

Abstract
Korean salamanders of the genus Hynobius are currently classified into 3 species, H. leechii, H. quelpaertensis, and H. yangi. To investigate the phylogenetic relationship of these species, we analyzed the partial sequence of mito-chondrial cytochrome b gene (907 bp) of 197 specimens from 43 regions in South Korea. Of these specimens, 93 were additionally examined with 12S rRNA (799 bp). Based on the partial sequence of the mitochondrial cytochrome b gene and 12S rRNA, 89 and 36 haplotypes were defined, respectively, consisting of six subclades (H. leechii, H. quelpaertensis, H. yangi, HC1, HC2, and HC3). Among the-se subclades, the three subclades (HC1, HC2, and HC3) were clearly separated from the 3 previously reported spe-cies in the genus Hynobius. Pairwise sequence divergence between the six subclades ranged from 6.3 to 11.2% in cytochrome b gene and 2.0 to 4.3% in 12S rRNA. These results indicate there may be more divergent populations than the three currently described. Moreover, the estima-tion of divergence time revealed that the Hynobius species in South Korea diverged during the Miocene epoch, ap-proximately 9 - 5 MYA. In addition, we confirmed the distri-bution of the three known species (H. leechii, H. quel-paertensis, and H. yangi) and determined the distributions of new, distinct groups (or subclades; HC1, HC1, and HC3). To more accurately establish the taxonomic status and population structure, further genetic, morphological, and ecological studies will be needed.
Keywords: 12S rRNA, Asian salamander, Hynobius, mitochondrial cytochrome b, phylogenetic relationship
INTRODUCTION
In Asia, the genus Hynobius, the largest genus in Hynobiids, consists of 32 species of salamanders (Amphibiaweb, 2010; Frost, 2009). Among the 32 species, three species are known to be distributed in South Korea: the Korean salamander (H. leechii), the Jeju salamander (H. quelpaertensis), and the Kori salamander (H. yangi) (Boulenger, 1887; Kim et al., 2003; Yang et al., 2001). Among those species, H. quelpaertensis and H. yangi are endemic to South Korea, while H. leechii is distributed in Northeast China, North Korea, and South Korea (Amphibiaweb, 2010; Kim et al., 2003; Yang et al., 2001). In South Korea, H. leechii inhabits most of the country except for Jeju Island and some parts of the southern coastal area. The Jeju salamander’s (H. quelpaertensis) range of distribution is identified as mainly Jeju island, as its name indicates, but it is also found in the southwestern coastal regions of the Korean peninsula, including Namhae, Jindo, and Haenam (Yang et al., 2001). The Kori salamander (H. yangi) so named because it was first discovered in the Kori area of Busan is reported to have a distribution quite restricted to within a 20 km2 region (Korea hydro & nuclear power CO., LTD, 2004).
Significant declines in amphibians have been reported worldwide. Nearly one-third of amphibian species are threat-ened and faced with extinction due to habitat loss, global warm-ing, and emerging diseases, such as chytridiomycosis (Batrachochytrium dendrobatidis) and Ranavirus (Iridoviridae) (Pasmans et al., 2004; 2008; Stuart et al., 2004). In South Ko-rea, major declines in amphibian populations have been pri-marily caused by human activities (Min et al., 2008) including road construction and progression of civilization. Furthermore, those factors are worked as potential physical barriers not only for amphibian population but other vertebrates (Lee et al., 2008).
The three species of Hynobius in South Korea are confronted with severe habitat destruction and fragmentation; especially, the Kori-salamander whose distribution is very limited. Con-struction of a nuclear power plant necessitated the translocation of the main population of this species from its type locality to an alternative habitat in 2006. Aside from economic growth and development, emerging disease is another great cause threat-ening amphibian populations (Stauart et al., 2004). Recently, Yang et al. (2009) revealed that chytridiomycosis, which can cause severe mortality rates in infected amphibians, was de-tected in wild populations in South Korea, indicating the need to pay more attention to amphibian conservation and protection. Despite the urgent need to understand these species better to efficiently protect and conserve them, very little information is available on the genus Hynobius thus far.
There have been several studies regarding the phylogenetic relationship of the Hynobius species in South Korea using a variety of methods, including mitochondrial DNA RFLP (Lee and Jung, 1993), chromosome banding technique (Ag NOR’s staining) (Cha and Lee, 1995), mitochondrial DNA sequence analysis (Lee et al., 1998), and isozyme electrophoresis (Kim et al., 2007; Yang et al., 1997; 2001). As a consequence of the previous studies, H. quelpaertensis and H. yangi were consid-ered unique species, separate from H. leechii (Kim et al., 2003; Yang et al., 2001).
Even though distributions of Korean Hynobius species are relatively well known, there is controversy due to the lack of diverse sampling sites in previous studies, especially in the southern parts of the Korean peninsula (Cha and Lee, 1995; Lee and Jung, 1993; Lee et al., 1998). Furthermore, it is very difficult to distinguish the three species by morphological char-acteristics alone. Thus, distributions along the southern coast and islands are unclear and need further investigation. Our study aims to: 1) clarify the species status, distribution, and marginal boundary of the species in Hynobius, 2) renew the distribution of each species in the genus Hynobius, and 3) examine molecular genetic variations and phylogenetic relation-ships among the species using mitochondrial DNA (mtDNA) cytochrome b (cyt-b) and 12S rRNA genes.
