

Abstract
Encephalocraniocutaneous lipomatosis (ECCL), also known as Haberland syndrome, is a rare syndrome with unknown etiology. The syndrome is characterized by a triad of unique cutaneous, ocular, and central nervous system (CNS) manifestations. The cutaneous hallmark, nevus psiloliparus (NP), along with overlying alopecia is a constant feature. Choristoma of the eyelid is the most common ocular manifestation, while intracranial lipoma is the predominant CNS finding. Genetic counseling is required to emphasize that the disorder, although congenital, is not inheritable. We present a 21-year-old female with cutaneous, ocular, and CNS features satisfying the diagnostic criteria for ECCL. To our knowledge, this is the first case of ECCL having a large temporal exostosis. The objective of this article is to better understand the phenotypic spectrum of this syndrome whose molecular basis is still unknown.
Keywords: Choristoma, encephalocraniocutaneous lipomatosis, exostosis, neurocutaneous, nevus psiloliparus
Introduction
Encephalocraniocutaneous lipomatosis (ECCL) is a unique congenital neurocutaneous disorder first described by Haberland and Perou in 1970.[1] Subsequently, Fishman et al. reported additional cases.[2] Hence, the syndrome is also known as Haberland syndrome or Fishman syndrome after the pioneering works done by these authors. To date, 54 cases have been reported in literature.[3] The syndrome is characterized by constellation of features involving eye, skin, and CNS. It is predominantly unilateral, but bilateral cases have also been reported.[4] The revised diagnostic criteria proposed by Ute Moog in 2009[3] divide the ECCL cases into definite and probable, taking into consideration major and minor criteria for cutaneous, eye, CNS, and other systems’ involvement.
Case Report
A 21-year-old female presented with skin-colored swelling over right frontotemporal region, present since birth. It was hard to touch with adjoining area of soft tissue. The overlying skin was devoid of hair. At birth, she was also noted to have a nodular lesion in the right upper eyelid and scleral mass in the right ocular globe. A few small skin nodules were also present over ipsilateral side of the face [Figure 1]. All these cutaneous and ocular lesions were increasing in size in proportion to patient's generalized body growth. The scleral mass was now interfering with her vision, so the patient wanted its removal. She also wanted surgical correction of the scalp swelling for cosmetic reasons. She had no history of seizures and neurological examination was unremarkable. Plain computed tomography (CT) and magnetic resonance imaging (MRI) brain were done to exclude intracranial and intraorbital extension of these lesions. The right frontotemporal scalp lesion was diagnosed as subcutaneous lipoma as it gave fat signal on T1W images and was suppressed on fat-saturated T1W [Figures 2A and B]. Also, on plain CT, it had fat HU value of −60 to −80 HU. A large right temporal exostosis was seen within this scalp lipoma [Figure 3]. However, no intracranial extension of this lesion was seen. The soft tissue in the right upper eyelid gave fat attenuation and signal at places, hence possibility of dermolipoma was considered [Figures 4A and B]. The scleral mass had no intraorbital extension. On MRI, a lipoma was found in the middle cranial fossa on the right side, adjacent to the temporal lobe and reaching up to right cerebellopontine cistern [Figures 5A and B]. MRI of the whole spine was also done, but no abnormality was detected. In view of the imaging and constellation of clinical features, the final diagnosis of ECCL was given.
Figure 1.
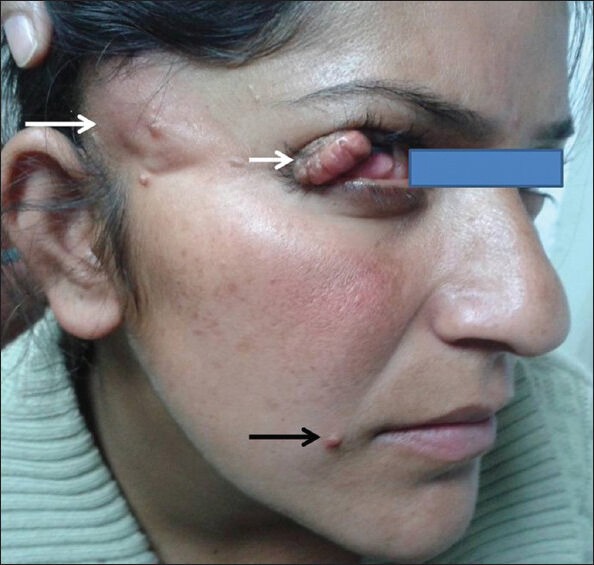
21-year-old female with patchy alopecia and possible nevus psiloliparus (NP) over right frontotemporal region (long white arrow) with dermolipoma of ipsilateral upper eyelid and scleral mass (short white arrow). Skin coloured papules are also seen (black arrow). (Photograph published with prior consent of the patient for on line and in print publication)
Figure 2 (A, B).
