

Abstract
Primary hepatic carcinoids are rare tumors that are often diagnosed at a locally advanced stage. Their primary nature can only be ascertained after thorough investigations and long-term follow-up to exclude another primary origin. As with secondary neuroendocrine liver tumors, surgical resection remains the mainstay of therapy. Despite their large size and often central location liver resection is often feasible, offering long-term survival and cure to most patients. In selected patients liver transplantation appears to be a good indication for tumors not amenable to liver resection. An aggressive surgical attitude is therefore warranted. We report a large and unusually fast-growing liver carcinoid that appeared only marginally resectable in a patient who remains free of disease four years after surgery.
1. Introduction
With less than 100 reports in the literature little is known of primary hepatic carcinoids. They often present as large centrally situated liver masses, characteristics that may discourage attempts at resection. Data is scarce on the outcome of surgical treatment but these tumors seem to be associated with a favorable prognosis, justifying an aggressive surgical approach. Presented here is a patient with a giant primary liver carcinoid that appeared only marginally resectable, who remains disease free four years after surgical resection.
2. Case Report
A 52-year-old female complained of fatigue and intermittent fever of one-year duration. There was no history of liver failure, hematemesis, flushing, or diarrhea. She had undergone a previous appendectomy. Abdominal ultrasound revealed a heterogeneous 8 cm right lobe focal liver lesion. Contrast enhanced CT performed one month later showed a solid 15 cm liver mass with a hypodense core that was suggestive of an atypical hemangioma. The patient came to our attention two months after the initial ultrasound with palpable hepatomegaly. MR showed a hypervascularized solitary liver lesion measuring 20 cm (Figure 1). Malignancy was further suspected by FDG-PET scan showing increased uptake. Laboratory data showed normal liver function and hepatitis B and C serologies were negative. Serum tumor markers including CEA, AFP, CA 12.5, CA 19.9, and NSE were within normal range while chromogranin A was moderately elevated. 24-hour urinary 5-HIAA excretion was normal. Chest CT showed no signs of malignancy. No primary tumor was found at gastroscopy and colonoscopy. Laparoscopy showed that the right liver lobe was completely occupied by a polylobulated firm white mass. The left lobe appeared healthy and liver biopsy confirmed this. There were no serosal surface tumor deposits and no lymphadenopathy at the porta hepatis. Differential diagnosis included hepatocellular carcinoma, cholangiocellular carcinoma, hypervascularized metastasis, angiosarcoma, hemangiopericytoma, and a neuroendocrine tumor.
Figure 1.
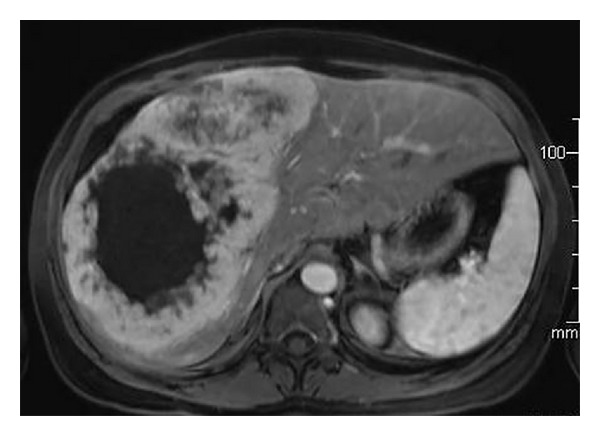
Transverse section of arterial phase of magnetic resonance: large right liver lobe tumor exhibiting early peripheral enhancement and a central cystic component.
During laparotomy thorough exploration of the abdominal cavity, small bowel, and mesentery was performed before proceeding to an extended right hepatectomy. The resected specimen weighed 2.2 kg and was almost entirely occupied by a 22 cm solid mass with a large central cystic component. Microscopically the tumor displayed a trabecular and pseudoglandular pattern and a highly vascular stroma. Tumor cells were uniform and displayed rare mitoses (<1 per mm³). At immunohistochemical examination tumor cells stained positive with chromogranin A and synaptophysin antibodies and were negative for hepatocyte, AFP, and CD56 antibodies. 10% nuclear reactivity for Ki-67 was present. There was intense expression of somatostatin subtypes I and II receptors but not subtype V.