MATERIALS AND METHODS
Materials
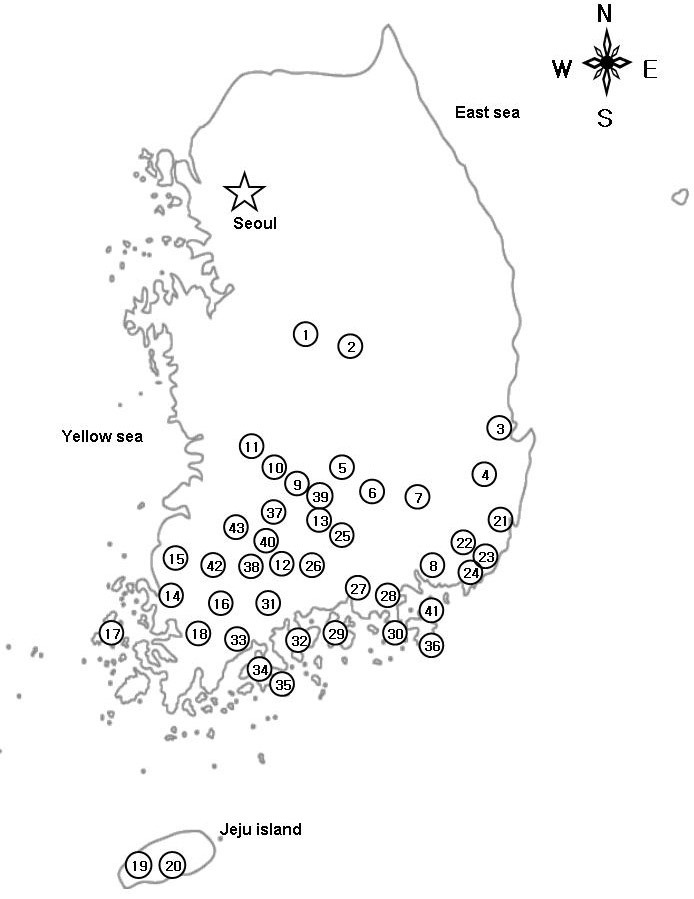
Adult salamanders were collected from 2007 to 2009. Sampling was mostly concentrated to the southern coastal regions, in-cluding several islands. A total of 197 specimens of Hynobius spp. were collected from 43 sites across the species range in the southern part of the Korean peninsula (Fig. 1). Map number, locality information, and number of individuals are listed in Sup-plementary materials. All specimens collected were euthanized by submersion in a 10% solution of MS-222 (Cat. No. A5040, Sigma-Aldrich, USA) and tissue was collected from the tongue for genetic analysis. All voucher specimens were deposited at the Conservation Genome Resource Bank for Korean Wildlife (CGRB) in Seoul National University.
Fig. 1. Sampling localities in the present study. For locality numbers, refer to Supplementary materials.
One hundred eighty two of the 197 Hynobius samples were used for analysis of mtDNA cyt-b DNA sequence variation. Additionally, we examined the 12S rRNA from 93 specimens. The sequences of mtDNA cyt-b and 12S rRNA were deposited in GenBank and the accession numbers are given in Supplementary materials. For phylogenetic analysis, we combined the mtDNA cyt-b and 12S rRNA segments and previously published sequence of H. leechii from China (GenBank accession number: NC_008079; Zhang et al., 2006) and H. quelpaertensis from Jeju island of South Korea (EF201847; Oh et al., 2007). Three species from the same genus (H. chinensis; NC_008088, H. amjiensis; NC_008076, H. arisanensis; NC_009335; Zhang et al., 2006) were used as outgroups to better resolve phylogenetic relationships among the Hynobius species collected in Korean peninsula.
DNA extraction, PCR amplification and sequencing
Whole genomic DNA was extracted from tissue samples using the DNeasy tissue kit (Cat. No. 69506, Qiagen, USA) following the manufacturer’s protocol. Six primers were applied to amplify the mtDNA cyt-b and 12S rRNA genes. The partial mtDNA cyt-b gene was amplified with a set of primers, 12F (5′ GGC CCA CCC AAT TCG AAA AAC 3′) and 12R (5′ GTT GTT CGA CTG GTT GTC CG 3′), previously designed from the complete nu-cleotide sequences from the four species in the same genus (Oh et al., 2007). Four primers were used to amplify the 12S rRNA; 1F (5′ GGT TTG GTC CTA GCC TTA CT 3′) and 1R (5′ CTT TTG CCA CAG AGA AGG GT 3′) from Oh et al. (2007), 12SL (5′ AAA GCA CGG CAC TGA AGA TGC 3′) and 12SR (5′ TTT CAT GTT TCC TTG CGG TAC 3′) from Wang et al. (2000). The PCR reaction consisted of 10-50 ng genomic DNA, 0.2 mM dNTPs, 1.5 mM MgCl2, 1 μM of each primer, 1 unit of Takara Taq™ (Cat. No. R001A, Takara, Japan), and 10 X PCR buffer in total volume of 30 μl. PCR conditions were as follows: 2 min at 94℃, 30 cycles of 10 s at 94℃, 15 s at 53-57℃, 1 min at 72℃, and an extra extension for 7 min at 72℃ (Oh et al., 2007). PCR products were checked on 1.5% agarose gels and purified with the Zymoclean Gel DNA Recovery Kit (Cat. No. D4002, Zymo research, USA). All samples were sequenced on an ABI3730XL (Applied Biosystmes, USA) according to the manufacturer’s instructions.
Phylogenetic analysis
The partial sequences of mtDNA cyt-b gene and 12S rRNA were edited and aligned using Sequencher 4.8 (Gene Codes, USA). Multiple alignments were made using CLUSTAL X 1.83 (Thompson et al., 1997). Haplotype and nucleotide diversities were calculated using DnaSP version 5.0 (Librado and Rozas, 2009). The pairwise sequence divergence between subclades was calculated by MEGA version 4 (Tamura et al., 2007) using p-distance.
DNA molecular phylogenies were reconstructed by maximum likelihood (ML) methodologies with PAUP 4.0b10* (Swofford, 2002). The best fit model of sequence evolution was chosen on the basis of Akaike information criterion (AIC) as performed in MODELTEST 3.7 (Posada and Crandall, 1998), and TVM + I + G model was selected; Base frequencies of 0.3431, 0.2339, 0.1296, and 0.2934 for A, C, G and T nucleotides respectively; Substitution rates of 1.9929, 30.5858, 2.9196, 0.8769, 30.58598, and 1 for A-C, A-G, A-T, C-G, C-T, and G-T nucleotide r-ate; Proportion of invariable sites = 0.5655; a gamma distribution shape parameter = 0.5821. The ML topologies were constructed using a heuristic search with a tree-bisection-reconnection (TBR) algorithm. The stability of internal nodes was assessed by bootstrap analysis (100 replicates).
Bayesian inferences (BI) were also performed using MR-BAYES version 3.1.2 (Ronquist and Huelsenbeck, 2003). The best fitting model substitution model for the nucleotide matrix was determined through the Akaike information criterion (AIC) implemented in MRMODELTEST version 2.02 (Nylander, 2004). The partition strategy by gene was applied and default settings were used for the prior distribution in the Bayesian inferences. Metropolis-coupled Markov chain Monte Carlo (MCMCMC) analyses were run with one cold chain and three heated chains for 4 million generations and sampled every 1,000 generations. Five independent MCMCMC runs were performed and 1,000 trees were discarded as burn-in.