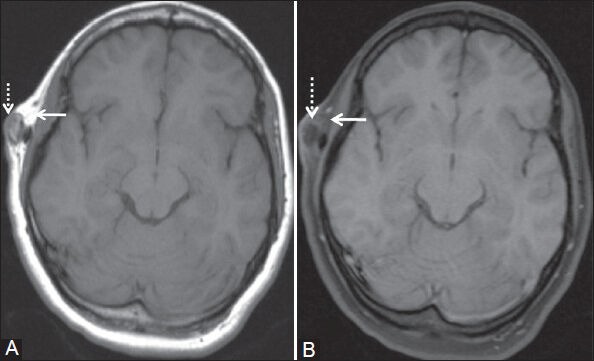
(A) T1W and (B) fat-saturated T1W axial images show subcutaneous lipoma over right frontotemporal region giving fat signal on T1W image and signal suppression on fat-saturated T1W image (white arrow). Calcified area representing exostosis is seen within this lesion which is hypointense on both the images (dashed white arrow)
Figure 3.
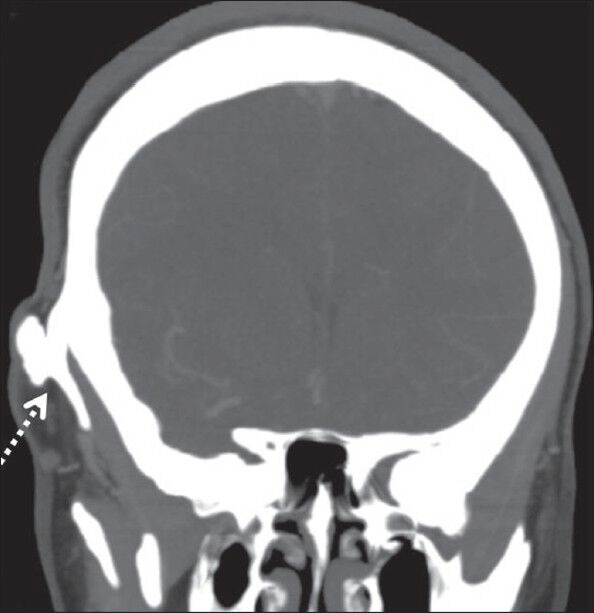
Coronal plain CT shows right temporal exostosis along its entire length (dashed white arrow)
Figure 4 (A, B).
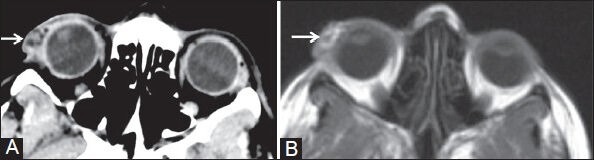
(A) Axial plain CT and (B) T1W MR images show patchy fatty areas in soft tissue mass of right upper eyelid (white arrow)
Figure 5 (A,B).
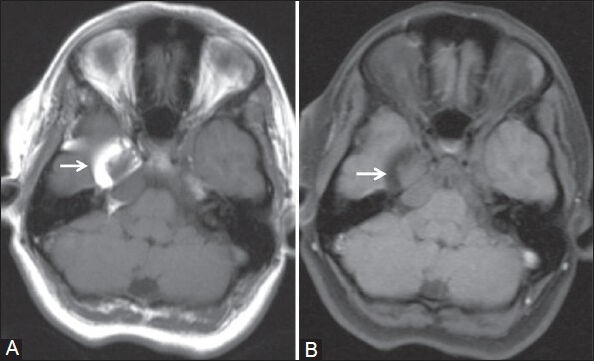
(A) T1W and (B) fat-saturated T1W axial images show lipoma in middle cranial fossa on the right side reaching up to ipsilateral cerebellopontine angle (white arrow)
The original diagnostic criteria for ECCL were proposed by Hunter in 2006.[5] The revised criteria were given by Ute Moog in 2009[3] for definite/proven and possible ECCL cases [Table 1], excluding the third group of probable ECCL as given by Hunter. In the revised criteria for ECCL, there are one major and three minor criteria involving ocular system, three major and five minor criteria involving cutaneous system, and three major and six minor criteria for CNS involvement. Three major criteria are also seen involving the other systems.
