 nm excitation and 510 ± 10 nm emission, orange fluorescence with 540 ± 10 nm excitation and 560 ± 10 nm emission, red fluorescence with 560 ± 20 nm excitation and 600 ± 20 nm emission, and far-red fluorescence with 595 ± 20 nm excitation and 640 ± 20 nm emission. KO* is a modified version of KO in which synonymous alternative codons were used near the translation start site to enhance bacterial expression (18). (Adapted with permission from refs. 17–19.)
3.3. Assaying cytotoxicity in bacteria
Adjust the concentrations of pQE-60NA-based FP expression plasmids to 1 ng/μL in sterile H2O. Transform these plasmids into competent DH10B cells harboring the pREP4 repressor plasmid.
Prepare LB + ampicillin + kanamycin plates, one without IPTG (repressing conditions) and one with 1 mM IPTG (derepressing conditions). For each plate, use a marker to demarcate sectors for the FPs that are being tested.
Spread equal volumes of a given transformation mixture on the appropriate sector of each plate, aiming to achieve a density of about 10 colonies/cm2 (see Note 10).
Incubate 12–15 h at 37°C. Compare colony sizes for repressing versus derepressing conditions. Large colonies under derepressing conditions indicate low cytotoxicity (see Note 11). An example of the colony size assay is shown in Fig. 3A.
Colony size is a meaningful measure only if different FPs are expressed at similar levels (see Note 12). Expression is analyzed by SDS-PAGE of cell lysates. For this purpose, grow transformed clones overnight at 37°C in 5-mL cultures of LB + ampicillin + kanamycin to generate saturated precultures. Dilute each preculture to an OD600 of 0.05 in 5 mL of LB + ampicillin + kanamycin in a 50-mL baffled flask. After growth for 2 h at 37°C in an orbital shaker to yield an OD600 of ~0.5, add 1 mM IPTG to induce FP expression, and incubate with shaking for 4 h at 37°C. Then centrifuge 1 OD600 unit of each culture in a microfuge tube, resuspend each cell pellet in 200 μL of 1X SDS-PAGE sample loading buffer, and boil for 10 min. Centrifuge this mixture at 16,000g for 10 min. Without disturbing the pellet, dilute a portion of each sample supernatant 1:4 in SDS-PAGE sample buffer, and load 20 μL in a lane of an SDS-polyacrylamide gel. Load 10 μL of PageRuler prestained protein ladder as molecular weight standards. Run the gel and stain with Coomassie Blue or a comparable dye. FP bands can be identified based on size and intensity, with reference to a control sample from cells carrying an empty expression vector. A typical gel image is shown in Fig. 3B.
Fig 3.

DsRed-Express2 and its derivatives are noncytotoxic when expressed at high levels in bacteria. (A) Representative colony size assay showing the bacterial cytotoxicities of several red and orange FPs. DH10B cells harboring the pREP4 repressor plasmid were transformed with pQE-60NA encoding either DsRed-Express2, E2-Red/Green, E2-Orange, mOrange2, KO, or KO* (see Figure 2 legend). In this assay, cells expressing DsRed-Express2, E2-Red/Green, and E2-Orange produced large colonies, indicating low cytotoxicity. Cells expressing mOrange2 and KO* produced very small or pinprick colonies, respectively, indicating cytotoxicity. Cells expressing KO produced large colonies, but these colonies were nearly colorless due to poor expression. (B) Quantitation of FP expression under derepressing conditions. Cells were grown to an OD600 of ~0.6 and then treated with 1 mM IPTG for 4 h. Whole-cell lysates were separated using SDS-PAGE followed by staining with Coomassie Blue. Control cells were transformed with the empty pQE-60NA vector. The image shows that KO epxression was very low, while KO* expression was comparable to that of other FPs. (Reproduced with permission from ref. 18.)
3.4. HeLa cell transient transfection assay for cytotoxicity
Culture HeLa cells in a 100-mm dish to 50–70% confluence.