Estimation of divergence times
PHYLTEST 2.0 (Kumar, 1996) was used to apply a relative rate test (Takezaki et al., 1995). The PHYLTEST examines the con-stancy of molecular clock for two lineages when an outgroup is given. We performed several tests for pairwise comparisons of the subclades observed in the phylogenetic trees. If the null hypothesis was not rejected, we estimated dates of pairwise divergence comparison of the subclades by calculating the uncorrected sequence divergence (%) between subclades. There is no reliable temporal calibration directly applicable to the genus Hynobius and we applied the evolutionary rate for Hynobiids from previous studies (1.28% per MYA; Matsui et al., 2007; Tomigawa et al., 2006; Weisrock et al., 2001).
RESULTS
Mitochondrial DNA sequence variation
The partial sequence (907 bp) of mtDNA cyt-b gene of 182 specimens, representing 89 haplotypes, divided into 6 subclades including 3 new distinct groups: Hynobius subclade1 (HC1), Hynobius subclade2 (HC2), and Hynobius subclade3 (HC3), for the genus Hynobius in South Korea. Two hundred forty five of 907 nucleotides were polymorphic and 299 were parsimony-informative.
The sequence divergence of the 89 haplotypes ranged from 6.3 to 11.2% (overall mean sequence divergence = 8.4%). Within the subclades, each subclade had an overall mean dis-tance of 1.9%, 2.0%, 0.1%, 1.0%, 0.5%, and 0.3% for H. leechii (HL), H. quelpaertensis (HQ), H. yangi (HY), Hynobius subclade1 (HC1), Hynobius subclade2 (HC2), and Hynobius subclade3 (HC3), respectively. HY, HC2, and HC3 had relative-ly low variability (less than 1.0%). Overall haplotype (h) and nucleotide diversities (π) were 0.964 ± 0.009 and 8.207% ± 0.092, respectively (Table 2). Overall haplotype diversity was slightly higher than that within the subclades (entire h = 0.964, h of each subclade = 0.433-0.970 nucleotide diversity was also higher than that in any particular subclades (overall π = 8.207%, π of each subclade = 0.148-2.011%). Among the Hynobius subclades, H. yangi had the lowest nucleotide and haplotype diversities (Table 1).
Table 2.
Matrix of pairwise sequence of mitochondrial DNA between different populations of Hynobius spp. in South Korea with the other three species of Hynobius from GenBank as outgroups (below diagonal: cytochrome b gene; above diagonal: 12S rRNA)
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
|---|---|---|---|---|---|---|---|---|---|
| [1] HL | 0.022 | 0.022 | 0.023 | 0.032 | 0.027 | 0.029 | 0.029 | 0.059 | |
| [2] HQ | 0.107 | 0.026 | 0.020 | 0.029 | 0.029 | 0.031 | 0.029 | 0.055 | |
| [3] HY | 0.063 | 0.106 | 0.029 | 0.033 | 0.030 | 0.036 | 0.037 | 0.062 | |
| [4] HC1 | 0.104 | 0.097 | 0.103 | 0.020 | 0.035 | 0.029 | 0.029 | 0.056 | |
| [5] HC2 | 0.109 | 0.094 | 0.109 | 0.068 | 0.043 | 0.039 | 0.040 | 0.068 | |
| [6] HC3 | 0.091 | 0.112 | 0.086 | 0.109 | 0.106 | 0.039 | 0.042 | 0.062 | |
| [7] H. chinensis (NC_008088) | 0.125 | 0.139 | 0.121 | 0.140 | 0.125 | 0.120 | 0.025 | 0.058 | |
| [8] H. amjiensis (NC_008076) | 0.128 | 0.138 | 0.120 | 0.140 | 0.141 | 0.132 | 0.114 | 0.061 | |
| [9] H. arisanensis (NC_009335) | 0.142 | 0.153 | 0.134 | 0.164 | 0.164 | 0.133 | 0.149 | 0.155 | |
Table 1.
Summary of sample sizes (N), number of haplotypes (n), nucleotide diversity (π), haplotype diversity (h), polymorphic sites (P), single-ton (S), and parsimony informative sites (PI) for the specimens used in this study
| Taxon | N | n | π | h | P | S | PI |
|---|---|---|---|---|---|---|---|
| Cytochrome b within species (907 bp) | |||||||
| HL | 29 | 17 | 1.921 (0.099) | 0.948 (0.023) | 57 | 10 | 47 |
| HQ | 39 | 25 | 2.011 (0.207) | 0.970 (0.013) | 69 | 10 | 59 |
| HY | 41 | 11 | 0.148 (0.059) | 0.433 (0.098) | 19 | 10 | 9 |
| HC1 | 37 | 21 | 1.014 (0.132) | 0.961 (0.014) | 54 | 18 | 34 |
| HC2 | 12 | 4 | 0.543 (0.116) | 0.712 (0.105) | 12 | 1 | 11 |
| HC3 | 24 | 11 | 0.297 (0.041) | 0.873 (0.054) | 12 | 4 | 8 |
| Hynobius_overall | 182 | 89 | 8.207 (0.092) | 0.964 (0.009) | 245 | 16 | 229 |
| 12S rRNA within species (799 bp, excluding gaps) | |||||||
| HL | 25 | 11 | 0.777 (0.040) | 0.910 (0.031) | 20 | 4 | 16 |
| HQ | 23 | 10 | 0.276 (0.028) | 0.830 (0.067) | 8 | 3 | 5 |
| HY | 12 | 2 | 0.038 (0.019) | 0.303 (0.147) | 1 | 0 | 1 |
| HC1 | 21 | 7 | 0.273 (0.067) | 0.752 (0.072) | 11 | 5 | 6 |
| HC2 | 5 | 2 | 0.076 (0.022) | 0.600 (0.175) | 1 | 0 | 1 |
| HC3 | 7 | 4 | 0.204 (0.065) | 0.810 (0.13) | 4 | 2 | 2 |
| Hynobius_overall | 93 | 36 | 2.080 (0.072) | 0.959 (0.008) | 75 | 8 | 67 |
In mtDNA 12S rRNA (799 bp) from 93 samples, we found 36 haplotypes composed of 6 major subclades. In 799 characters, seventy-five were polymorphic sites, of which 67 were parsimo-nious-informative. The sequence divergence between each subclade ranged from 2.0 to 4.3% (overall mean sequence divergence = 2.1%). Overall haplotype (h) and nucleotide diver-sities (π) were 0.959 ± 0.008 and 2.080% ± 0.072, respectively (Table 1). In each subclade, haplotype diversity ranged from 0.303 to 0.910. HL displayed the highest haplotype diversity (0.910 ± 0.031), as well as the highest nucleotide diversity (0.777% ± 0.040). For each subclade, nucleotide diversity ranged from 0.038 to 0.777%. Similar to results of the mtDNA cyt-b gene, H. yangi had the lowest nucleotide and haplotype diversities (Table 1).