Final diagnosis was a well-differentiated nonsecreting neuroendocrine tumor. Further investigations including In111-DTPA-octreotide scan, EUS of the pancreas, thyroid US and Tc-scintigraphy, and small bowel barium study failed to find a primary neuroendocrine tumor. Review of the pathology of the previous appendectomy was also noncontributive. Postoperative screening for recurrence and detection of a possible primary tumor has included abdominal MR, chest X-ray, and serum chromogranin A assay every 6 months. Octreotide and FDG-PET scan are scheduled yearly. At 48-month follow-up the patient shows no signs of liver recurrence or appearance of a primary tumor or secondary extrahepatic tumor. She is asymptomatic and fully functional.
3. Discussion
Carcinoid tumors also known as well-differentiated neuroendocrine tumors (NET) derive from neuroectodermal cells that are dispersed throughout the digestive tract but are also found in organs such as the adrenals, bronchi, thymus, thyroid, and paravertebral ganglia. Fifty-four percent of carcinoids occur within the gastrointestinal tract, mostly in the appendix and small bowel (16.7% and 44.7% resp.) but also in the rectum (19.6%), colon (10.6%), and stomach (7.2%) [1]. Carcinoids also occur in the lung (30.1%), pancreas (2.3%), genitals (1.2%), biliary tract (1.1%), and head and neck (0.4%). In the United States the incidence of carcinoid tumors is 6.25 cases per 100000 per year [2].
The 2010 World Health Organization carcinoid and pancreatic neuroendocrine tumor grading system takes into account the number of mitoses per 10 high power microscopic fields or the percentage of tumor cells that immunolabel positively for Ki-67 antigen. These measures reflect the rate of proliferation and correlate with prognosis. Carcinoids are classified into three types: (1) well-differentiated tumors of low grade malignancy with an indolent development and a good prognosis, (2) moderately differentiated or intermediate grade neoplasms, and (3) poorly differentiated or high grade epithelial neoplasms that carry a poor prognosis (Table 1).
Table 1.
Histopathological classification of neuroendocrine tumors.
| Histological classification | Well-differentiated (low grade, G1) | Moderately differentiated (intermediate grade, G2) | Poorly differentiated (high grade, G3) |
|---|---|---|---|
| Appearance | Monomorphic population of small round cells | Undefined | Cellular pleomorphism |
| Prognosis | Prolonged survival | Intermediate | Poor |
| Mitotic rate* | <2 | 2–20 | >20 |
| Ki-67 index** | <3% | 3–20% | >20% |
| Necrosis | Absent | Undefined | Present |
*Per 2 mm2; **percentage of tumor cells that immunolabel positively for Ki-67 antigen.
Less than 100 patients with primary hepatic carcinoid tumors (PHCT) have been reported, mostly as single cases [3]. The two largest series comprise 11 and 8 patients [4, 5]. The tumor occurs mostly in middle age (mean 49.8 years) with a slight female predominance (58.5%). Carcinoid tumors typically grow slowly and become clinically evident only at an advanced stage. Symptoms include abdominal pain (44%), abdominal mass (14.3%), and fatigue (7.1%). Carcinoid syndrome, characterized by flushing, abdominal pain, diarrhea, wheezing, and right heart failure is present in a mere 16.7% of patients. Cushing and Zollinger-Ellison syndromes are present in 2.4% and 6% of patients, respectively. Hence most PHCTs are nonsecreting although few studies actually assessed the secretion of serotonin, histamine, bradykinin, gastrin, vasoactive intestinal peptide, insulin, glucagon, or prostaglandins in the systemic circulation. When reported, the most frequently secreted hormones were gastrin (10.1%) and chromogranin A (7.2%) [6].