Remove the culture medium by aspiration. Wash the cells with 10 mL sterile PBS. Remove the PBS, and dissociate the HeLa cells by incubating for 4 min at 37°C with 2 mL of Trypsin-EDTA solution. Resuspend the cells in 5 mL of culture medium at 37°C (see Note 13).
Add the cell suspension to a 15-mL conical tube and centrifuge 5 min at 2,000g.
Discard the supernatant, and resuspend the cell pellet in 5 mL of fresh culture medium.
Determine the cell density using a hemocytometer.
Add 0.5–1 x 105 cells to each well of several 24-well plates. To test each FP in triplicate, use 15 culture wells per FP plus 5 culture wells for the “No DNA” control. Allow the cells to grow for 16–24 h to attain ~30% confluence.
Dilute the FP expression plasmids to 400 ng/μL.
For each FP, dilute 30 μL of plasmid in 750 μL of OptiMEM. Mix gently. For the “No DNA” control, use 30 μL of sterile H2O.
In separate tubes, add 15 μL of Lipofectamine 2000 to 750 μL of OptiMEM for each FP. Mix gently and incubate at room temperature for 5 min.
For each FP, combine the plasmid mixture with the Lipofectamine mixture. Mix gently and incubate at room temperature for 20 min.
For a given FP, add 100 μL of the plasmid/Lipofectamine mixture to each of 15 culture wells. For the “No DNA” control, add 100 μL of the H2O/Lipofectamine mixture to each of 5 culture wells.
Incubate the cells for 24 h. Remove the Lipofectamine-containing medium and replace with fresh culture medium.
Analyze the cells by flow cytometry at daily intervals for 5 days, beginning at 24 h post-transfection. To prepare cells for analysis on a given day, select three wells per FP and one well for the “No DNA” control. For each well, remove the medium, wash the cells with 1 mL of PBS, then replace the PBS with 200 μL of Trypsin-EDTA and incubate at 37°C for 4 min. Remove the Trypsin-EDTA, and suspend the cells in 2 mL of culture medium.
Perform flow cytometry with the suspended cells using suitable laser lines and emission filters. For red FPs, 561-nm lasers and PE filter sets (585/15 nm) are commonly used. Based on side scatter area (SSC-A) and forward scatter area (FSC-A) profiles, gate on living cells. Based on side scatter width (SSC-W) and forward scatter width (FSC-W), gate on single, non-aggregated cells. Gate fluorescent cells based on the “No DNA” control sample.
Analyze the fluorescence data with FlowJo software (Treestar Inc., Ashland, OR). For each sample, including the “No DNA” control, record the average population brightness of living fluorescent cells. To determine the signal / background ratio, divide the average brightness of the fluorescent cells by the average brightness of the “No DNA” cells. Data for representative orange, red, and far-red FPs are shown in Fig. 4.
Fig 4.

E2-Crimson is noncytotoxic to HeLa cells under conditions of standard high-level expression. (A) HeLa cells were transiently transfected in 24-well plates for constitutive high-level expression of the indicated FPs. For each FP, three wells per day were analyzed by flow cytometry, and the average brightness of the viable fluorescent cells was measured relative to untransfected cells. Maintenance of a high percentage of the original fluorescence indicates low cytotoxicity. (B) Fluorescence intensity distributions for the same data were analyzed for 24 h (blue) and 120 h (red) post-transfection. Each data point is a binned value that represents the percentage of cells with fluorescence in a range centered about the data point. Cells expressing E2-Crimson maintained a nearly identical population distribution over the course of the experiment, while cells expressing the other FPs showed dramatic population shifts in favor of dimmer cells. (Reproduced with permission from ref. 19.)
3.5. Generation of lentiviral vectors and HeLa cell lentiviral transduction assay for cytotoxicity
Generate lentiviral particles for FP-encoding vectors according to the Lenti-X Expression Kit protocol. In parallel, generate control viral particles by transfecting 293-T cells with pLVX-Puro.
Aliquot the viral particles and store them at −80°C (see Note 14).
Determine the viral titer for each sample using the QuickTiter Lentivirus Quantitation Kit (see Note 15).