The concatenated fragment of mtDNA cyt-b gene and 12S rRNA represented 1,706 bp in length. In concatenated mtDNA cyt-b (907 bp) and 12S rRNA (799 bp) gene sequences from 93 samples, we found 65 haplotype, distinguished by 6 subclades. In 1,706 characters, 311 were variable sites and of those, 281 were parsimonious-informative. Overall haplotype diversity (h) was 0.986 ± 0.005, while haplotype diversity of each subclade ranged from 0.561 to 1.00. Overall nucleotide diversity (π) was 5.350% ± 0.092 and each subclade ranged from 0.037 to 1.424% (data not shown).
Phylogenetic relationship
For the phylogenetic relationship and reconstruction of the phy-logenetic trees of Hynobius species in South Korea, combined fragment of mtDNA cyt-b and 12S rRNA genes were performed. The ML and BI showed almost the same topology with minor differences in the details. The ML reconstruction for phylogenet-ic relationships among the haplotypes is shown in Fig. 2. The phylogenetic analyses identified six subclades belonging to two monophyletic major clades (Clades I and II). Among the six distinct subclades, three corresponded to the species previous-ly reported [H. leechii (HL), H. quelpaertensis (HQ), and H. yangi (HY)], but the others, named as Hynobius subclade1 (HC1), Hynobius subclade2 (HC2), and Hynobius subclade3 (HC3), were distinct.
Fig. 2. ML tree based on 1,706 bp of the concatenated mitochon-drial genes (cyt-b + 12S rRNA). Nodal values indicate ML bootstrap value and BI posterior probability.
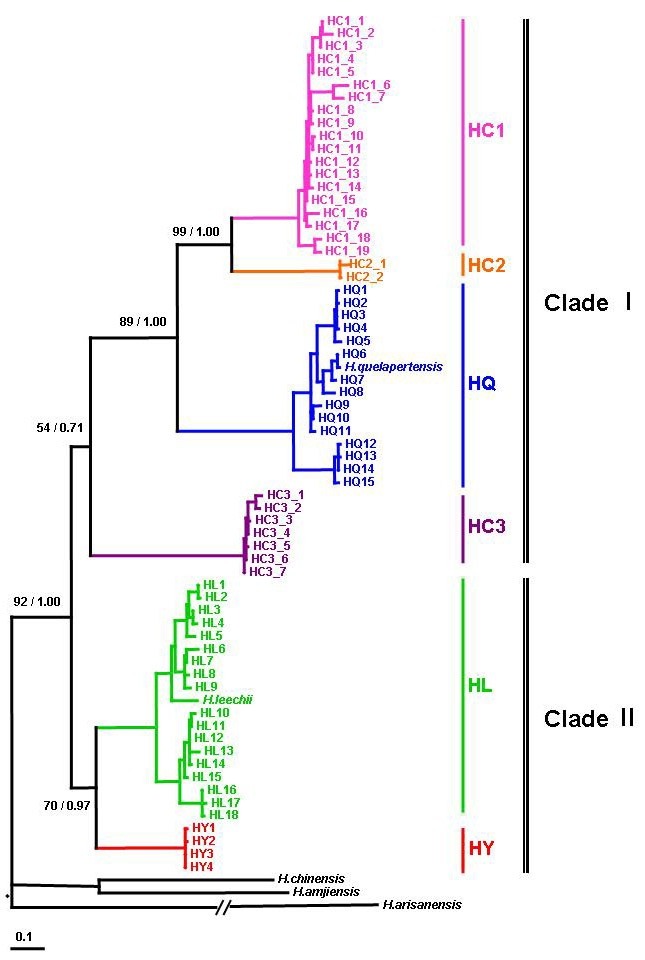
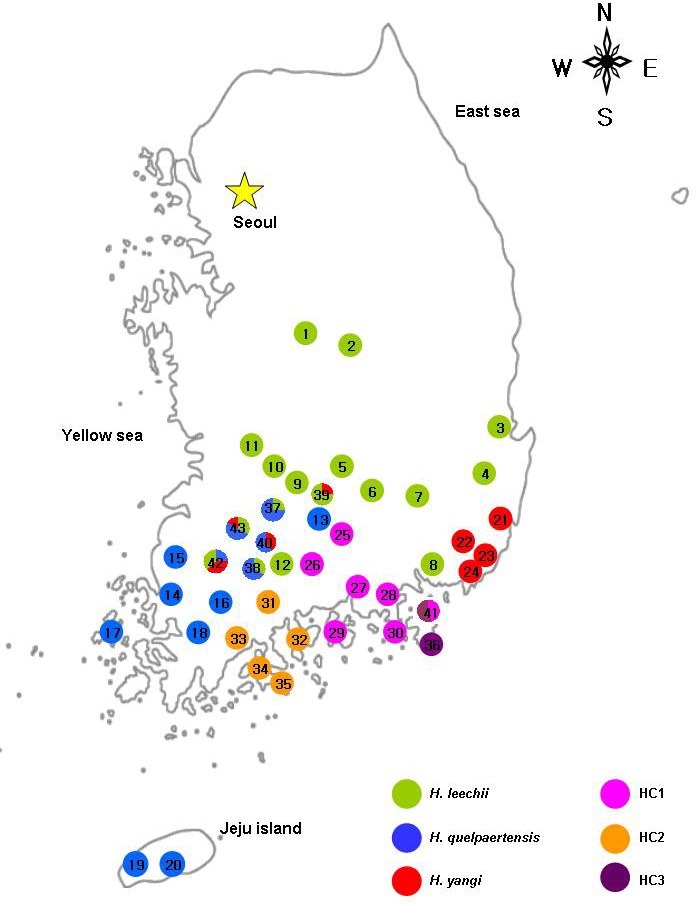
Clade I was comprised of four subclades: HQ, HC1, HC2, and HC3. HC3 was basal divergent to the rest of the subclades and HQ had a sister relationship with HC1 and HC2. In Clade I, the subclade HC2 included five specimens from Suncheon, displaying two haplotypes. Subclade HC1, distributed in the southeastern part of the Korean peninsula (Fig. 3), showed 19 haplotypes. Subclade HQ (H. quelpaertensis) covered the en-tire range of distribution in the southwestern part of the Korean peninsula as well as Jeju Island (Fig. 3), and represented 16 haplotypes. The distinct subclades, HL (H. leechii) and HY (H. yangi), belonged to Clade II and the sister relationship between HL and HY was well supported (BP = 70, PP = 0.97) having three exclusive transitions in common (position cyt-b 413: C → T, position cyt-b 767, 900: A → G). Subclade HL displayed 18 haplotypes covering the entire range for species. Subclade HY revealed four haplotypes which were distributed in Busan and the central-southern areas of Hamyang, Damyang, Namwon, and Gokseong.