Ascertaining that a carcinoid liver tumor is a primary rather than a secondary deposit is challenging. A single large centrally situated tumor is suggestive of a primary tumor whereas neuroendocrine liver metastases present typically as multiple diffuse liver masses [5]. The pancreas is the most common primary site (35%) of neuroendocrine liver metastases [7]. However in 11–14% of patients with liver carcinoids no primary tumor is found. Thorough pre- and intraoperative investigations are required before concluding to a primary liver carcinoid [8]. These include computerized tomography, magnetic resonance, CT or MR enteroclysis, somatostatin scintigraphy, PET scan, gastroscopy, colonoscopy, endoscopic ultrasound of the pancreas, bronchoscopy, video capsule endoscopy or balloon enteroscopy, and operative exploration. In patients having previously undergone appendectomy the pathology report should be reviewed to exclude a primary tumor. Even when after thorough investigation a primary tumor is not identified, long-term reevaluation with conventional imaging, octreotide scan, and possibly PET scan is useful to detect a small primary tumor that may have initially been overlooked.
The best practice for neuroendocrine tumors metastatic to the liver remains surgical resection of both the primary tumor and liver metastases whenever possible. Recent publications indicate an advantage for aggressive liver surgery for locally advanced and metastatic NET in terms of duration and quality of life [9]. Surgical resection even seems to benefit patients with positive resection margins [10]. A Mayo Clinic study on secondary liver NET showed no difference in survival between patients having complete resection and those having 90% resection of their liver secondaries. Supported by a 75% four-year survival rate the authors considered resection to be indicated if the primary tumor and at least 90% of the metastatic tumor burden to the liver could be resected or ablated [11]. Reports indicate 5-year survival rates in the range of 47–92% after resection of NET liver metastases. This contrasts with a 20–30% 5-year survival in historical controls having not undergone liver resection [12]. However recurrences, mainly in the liver, remain high (78%–84%), occurring after a median of 19 months [13, 14]. More recently liver transplantation (LT) has been proposed in selected patients that were not amenable to partial liver resection. Initial results were disappointing owing to the lack of patient selection. Noncarcinoid tumors, nongastrointestinal carcinoids, high-grade tumors, or tumors not drained by the portal vein are considered to be associated with worse outcomes [15, 16]. A retrospective analysis of the United Network for Organ Sharing database on LT performed in the US between 1988 and 2008 included 150 patients with metastatic NET who had an overall 1-, 3- and 5-year survival rate of 81%, 65%, and 49%, respectively [17]. Carcinoid and noncarcinoid tumors had similar prognoses. LT for NET afforded similar survival as LT for hepatocellular carcinoma (HCC), a well-established and accepted treatment of HCC. Tumor recurrences for metastatic NET were 31% which is higher than tumor recurrence for HCC (10–15%).
Because there are only sporadic reports, there are currently no established standards for the treatment of PHCT. Primary hepatic carcinoids are associated with a resectability rate of 70%, a 74–78% 5-year survival rate, and an 18% 5-year recurrence rate [18, 19]. Better long-term disease free survival can therefore be expected after resection of PHCT than after resection of NETs of other primary origins. Postoperative intra-abdominal fluid collections and liver related complications (insufficiency or portal vein thrombosis) did have a negative impact on overall survival in one series [5]. Data are even scarcer concerning LT for PHCT. Five patients have been reported [5, 20–22] including 2 males and 3 females ranging from 35 to 50 years of age. Four patients are alive and disease free after 38, 45, 95, and 120 months. One patient had a liver and mesentery recurrence after 54 months. Based on this small data set it appears that PHCT treated by LT could have a better prognosis than liver NET metastases treated by LT [5, 20].
In patients with unresectable disease a variety of palliative options exist but data on these is very limited. Systemic 5-fluorouracil downstaged disease in 1 of 3 patients [23]. Hepatic artery embolization may also be effective because hepatic carcinoids derive their vascular supply from the hepatic artery [24]. Octreotide, a somatostatin analog, can effectively alleviate symptoms resulting from hormone secretion but may also have a direct antiproliferative effect [25]. Yttrium-90 targeted radionuclides coupled with octreotide have also shown some therapeutic effect [26].