Culture HeLa cells to 50–70% confluence. Split the cells as described in Subheading 3.4. into 6-well dishes. Add 2.5 x 105 cells per well, with 3 wells per FP and 1 well for the control virus. Incubate 16–24 h at 37°C until the cells are ~50% confluent.
Remove the culture medium and replace it with exactly 2 mL of fresh culture medium per well.
Add 1.6 μL of 10 mg/mL polybrene to each well for a final concentration of 8 μg/mL (see Note 16).
Add equivalent titers of viral particles to each well, resulting in three replicates per FP. Incubate 24 h at 37°C.
Remove the culture medium and excess viral particles, wash each well with 2 mL of culture medium, and add 2 mL of fresh culture medium. This time point is defined as Day 0.
Incubate 72 h at 37°C to allow expression of the FPs.
At the end of this incubation (on Day 3), remove the culture medium and wash the cells in each well with 2 mL of PBS. Add 0.5 mL of Trypsin-EDTA to each well, and incubate 4 min at 37°C.
Remove the Trypsin-EDTA, and resuspend the cells in 2 mL of fresh culture medium per well. To allow continued growth, add 0.5 mL of cell suspension to 2 mL of culture medium per well in fresh 6-well plates.
Analyze the remaining cells by flow cytometry as described in Subheading 3.4. Record the average brightness of viable fluorescent cells and control cells. Also record the percentage of viable cells that are fluorescent.
Repeat the passage and flow cytometry analysis on Days 5 and 7. Do a final analysis on Day 10. Results for a monomeric green FP and several representative red FPs are shown in Fig. 5.
Fig 5.
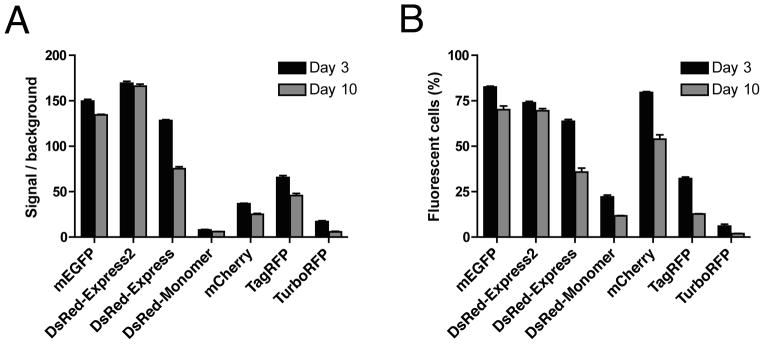
Fluorescent protein cytotoxicities after lentiviral transduction. (A) HeLa cells were transduced with lentiviral particles encoding the indicated FPs or with a control lentiviral particles lacking an FP gene. At 3 and 10 days after transduction, cells were analyzed by flow cytometry. Plotted are the average fluorescence signals from viable fluorescent cells relative to the control. (B) The percentage of viable cells that were fluorescent was also recorded at 3 and 10 days after transduction. In these experiments, DsRed-Express2 and monomeric EGFP (mEGFP, which is EGFP with the A206K mutation) maintained almost all of the original fluorescence signal and showed only a small decline in the percentage of fluorescent cells. By contrast, all of the other red FPs showed dramatic reductions in average fluorescence and in the percentage of fluorescent cells, indicating that those other FPs were cytotoxic. (Reproduced with permission from ref. 17.)
Footnotes
E. coli strain DH5α is optimal for use in the bacterial lysis assay. In our experience, other strains do not lyse as efficiently in BPER II.
Dissolve all ingredients except β-mercaptoethanol in 90% of the total volume. Add fresh β-mercaptoethanol immediately before use.
Screening colonies as early as possible for brightness allows for selection of mutants that are both bright and rapidly maturing.
Restreak promising candidates on adjacent sectors of the same plate to examine relative brightness. As a control, streak bacteria expressing the template FP.
Due to slight variability in the procedure, each plate should include clones carrying the template FP for direct comparison with mutants.
When expressing extremely cytotoxic FPs, grow cells containing the pREP4 plasmid under repressing conditions and then derepress expression with IPTG. In this case, allow the cells to grow for 3–4 h, add 1 mM IPTG, and continue the incubation for 6–12 h.