Table 1.
Revised diagnostic criteria for ECCL

Application of criteria to the diagnosis of ECCL[3]
Definite case
Three systems involved, major criteria in ≥2, or
Three systems involved, proven NP or possible NP + ≥1 of the minor skin criteria 2-5, or
Two systems involved with major criteria, one of which is proven NP or possible NP + ≥1 of the minor skin criteria 2-5.
Probable case
Two systems involved, major criteria in both
Two systems involved, proven or possible NP.
Our case fulfilled the criteria for definite diagnosis for ECCL with three main systems’ involvement. The two major criteria in our case were eyelid choristoma and intracranial lipoma. The minor criteria were possible NP (in the absence of histopathologic confirmation), patchy non-scarring alopecia, subcutaneous lipoma in right frontotemporal region, and small nodular skin tags over ipsilateral face. Additionally, our case had a large temporal calvarial exostosis not reported earlier in the literature.
Discussion
ECCL is a unique, non-progressive congenital syndrome involving mainly the cutaneous, ocular, and neurologic systems. The first case was described by Haberland and Perou in 1970.[1] It is a rare syndrome and only 54 cases have been delineated since 1970.[3] The cases are sporadic with no evidence of genetic transmission or chromosomal abnormality and believed to show mosaic inheritance of a lethal autosomal mutation.[6] No epidemiological data regarding its frequency are available. There is no clear geographic, gender, or racial predilection. The etiology of this syndrome is not clear. The probable pathogenesis is dysgenesia of the cephalic neural crest and the anterior neural tube.[7] It predominantly affects the ectodermal and mesodermal derivatives. The clinical spectrum is broad, but a group of clinical features is regarded as characteristic for the disorder. The pattern of skin and ocular anomalies is variable in their extent and severity, but consistent and recognizable. The neurodevelopment spectrum is very wide. It varies from completely normal mental status to severe mental retardation.[8] The patient may also have seizures, facial paresis, hemiplegia, spasticity of contralateral limb, sensorineural hearing loss, and behavioral changes.[9] There are only a few cases reported with normal mental development and no seizures.[10]
A distinct type of mesodermal nevus, nevus psiloliparus (NP), is the cutaneous hallmark and is evident since birth. It is a skin-colored, cutaneous soft tumor characterized by the presence of mature fat and paucity or absence of scalp hair follicles. Even though NP is the dermatologic hallmark, it is not a pathognomonic sign and literature shows this nevus in two otherwise healthy persons also.[8] These subcutaneous fatty masses are typically seen in frontotemporal or zygomatic regions. Other cutaneous abnormalities include skin-colored papules or nodules which represent angiolipomas, fibrolipomas, connective tissue nevi, or mixed hamartoma of fat, cartilage, or other connective tissue.[11,12] These cutaneous neoplasms are predominantly seen on scalp, face, and neck.
The most prominent ocular finding is choristoma which is a congenital benign neoplasm in an abnormal location. It includes epibulbar or limbal dermoid or lipodermoid. Other ocular anomalies are corneal and scleral anomalies, colobomas, aniridia, microphthalmia, and globe calcification. Still other anomalies reported are a small tag in the anterior chamber, a persistent posterior hyaloid system, a dysplastic iris, papilledema, and epicanthus inversus with hypertelorism.[11,12]
The most common CNS malformation is intracranial lipoma. Cerebellopontine cistern is the most common location for these tumors. Spinal lipomas are also seen. Focal vascular defects leading to cerebral atrophy, porencephalic cysts, dilated ventricles, and calcifications have been described. Vascular anomalies like excessive or abnormal vessels and leptomeningeal angiomatosis are seen. Also, congenital anomalies of meninges like arachnoid cysts are noted.[3] The CNS anomalies do not correlate with the extent of neurological symptoms.