In conclusion primary hepatic carcinoid tumors are rare and their primary nature can only be ascertained after thorough investigations and long-term follow-up to exclude another primary origin. Their large size and often central situation within the liver should not deter surgeons from attempting resection because long-term survival and cure can be expected. In selected patients not amenable to partial liver resection liver transplantation can be considered.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Maggard MA, O’Connell JB, Ko CY. Updated population-based review of carcinoid tumors. Annals of Surgery. 2004;240(1):117–122. doi: 10.1097/01.sla.0000129342.67174.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Modlin IM, Sandor A. An analysis of 8305 cases of carcinoid tumours. Cancer. 1997;79(4):813–829. doi: 10.1002/(sici)1097-0142(19970215)79:4<813::aid-cncr19>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 3.Lin C-W, Lai C-H, Hsu C-C, et al. Primary hepatic carcinoid tumor: a case report and review of the literature. Cases Journal. 2009;2(1, article 90) doi: 10.1186/1757-1626-2-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang Y-Q, Xu F, Yang J-M, Huang B. Primary hepatic neuroendocrine carcinoma: clinical analysis of 11 cases. Hepatobiliary and Pancreatic Diseases International. 2010;9(1):44–48. [PubMed] [Google Scholar]
- 5.Fenwick SW, Wyatt JI, Toogood GJ, Lodge JPA. Hepatic resection and transplantation for primary carcinoid tumors of the liver. Annals of Surgery. 2004;239(2):210–219. doi: 10.1097/01.sla.0000109155.89514.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gravante G, de Liguori Carino N, Overton J, Manzia TM, Orlando G. Primary carcinoids of the liver: a review of symptoms, diagnosis and treatments. Digestive Surgery. 2008;25(5):364–368. doi: 10.1159/000167021. [DOI] [PubMed] [Google Scholar]
- 7.Kongkam P, Al-Haddad M, Attasaranya S, et al. EUS and clinical characteristics of cystic pancreatic neuroendocrine tumors. Endoscopy. 2008;40(7):602–605. doi: 10.1055/s-2007-995740. [DOI] [PubMed] [Google Scholar]
- 8.Cerwenka H. Neuroendocrine liver metastases: contributions of endoscopy and surgery to primary tumor search. World Journal of Gastroenterology. 2012;18(10):1009–1014. doi: 10.3748/wjg.v18.i10.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Norton JA, Warren RS, Kelly MG, et al. Aggressive surgery for metastatic liver neuroendocrine tumors. Surgery. 2003;134(6):1057–1065. doi: 10.1016/j.surg.2003.07.025. [DOI] [PubMed] [Google Scholar]
- 10.Glazer ES, Tseng JF, Al-Refaie W, et al. Long-term survival after surgical management of neuroendocrine hepatic metastases. HPB. 2010;12(6):427–433. doi: 10.1111/j.1477-2574.2010.00198.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sarmiento JM, Que FG, Grant CS, et al. Concurrent resections of pancreatic islet cell cancers with synchronous hepatic metastases: outcomes of an aggressive approach. Surgery. 2002;132(6):976–983. doi: 10.1067/msy.2002.128615. [DOI] [PubMed] [Google Scholar]
- 12.Chamberlain RS, Canes D, Brown KT, et al. Hepatic neuroendocrine metastases: does intervention alter outcomes? Journal of the American College of Surgeons. 2000;190(4):432–445. doi: 10.1016/s1072-7515(00)00222-2. [DOI] [PubMed] [Google Scholar]
- 13.Scigliano S, Lebtahi R, Maire F, et al. Clinical and imaging follow-up after exhaustive liver resection of endocrine metastases: a 15-year monocentric experience. Endocrine-Related Cancer. 2009;16(3):977–990. doi: 10.1677/ERC-08-0247. [DOI] [PubMed] [Google Scholar]