Fig. 3. Distributional changes of three species (H. leechii, H. quelpaertensis, and H. yangi) and distributions of three distinct subclades in the genus Hynobius of South Korea.
To investigate possible relationships among haplotypes for each subclade, the Median-joining network method (Bandelt et al., 1999) was performed using the program Network 4.0. There were no notable results, except for the case of subclade HL in which Pohang and Gyeongju, which have small sample sizes (Fig. 2; HL1, HL2, HL3, and HL4), were slightly differentiated from other HL haplotypes (data not shown).
Estimations of divergence times
The relative rate tests indicated that the null hypothesis of rate constancy could not be rejected for any pairs of subclades. The uncorrected pairwise divergence (p-distance) between the six subclades ranged from 6.3 to 11.2% in mtDNA cyt-b gene (Table 2). The largest p-distance of 11.2% was observed between HQ and HC3; whereas HL and HY represented the smallest divergence of 6.3%. Adopting the molecular calibration of 1.28% per 1 million years ago (MYA) in salamanders (Matsui et al., 2007; Tomigawa et al., 2006; Weisrock et al., 2001), the estimation of divergence times between Clade I and Clade II were approximately 9 MYA. In Clade I, the subclades diverged from each other about 9 to 5 MYA and Clade II, HL and HY diverged about 5 MYA.
DISCUSSION
The taxonomic understanding of the genus Hynobius is contin-ually improving. Since 2000, there have been some reports regarding new species in the genus Hynobius (H. fuca and H. glacialis, Lai and Lue, 2008; H. guabangshanensis, Shen et al., 2004; H. katoi, Matsui et al., 2004). Recently, in South Korea, H. quelpaertensis and H. yangi were identified as new species (Kim et al., 2003; Yang et al., 2001). Morphological comparison revealed that the three species, H. leechii, H. quelpaertensis, and H. yangi, have the different shape and number of vomerine teeth and coccyx. H. leechii has 31 to 36 of vomerine teeth and 26 to 30 of coccyx while H. quelpaertensis has large number of vomerine teeth (37-42) than H. leechii. In addition, H. yangi displayed the ‘V’ shape of vomerine teeth and had fewer num-bers of coccyx (25 to 26) (Kim et al., 2003; Yang et al., 1997; 2001). Even though H. yangi has the smallest SVL (Snout-Vent Length) among the three species, in general it is hard to distinguish the three species in the genus Hynobius in South Korea using external morphology alone due to similar color patterns. In a situation where it is very difficult to identify species using morphological characteristics, genetic tools can be helpful in resolving the issue of explicit species status (Fu and Zeng, 2008; Fu et al., 2003).
In present study, we identified that three known species of the Korean Hynobius (H. leechii, H. quelpaertensis, and H. yangi) were well differentiated as well as the three cryptic subclades (HC1, HC2, and HC3) were observed with a relative large se-quence divergence (Fig. 2 and Table 2). Except for some regions the six subclades were allopatrically distributed (Fig. 3). This allopatrical distribution could have been affected through geographic isolation and/or behavior characteristic. Salaman-ders are known as having low vagility and physical barriers such as mountains, deserts, and rivers can prevent their dispersal (Martínez-Solano et al., 2007; Zhang et al., 2006). Beside, the Korean peninsular consists of many mountains and rivers (Kim et al., 2008), which might have caused so diverged Hynobius subclades in South Korea. Unfortunately, we can not discuss the process and specific events that are involved with the spe-ciation of the Korean Hynobius examined in the present study.
With the reconstruction of a phylogenetic tree based on ML and BI analyses, two monophyletic clades were observed in the Korean Hynobius species (Fig. 2); one consisted of the previ-ously recognized species, H. leechii and H. yangi, while the other clade was comprised of H. quelpaertensis and three other distinct subclades (HC1, HC2, and HC3) first found in the present study. In the phylogenetic tree, H. leechii and H. yangi demonstrated a sister group relationship and were a monophy-letic clade (Clade II, Fig. 2), while H. quelpaertensis and the three other distinct subclades represented monophyletic rela-tionships (Clade I, Fig. 2). These results coincide with those of Yang et al. (1997), implying the presence of two monophyletic clades of the Korean Hynobius species: one composed of H. leechii and H. yangi which have a sister group relationship whereas the other consisting of H. quelpaertensis.
The distinct subclades (HC1, HC2, and HC3) were identified mostly in peripheral populations at the end of the distribution area and islands. Those subclades were allopatrically or sympatrically distributed in these regions (Fig. 3). For examples, subclade HC2, found in Suncheon, Yeosu, Boseong, and Goheung (Podu-myeon and Naro islands) was clearly isolated in the distribution from other subclades (Fig. 3, marked with a orange circle). Subclade HC3 was identified on Geoje Island, Dongbu-myeon, and Geoje-myeon (Fig. 3, marked with a pur-ple circle) but the haplotypes in subclade HC1 were observed in Geoje-myeon. Subclade HC1 was widely distributed in Gyeong- sangnam-do, allopatrically located in the southeastern part of South Korea (Hadong, Namhae, Goseong, Sacheon, Sancheong, and Tongyeong) except for one region, Geoje-myeon in Geoje (Fig. 3, marked with a pink circle).
Up to now there have been several studies describing the Korean Hynobius species to be more divergent than compared to their current taxonomic status. Lee and Jung (1993), using restriction fragment length polymorphism (RFLP) analysis, re-ported that Hynobius salamanders in Hadong have low genetic similarities with other inland populations (proportion of shared restriction fragments (F) between Hadong population and oth-ers: 0.458-0.569, average F = 0.669). Along with Lee and Jung’s (1993) findings, our results of mtDNA sequence variation de-fined the Hadong population as a distinct subclade, HC1, split from HC2 with good nodal supports (bootstrap value = 99% and posterior probability = 1.00). Based on allozyme analysis using 23 loci, Yang et al. (1997) designated the salamanders in Geoje and Namhae as genetically distinct forms, and the Rog-er’s genetic similarity coefficient between Geoje population and H. quelpaertensis as 0.858. In the UPGMA dendrogram, those types were not clustered with any Hynobius species in South Korea. However, Yang et al. (1997) determined the population from Namhae and Geoje to be H. quelpaertensis. Our study confirmed the Geoje and Namhae populations as distinct subclades (HC3 and HC1). In addition, pairwise dis-tances between HC3, HC1, and other subclades showed high values, ranging from 6.8 to 11.2%. The both studies revealed the highly divergent groups, implying that these groups could be considered as a cryptic species with more detailed studies.