Skeletal anomalies like macrocephaly, jaw tumors, non-ossifying fibromas, and asteolytic bone lesions have been described. These lesions are progressive unlike other features of the disorder. Coarctation of aorta is the prominent feature in cardiovascular system.[3]
The differential diagnoses to be considered are Proteus syndrome, oculoectodermal syndrome (OES), epidermal nevus syndromes (ENS), oculocerebrocutaneous syndrome (Delleman syndrome), nevus sebaceous syndrome of Jadassohn, and oculo-auriculo-vertebral spectrum (OAV spectrum or Goldenhar's syndrome) that have a few overlapping features.[3,9,11] The closest differential is Proteus syndrome. However, it has a progressive course. The cerebriform connective tissue and epidermal nevus are specific criteria for this syndrome, while ECCL is characterized by its non-progressive nature with features evident at birth, except for possible skeletal cysts, jaw tumors, and vascular lesions. So, both syndromes should be considered as separate entities. Another differential is OES in which the ocular and cutaneous manifestations are similar but intracranial lipomas are not present. ENS also simulate ECCL in congenital anomalies involving the ocular, CNS, skeletal, and other systems. However, the main differentiating feature is epidermal nevus that follows the lines of Blaschko.
The treatment of ECCL is nonspecific. It includes anticonvulsants for seizures and surgical corrections of ocular and cutaneous lesions. Neurosurgery is required if the patient is refractory to medical treatment.
Conclusion
The sporadic cases of ECCL are underdiagnosed or misdiagnosed. Awareness about the broad clinical spectrum of this rare syndrome is warranted for early diagnosis. The affected family should be reassured that this genetic syndrome would not be transmitted to the offspring.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Haberland C, Perou M. Encephalocraniocutaneous lipomatosis: A new example of ectomesodermal dysgenesis. Arch Neurol. 1970;22:144–55. doi: 10.1001/archneur.1970.00480200050005. [DOI] [PubMed] [Google Scholar]
- 2.Fishman MA, Chang CS, Miller JE. Encephalocraniocutaneous lipomatosis. Pediatrics. 1978;61:580–2. [PubMed] [Google Scholar]
- 3.Moog U. Encephalocraniocutaneous lipomatosis. J Med Genet. 2009;46:721–9. doi: 10.1136/jmg.2009.066068. [DOI] [PubMed] [Google Scholar]
- 4.Brassesco MS, Valera ET, Becker AP, Castro-Gamero AM, De Aboim Machado A, Santos AC, et al. Low-grade astrocytoma in a child with encephalocraniocutaneous lipomatosis. J Neurooncol. 2010;96:437–41. doi: 10.1007/s11060-009-9978-1. [DOI] [PubMed] [Google Scholar]
- 5.Hunter AG. Oculocerebrocutaneous and encephalocraniocutaneous lipomatosis syndromes: Blind men and an elephant or separate syndromes? Am J Med Genet A. 2006;140:709–26. doi: 10.1002/ajmg.a.31149. [DOI] [PubMed] [Google Scholar]
- 6.Gokhale NR, Mahajan PM, Belgaumkar VA, Pradhan SN, Uttarwar NS. Encephalocraniocutaneous lipomatosis: A rare neurocutaneous syndrome. Indian J Dematol Venereol Leprol. 2007;73:40–2. doi: 10.4103/0378-6323.30651. [DOI] [PubMed] [Google Scholar]
- 7.Koishi GN, Yoshida M, Alonso N, Matushita H, Goldenberg D. Encephalocraniocutaneous Lipomatosis (Haberland's Syndrome) – A Case Report of a Neurocutaneous Syndrome and a Review of the Literature. Clinics (Sao Paulo) 2008;63:406–8. doi: 10.1590/S1807-59322008000300020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim DH, Park SB, Lee Y, Im M, Seo YJ, Choi SH, et al. Encephalocraniocutaneous Lipomatosis without Neurologic Anomalies. Ann Dermatol. 2012;24:476–8. doi: 10.5021/ad.2012.24.4.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan CC, Chen JS, Chu CY. Haberland Syndrome. Dermatol Sin. 2005;23:41–5. [Google Scholar]
- 10.Parazzini C, Triulzi F, Russo G, Mastrangelo M, Scotti G. Encephalocraniocutaneous Lipomatosis: Complete neuroradiologic evaluation and follow-up of two cases. AJNR Am J Neuroradiol. 1999;20:173–6. [PubMed] [Google Scholar]
- 11.MacLaren MJ, Kluijt I, Koole FD. Ophthalmologic abnormalities in encephalocraniocutaneous lipomatosis. Doc Ophthalmol. 1995;90:87–98. doi: 10.1007/BF01203299. [DOI] [PubMed] [Google Scholar]
- 12.Chittenden HB, Harman HE, Robinson F, Higgins EM. A case of encephalocraniocutaneous lipomatosis. Br J Ophthalmol. 2002;86:934–5. doi: 10.1136/bjo.86.8.934. [DOI] [PMC free article] [PubMed] [Google Scholar]