Calculate s.e.m. by dividing the standard deviation by the square root of the number of samples.
In our experience, mutation to charged residues such as lysine can help to increase solubility. These mutations may not be discovered through random mutagenesis by error-prone PCR due to the limited number of codon substitutions that can be obtained from single point mutations.
To determine the number of colonies that should be screened in this assay, determine the size of your library by calculating the total number of possible mutant combinations. For example, if 2 mutations are screened at each of 3 positions, the library size is 23 or 8 potential clones. A ten-fold overscreening is generally sufficient to identify all of the relevant clones. In the example given above, 80 colonies would be chosen for the bacterial lysis assay.
A high density of colonies can bias the results toward small colonies. For this reason, low density is critical for proper evaluation. Plating the right number of cells may require trial and error.
If colonies are not visible to the naked eye, they can sometimes be seen with the slide projector assay as fluorescent “pinprick” colonies.
For certain FPs, bacterial expression from the endogenous start codon may be very low, yielding large, nearly colorless colonies under derepressing conditions. This effect is likely due to formation of secondary structure near the 5’ end of the mRNA (20, 21). In such a case, the FP cannot be tested using this assay. However, we have successfully introduced silent mutations within the first few codons of FP genes, thereby increasing bacterial expression and allowing for analysis of cytotoxicity. As an example, the original KO and the expression-enhanced KO* (18) are shown in Fig. 3.
Trypsin-EDTA and all media should be pre-warmed to 37°C before use. This pre-warming is assumed for all steps that describe Trypsin-EDTA treatments, washes in culture medium, and changes of culture medium.
Freezing and thawing lentiviral particles may reduce their efficacy. For this reason, aliquots are recommended for storage.
This method measures the total titer of the viral particles, and is not a measure of viral particles that are competent for transduction.
4–10 μg/mL of polybrene is typically used for lentiviral transduction. Therefore, it is usually unnecessary to add more polybrene to adjust for the change in volume caused by addition of viral particles. However, if the viral particles are very dilute and more than 1 mL of viral particle solution is added, adjust the polybrene concentration to 8 μg/mL.
References
- 1.Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802–5. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 2.Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nat Methods. 2005;2:905–9. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- 3.Shaner NC, Campbell RE, Steinbach PA, Giepmans BNG, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2005;22:1567–72. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 4.Wang L, Jackson WC, Steinbach PA, Tsien RY. Evolution of new nonantibody proteins via iterative somatic hypermutation. Proc Natl Acad Sci USA. 2004;101:16745–9. doi: 10.1073/pnas.0407752101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Strongin DE, Bevis B, Khuong N, Downing ME, Strack RL, Sundaram K, Glick BS, Keenan RJ. Structural rearrangements near the chromophore influence the maturation speed and brightness of DsRed variants. Protein Eng Des Sel. 2007;20:525–34. doi: 10.1093/protein/gzm046. [DOI] [PubMed] [Google Scholar]
- 6.Merzlyak EM, Goedhart J, Shcherbo D, Bulina ME, Shcheglov AS, Fradkov AF, Gaintzeva A, Lukyanov KA, Lukyanov S, Gadella TW, Chudakov DM. Bright monomeric red fluorescent protein with an extended fluorescence lifetime. Nat Methods. 2007;4:555–7. doi: 10.1038/nmeth1062. [DOI] [PubMed] [Google Scholar]