We found a considerable amount of genetic variability within the genus Hynobius in South Korea. In mtDNA cyt-b gene, the interspecific sequence divergence in several Hynobius species varied from 3.2 to 9.8% in p-distance (Lai and Lue, 2008). Lai and Lue (2008) displayed a p-distance between new Hynobius species (H. formosanus and H. glasialis) and other Hynobius species 9.8-11.1%. Furthermore, the same locus for other Hynobius species, Lee et al. (1998) proposed a 7.8% in K2P between H. leechii and H. yangi. In the present study, the pair-wise comparison showed that the sequence divergence (p-distance) between H. leechii and HC1 (10.4%); H. leechii and HC2 (10.9%); H. leechii and HC3 (9.1%) is higher than that between H. leechii and H. yangi (p-distance = 6.3%; K2P = 6.8%, data not shown). These rates are higher than levels ordi-narily found between different species in the genus Hynobius [K2P > 7.8%, Lee et al. (1998); 12-13% in p-distance between H. amjiensis and H. yiwuensis, Fu et al. (2003); 5.9-6.5% between H. formosanus and H. glasialis, 9.9-10.6% in p-distance between H. arisanensis and H. fuca Lai and Lue (2008)]. According to the high pairwise sequence divergence, we assumed the presence of more divergent populations of the genus Hynobius in South Korea.
Because of similar morphological characters and small sam-ple size of Korean Hynobius species in previous studies, distri-butions of the three reported species (H. leechii, H. quelpaertensis, and H. yangi) were unclear, notably in the southern part of the Korean peninsula. Therefore, the present study highlight-ed the current distribution of those species previously described in the literature especially in peripheral areas and islands in the southern part of the Korean peninsula. With the results of the partial mtDNA cyt-b gene for 197 specimens from 43 regions, the present study revealed that the three species were distrib-uted throughout a wider range than had been previously re-ported. Moreover, we found the three species (H. leechii, H. quelpaertensis, and H. yangi) to be sympatric in Hamyang, Damyang, Namwom, and Gokseong (Fig. 3).
H. quelpaertensis mainly occurred in Jeju Island and several areas in southwestern parts of the Korean peninsula: Buan, Jangseong, Haenam, Jindo, Namhae, and Geoje (Kim, 2009; Yang et al., 1997; 2001). According to our results, H. quelpaer-tensis is widely distributed in Jeolla-do: Muan, Jangseong, Hwasun, Yeongam, Heuksan island, Gokseong, Damyang, and Namwon. In Gokseong, we found the sympatric distribution of H. leechii and H. quelpaertensis (Gyeom-myeon); H. yangi and H. quelpaertensis (Jukgok-myeon). Furthermore, in Damyang and Namwon the three species, H. leechii, H. quelpaertensis, and H. yangi, were distributed sympatrically. We also identified the H. quelpaertensis in Hamyang (Byeonggok-myeon) in Gyeongsangnam-do. However, in Namhae and Geoje, previ-ously described the presence of H. quelpaertensis, we found the distinct subclades of HC1 and HC3. Along with the conse-quences of Yang et al. (1997) and our results, we suggested that the salamanders in those areas be considered as HC1 and HC3.
H. yangi has been known as having a restricted habitat within 20 km2 from its type locality (Korea hydro & nuclear power CO., LTD, 2004). However, our study is the first to report populations of H. yangi outside of this area. It is necessary to confirm whether or not H. yangi exists in regions between its type locali-ty and the collection sites of this study. To elucidate the species range and the accurate population structure of H. yangi, more detailed research with extensive sampling and molecular stud-ies using microsatellite or nuclear DNA sequences will be indis-pensable. Nuclear power plant construction has been required translocation projects since 2006, which resulted in this species being the most vulnerable and well-known of the Korean Hynobius species. When compared with genetic diversity indices in other Hynobius species, the estimated genetic diversity (π = 0.148%) of H. yangi was significantly lower (Table 1). Even though the existence of this species in area outside the distribu-tion range recognized previously was documented in this study, the very limited distribution, significantly low genetic diversity and anthropogenic activities demand more attention with regard to its conservation. With more intensive research this species may be designated as a protected species.
According to the estimation of divergence time, the Hynobius species in South Korea diverged from a common ancestor approximately 9 MYA (late Miocene epoch) and H. leechii from H. quelpaertensis around 5.3 MYA (early Pliocene Epoch). However, Kim et al. (2007) estimated the divergence time be-tween H. leechii and H. quelpaertensis at about 4.3 MYA based on allozyme data. This difference may result from characteris-tics of mtDNA, high mutation fixation and fast evolution (Wan et al., 2004).
The extension of sedimentary basin was terminated, causing rising of block in South Korea during the Miocene (Son et al., 2007) when the Korean Hynobius species diverged. Later in Pliocene, the Amurian plate constructed top-to-west thrusts, and the Korea Strait shelf experienced tectonic tilting and sub-sidence (Park et al., 2000; Son et al., 2007). The asymmetrical tectonic movement since Miocene has produced the high mountains in the Taebaek mountain range (located along the eastern edge of the Korean peninsula), and the low mountains and hill areas on the west coast side of the Korean peninsula (Kim et al., 2008). Therefore, we presume that the uplifting of the mountain range may have acted as a physical barrier isolat-ing Hynobius populations leading to the diversification of the Korean Hynobius species. For an enhanced understanding of the evolutionary history of the genus Hynobius in South Korea, more complex research and geographical studies are needed.
Salamanders have very conserved morphological characters, and their genetic diversity, based on DNA sequences, is often higher than revealed by morphology alone. As a consequence, genetic studies are essential for revealing potential cryptic spe-cies (Fu et al., 2003; Martínez-Solano et al., 2007). In the pre-sent study, using mtDNA cyt-b gene and 12S rRNA, we suc-cessfully determined species identification and phylogenetic relationships among Hynobius species in South Korea. We reviewed the distribution of H. leechii, H. quelpaertensis, and H. yangi and found sympatry among the three species in certain regions in South Korea.