- 14.Sarmiento JM, Heywood G, Rubin J, Ilstrup DM, Nagorney DM, Que FG. Surgical treatment of neuroendocrine metastases to the liver: a plea for resection to increase survival. Journal of the American College of Surgeons. 2003;197(1):29–37. doi: 10.1016/S1072-7515(03)00230-8. [DOI] [PubMed] [Google Scholar]
- 15.Rosenau J, Bahr MJ, Von Wasielewski R, et al. Ki67, e-cadherin, and p53 as prognostic indicators of long-term outcome after liver transplantation for metastatic neuroendocrine tumors. Transplantation. 2002;73(3):386–394. doi: 10.1097/00007890-200202150-00012. [DOI] [PubMed] [Google Scholar]
- 16.Le Treut YP, Delpero JR, Dousset B, et al. Results of liver transplantation in the treatment of metastatic neuroendocrine tumors: a 31-case French multicentric report. Annals of Surgery. 1997;225(4):355–364. doi: 10.1097/00000658-199704000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gedaly R, Daily MF, Davenport D, et al. Liver transplantation for the treatment of liver metastases from neuroendocrine tumors: an analysis of the UNOS database. Archives of Surgery. 2011;146(8):953–958. doi: 10.1001/archsurg.2011.186. [DOI] [PubMed] [Google Scholar]
- 18.Mizuno Y, Ohkohchi N, Fujimori K, et al. Primary hepatic carcinoid tumor: a case report. Hepato-Gastroenterology. 2000;47(32):528–530. [PubMed] [Google Scholar]
- 19.Knox CD, Anderson CD, Lamps LW, Adkins RB, Pinson CW. Long-term survival after resection for primary hepatic carcinoid tumor. Annals of Surgical Oncology. 2003;10(10):1171–1175. doi: 10.1245/aso.2003.04.533. [DOI] [PubMed] [Google Scholar]
- 20.de Liguori Carino N, Manzia TM, Tariciotti L, Berlanda M, Orlando G, Tisone G. Liver transplantation in primary hepatic carcinoid tumor: case report and literature review. Transplantation Proceedings. 2009;41(4):1386–1389. doi: 10.1016/j.transproceed.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 21.Alekseev D, Goralczyk A, Lorf T, Ramadori G, Obed A. Ten years survival with excellent outcome after living donor liver transplantation from 70 years old donor for primary hepatic neuroendocrine carcinoma: case report. International Journal of Surgery Case Reports. 2012;3(1):34–36. doi: 10.1016/j.ijscr.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arnold JC, O’Grady JG, Bird GL, Calne RY, Williams R. Liver transplantation for primary and secondary hepatic apudomas. British Journal of Surgery. 1989;76(3):248–249. doi: 10.1002/bjs.1800760311. [DOI] [PubMed] [Google Scholar]
- 23.Andreola S, Lombardi L, Audisio RA, et al. A clinicopathologic study of primary hepatic carcinoid tumors. Cancer. 1990;65(5):1211–1218. doi: 10.1002/1097-0142(19900301)65:5<1211::aid-cncr2820650530>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 24.Krishnamurthy SC, Dutta V, Pai SA, et al. Primary carcinoid tumor of the liver: report of four resected cases including one with gastrin production. Journal of Surgical Oncology. 1996;62(3):218–221. doi: 10.1002/(SICI)1096-9098(199607)62:3<218::AID-JSO13>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 25.Wängberg B, Nllsson O, Johanson V, et al. Somatostatin receptors in the diagnosis and therapy of neuroendocrine tumors. Oncologist. 1997;2(1):50–58. [PubMed] [Google Scholar]
- 26.Otte A, Mueller-Brand J, Dellas S, Nitzsche EU, Herrmann R, Maecke HR. Yttrium-90-labelled somatostatin-analogue for cancer treatment. The Lancet. 1998;351(9100):417–418. doi: 10.1016/s0140-6736(05)78355-0. [DOI] [PubMed] [Google Scholar]