- 7.Shaner NC, Lin MZ, McKeown MR, Steinbach PA, Hazelwood KL, Davidson MW, Tsien RY. Improving the photostability of bright monomeric orange and red fluorescent proteins. Nat Methods. 2008;5:545–51. doi: 10.1038/nmeth.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shcherbo D, Merzlyak EM, Chepurnykh TV, Fradkov AF, Ermakova GV, Solovieva EA, Lukyanov KA, Bogdanova EA, Zaraisky AG, Lukyanov S, Chudakov DM. Bright far-red fluorescent protein for whole-body imaging. Nat Methods. 2007;4:741–6. doi: 10.1038/nmeth1083. [DOI] [PubMed] [Google Scholar]
- 9.Shcherbo D, Murphy CS, Ermakova GV, Solovieva EA, Chepurnykh TV, Shcheglov AS, Verkhusha VV, Pletnev VZ, Hazelwood KL, Roche PM, Lukyanov S, Zaraisky AG, Davidson MW, Chudakov DM. Far-red fluorescent tags for protein imaging in living tissues. Biochem J. 2009;418:567–74. doi: 10.1042/BJ20081949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kredel S, Oswald F, Nienhaus K, Deuschle K, Rocker C, Wolff M, Heilker R, Nienhaus GU, Wiedenmann J. mRuby, a bright monomeric red fluorescent protein for labeling of subcellular structures. PLoS One. 2009;4:e4391. doi: 10.1371/journal.pone.0004391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karasawa S, Araki T, Nagai T, Mizuno H, Miyawaki A. Cyan-emitting and orange-emitting fluorescent proteins as a donor/acceptor pair for fluorescence resonance energy transfer. Biochem J. 2004;381:307–12. doi: 10.1042/BJ20040321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, Imamura T, Ogawa M, Masai H, Miyawaki A. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 2008;132:487–98. doi: 10.1016/j.cell.2007.12.033. [DOI] [PubMed] [Google Scholar]
- 13.Shaner NC, Patterson GH, Davidson MW. Advances in fluorescent protein technology. J Cell Sci. 2007;120:4247–60. doi: 10.1242/jcs.005801. [DOI] [PubMed] [Google Scholar]
- 14.Bevis BJ, Glick BS. Rapidly maturing variants of the Discosoma red fluorescent protein (DsRed) Nat Biotechnol. 2002;20:83–7. doi: 10.1038/nbt0102-83. [DOI] [PubMed] [Google Scholar]
- 15.Kredel S, Nienhaus K, Oswald F, Wolff M, Ivanchenko S, Cymer F, Jeromin A, Michel FJ, Spindler KD, Heilker R, Nienhaus GU, Wiedenmann J. Optimized and far-red-emitting variants of fluorescent protein eqFP611. Chem Biol. 2008;15:224–33. doi: 10.1016/j.chembiol.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 16.Tao W, Evans BG, Yao J, Cooper S, Cornetta K, Ballas CB, Hangoc G, Broxmeyer HE. Enhanced green fluorescent protein is a nearly ideal long-term expression tracer for hematopoietic stem cells, whereas DsRed-Express fluorescent protein is not. Stem Cells. 2007;25:670–8. doi: 10.1634/stemcells.2006-0553. [DOI] [PubMed] [Google Scholar]
- 17.Strack RL, Strongin DE, Bhattacharyya D, Tao W, Berman A, Broxmeyer HE, Keenan RJ, Glick BS. A noncytotoxic DsRed variant for whole-cell labeling. Nat Methods. 2008;5:955–7. doi: 10.1038/nmeth.1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strack RL, Bhattacharyya D, Glick BS, Keenan RJ. Noncytotoxic orange and red/green derivatives of DsRed-Express2 for whole-cell labeling. BMC Biotechnol. 2009;9:32. doi: 10.1186/1472-6750-9-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Strack RL, Hein B, Bhattacharyya D, Hell SW, Keenan RJ, Glick BS. A Rapidly Maturing Far-Red Derivative of DsRed-Express2 for Whole-Cell Labeling. Biochemistry. 2009;8:8279–81. doi: 10.1021/bi900870u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pfleger BF, Fawzi NJ, Keasling JD. Optimization of DsRed production in Escherichia coli: effect of ribosome binding site sequestration on translation efficiency. Biotechnol Bioeng. 2005;92:553–8. doi: 10.1002/bit.20630. [DOI] [PubMed] [Google Scholar]
- 21.Sörensen M, Lippuner C, Kaiser T, Mißlitz A, Aebischer T, Bumann D. Rapidly maturing red fluorescent protein variants with strongly enhanced brightness in bacteria. FEBS Lett. 2003;552:110–4. doi: 10.1016/s0014-5793(03)00856-1. [DOI] [PubMed] [Google Scholar]