In addition, we defined the sister group relationship of H. leechii (HL) and H. yangi (HY) and revealed three distinct subclades (HC1, HC2, and HC3) whose pairwise sequence divergence was > 7.3% in mtDNA cyt-b gene and > 2.0% in 12S rRNA. Vieites et al. (2009) classify the candidate species into three categories: Unconfirmed Candidate Species (UCS), Confirmed Candidate Species (CCS), and Deep Conspecific Lineage (DCL). Among those categories, UCS is defined as that in which “…The genetic differentiation must be above a threshold value typical for comparisons among closely related species in the group of animals under study. Data deficient for morphology, ecology, and distribution”. Therefore, the large sequence divergence of three distinct subclades (HC1, HC2, and HC3) might be UCS for new taxa (H. sp1, H. sp2, and H. sp3) of the genus Hynobius in South Korea. However, to en-sure a complete evolutionary history and better determine how many species of the genus Hynobius are present in South Korea, further studies are necessary using morphological, ecological, and other genetic tools, such as microsatellites and nuclear DNA sequences.
Note: Supplementary information is available on the Molecules and Cells website (www.molecells.org).
Acknowledgments
This research was supported by the Korea Research Foundation Grant funded by the Korean Government (Ministry of Education and Human Resources Development; KRF-2007-313-C00503).
References
- 1.Bandelt H.J., Forster P., Röhl A. Median-joining net-works inferring intraspecific phylogenies. Mol. Biol. Evol. (1999);16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- 2.Bonett R.M., Chippindale P.T. Speciation, phylo-geography and evolution of life history and morphology in plethodontid salamanders of the Eurycea multiplicata complex. Mol. Ecol. (2004);13:1189–1203. doi: 10.1111/j.1365-294X.2004.02130.x. [DOI] [PubMed] [Google Scholar]
- 3.Boulenger G.A. Description of a new tailed batachian from Corea. Ann. Mag. Nat. Hist. Ser. (1887);5:19–67. [Google Scholar]
- 4.Cha S.H., Lee H.Y. NORs variation of Hynobius leechii. Korean J. Genet. (1995);17:87–98. [Google Scholar]
- 5.Frost D.R. Amphibian Species of the World: an online refer-ence. Version 5.3. American Museum of Natural History; New York, USA.: (2009). [(12 February, 2009)]. [Google Scholar]
- 6.Fu J., Zeng X.M. How many species are in the genus Batrachuperus? A phylogeographical analysis of the stream salamanders (family Hynobiidae) from Southwestern China. Mol. Ecol. (2008);17:1469–1488. doi: 10.1111/j.1365-294X.2007.03681.x. [DOI] [PubMed] [Google Scholar]
- 7.Fu J., Hayes M.H., Liu Z.J., Zeng X.M. Genetic divergence of the Southeastern Chinese salamanders of the genus Hynobius. Dong Wu Xue Bao. (2003);49:585–591. [Google Scholar]
- 8.Kim J.B. Taxonomic list and sitribution of Korean Amphibians. Korean J. Herpetol. (2009);1:1–13. [Google Scholar]
- 9.Kim J.B., Min M.S., Matsui M. A new species of lentic breeding Korean salamander of the genus Hynobius (Amphibia, Urodela). Zool. Sci. (2003);20:1163–1169. doi: 10.2108/zsj.20.1163. [DOI] [PubMed] [Google Scholar]
- 10.Kim J.B., Matsui M., Nishikawa K. Genetic relation-ship among salamanders of the genus Hynobius (Amphibia, Caudata) from Korea and Southwestern Japan. Zool. Sci. (2007);24:1128–1133. doi: 10.2108/zsj.24.1128. [DOI] [PubMed] [Google Scholar]
- 11.Kim J.W., Lee M.B., Kong W.S., Kim T.H., Kang C.S., Park B.I., Park H.D., Sung H.H., Son M.Y., Yang H.K., et al. Physical Geography of Korea. Seoul National University Press; Seoul, South Korea: (2008). pp. 19–29. [Google Scholar]
- 12.Korea hydro & nuclear power CO., LTD. Status for strategy of Hynobius yangi. (2004)
- 13.Kumar S. PHYLTEST. Version 2.0. Pennsylvania State University; University Park: (1996). [Google Scholar]
- 14.Lai J.S., Lue K.Y. Two new Hynobius (Caudata: Hynobiidae) salamanders from Taiwan. Herpetologica. (2008);64:63–80. [Google Scholar]
- 15.Lee H.Y., Jung Y.K. Genetic differentiation of Korean salamander. Korean J. Zool. (1993);36:14–20. [Google Scholar]
- 16.Lee H.Y., Kim Y.R., Yang D.E., Yang S.Y. The genetic differentiation of the mitochondrial cytochrome b gene of Korean salamander. Korean J. Genet. (1998);20:155–162. [Google Scholar]
- 17.Lee M.Y., Lissovsky A.A., Park S.K., Obolenskaya E.V., Dokuchaev N.E., Zhang Y.P., Yu L., Kim Y.J., Voloshina I., Myslenkov A., et al. Mitochondrial cytochrome b sequence variations and population structure of Siberian chipmunk (Tamias sibiricus) in Northeastern Asia and population substructure in South Korea. Mol. Cells. (2008);26:566–575. [PubMed] [Google Scholar]
- 18.Librado P., Rozas J. DnaSP v5: a software for com-prehensive analysis of DNA polymorphism data. Bioinformatics. (2009);25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 19.Martínez-Solano I., Jockusch E.L., Wake D.B. Extreme population subdivision throughout a continuous range: phylogeography of Batrachoseps attenuatus (Caudata: Plethodontidae) in Western North America. Mol. Ecol. (2007);16:4335–4355. doi: 10.1111/j.1365-294X.2007.03527.x. [DOI] [PubMed] [Google Scholar]
- 20.Matsui M., Kokuryo Y., Misawa Y., Nishikawa K. A new species of salamander of the genus Hynobius from Central Honshu, Japan (Amphibia, Urodela). Zool. Sci. (2004);21:661–669. doi: 10.2108/zsj.21.661. [DOI] [PubMed] [Google Scholar]
- 21.Matsui M., Tominaga A., Hayashi T., Misawa Y., Tanabe S. Phylogenetic relationships and phylogeography of Hynobius tokyoensis (Amphibia: Caudata) using complete se-quences of cytochrome b and control region genes of mitochondrial DNA. Mol. Phylogenet. Evol. (2007);44:204–216. doi: 10.1016/j.ympev.2006.11.031. [DOI] [PubMed] [Google Scholar]
- 22.Min M.S., Park S.K., Che J., Sik D.S., Lee H. Genetic diversity among local populations of the gold-spotted pond frog, Rana plancyi chosenica (Amphibia: Ranidae), assessed by mitochondrial cytochrome b gene and control region sequences. Korean J. Syst. Zool. (2008);24:25–32. [Google Scholar]
- 23.Nylander J.A.A. MRMODELTEST, Version 2.1. Computer program distributed by the author. Uppsala University; Uppsala, Sweden: (2004). [Google Scholar]
- 24.Oh D.J., Chang M.H., Oh H.S., Jung Y.H. The complete mitochondrial DNA sequence of the Jeju salamander, Hynobius quelpaertensis, and the phylogenetic relationships among the Hynobiidae. Korean J. Genet. (2007);29:331–341. [Google Scholar]
- 25.Park S.C., Yoo D.G., Lee C.W., Lee E.I. Last glacial sea-level changes and paleogeography of the Korea (Tsushima) Strait. Geo-Marine Lett. (2000);20:64–71. [Google Scholar]
- 26.Pasmans F., Zwart P., Hyatt A.D. Chytridiomycosis in the Central American bolitoglossine salamander (Bolitoglossa defleini). Vet. Rec. (2004);154:153. doi: 10.1136/vr.154.5.153. [DOI] [PubMed] [Google Scholar]
- 27.Pasmans F., Blahak S., Martel A., Pantchev N., Zwart P. Ranavirus-associated mass mortality in imported red tailed knobby newts (Tylototriton kweichpwensis): a case report. Vet. J. (2008);176:257–259. doi: 10.1016/j.tvjl.2007.02.028. [DOI] [PubMed] [Google Scholar]
- 28.Posada D., Crandall K.A. MODELTEST: Testing the model of DNA substitution. Bioinformatics. (1998);14:817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- 29.Poyarkov N.A., Kuzmin S.L. Phylogeography of the Siberian Newt Salamandrella keyserlingii by mitochondrial DNA sequence analysis. Russ. J. Genet. (2008);44:1089–1100. [PubMed] [Google Scholar]
- 30.Ronquist F., Huelsenbeck J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. (2003);19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- 31.Shen Y.H., Deng X.J., Wang B. A new hynobiid spe-cies Hynobius guabangshanensis from Hunan province, China (Amphibia: Hynobiidae). Dong Wu Xue Bao. (2004);50:209–215. [Google Scholar]
- 32.Skerratt L.F., Berger L., Spear R., Cashins S., McDonald K.R., Phillott A.D., Hines H.B., Kenyon N. Spread of chytridiomycosis has caused the rapid global decline and extinction of frogs. Ecohealth. (2007);4:125–134. [Google Scholar]
- 33.Son M., Kim J.S., Chong H.Y., Lee Y.H., Kim I.S. Characteristics of the Cenozoic crustal deformation in SE Korea and their tectonic implications. Korean J. Petrol. Geol. (2007);13:1–16. [Google Scholar]
- 34.Stuart N.S., Chanson J.S., Cox N.A., Young B.E., Rodrigues A.S.L., Fischman D., Waller R.W. Status and trends of amphibian declines and extinction worldwide. Science. (2004);306:1783–1786. doi: 10.1126/science.1103538. [DOI] [PubMed] [Google Scholar]
- 35.Swofford D.L. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Sinauer Associates; Sunderland, Massachusetts, USA: (2002). [Google Scholar]
- 36.Takezaki N., Rzhetsky A., Nei M. Phylogenetic test of the molecular clock and linearized trees. Mol. Biol. Evol. (1995);12:823–833. doi: 10.1093/oxfordjournals.molbev.a040259. [DOI] [PubMed] [Google Scholar]
- 37.Thompson J.D., Gibson T.J., Plewniak F., Jeanmougin F., Higgins D.G. The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. (1997);25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tominaga A., Matsui M., Nishikawa K., Tanabe S. Phylogenetic relationships of Hynobius naevius (Amphibia: Cau-data) as revealed by mitochondrial 12S and 16S rRNA genes. Mol. Phylogenet. Evol. (2006);38:677–684. doi: 10.1016/j.ympev.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 39.Vieites D.R., Wollenberg K.C., Andreone F., Köhler J., Glaw F., Vences M. Vast underestimation of Madagascar’s biodiversity evidenced by an integrative amphibian inventory. Proc. Natl. Acad. Sci. USA. (2009);106:8267–8272. doi: 10.1073/pnas.0810821106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wan Q.H., Wu H., Fugihara T., Fang S.G. Which genetic marker for which conservation genetics issue? Electrophoresis. (2004);25:2165–2176. doi: 10.1002/elps.200305922. [DOI] [PubMed] [Google Scholar]
- 41.Wang H.Y., Tsai M.P., Tu M.C., Lee S.C. Universal primers for amplification of the complete mitochondrial 12S rRNA gene in vertebrates. Zool. Stud. (2000);39:61–66. [Google Scholar]
- 42.Weisrock D.W., Macey J.R., Ugurtas I.H., Larson A., Papenfuss T.J. Molecular phylogenetics and historical biogeography of the “true” salamander clade: Rapid branching of numerous highly divergent lineages in Mertensiella luschani associated with the rise of Anatolia. Mol. Phylogenet. Evol. (2001);18:434–448. doi: 10.1006/mpev.2000.0905. [DOI] [PubMed] [Google Scholar]
- 43.Yang S.Y., Kim J.B., Min M.S., Suh J.H., Suk H.Y. Genetic and phenetic differentiation among three forms of Kore-an salamander Hynobius leechii. Korean J. Biol. Sci. (1997);1:247–257. [Google Scholar]
- 44.Yang S.Y., Kim J.B., Min M.S., Suh J.H., Kang Y.J. Monograph of Korean Amphibia. Academy book; Seoul, Korea: (2001). [Google Scholar]
- 45.Yang H.J., Baek H.J., Spear R., Webb R., Park S.K., Kim T.H., Lasater K.C., Shin S.P., Son S.H., Park J.H., et al. First detection of the amphibian chytrid fungus, Batrachochytridium dendrobatidis, in wild populations of Korean amphibians. Dis. Aquat. Org. (2009);86:9–13. doi: 10.3354/dao02098. [DOI] [PubMed] [Google Scholar]
- 46.Zeng X., Fu J. Low genetic diversity in Chinese Hynobius leechii, with comments in the validity of Hynobius mant-churicus. Amphibia-Reptile. (2004);25:119–122. [Google Scholar]
- 47.Zhang P., Chen Y.Q., Zhou H., Liu Y.F., Wang X.L., Papenfuss T.J., Wake D.B., Qu L.H. Phylogeny, evolution, and biogeography of Asiatic salamanders (Hynobiidae). Proc. Natl. Acad. Sci. USA. (2006);103:7360–7365. doi: 10.1073/pnas.0602325103. [DOI] [PMC free article] [PubMed] [Google Scholar]