

Abstract
The effectiveness of an apoptosis-targeting therapy may be limited in tumor cells with defects in apoptosis. Recently, considerable attention in the field of cancer therapy has been focused on the mammalian rapamycin target (mTOR), inhibition of which results in autophagic cell death. In our study using multidrug-resistant v-Ha-ras-transformed NIH3T3 (Ras-NIH 3T3/Mdr) cells, we demonstrated that rapamycin-induced cell death may result from 2 different mechanisms. At high rapamycin concentrations (≥ 100 nM), cell death may occur via an autophagy-dependent pathway, whereas at lower concentrations (≤10 nM), cell death may occur after G1-phase cell cycle arrest. This effect was accompanied by upregulation of p21Cip1 and p27Kip1 expression via an autophagy-independent pathway. We also tested whether inhibition of mTOR with low concentrations of rapamycin and ectopic Beclin-1 expression would further sensitize multidrug resistance (MDR)-positive cancer cells by upregulating autophagy. Rapamycin at low concentrations might be insufficient to initiate autophagosome formation in autophagy but Beclin-1 overexpression triggered additional processes downstream of mTOR during G1 cell cycle arrest by rapamycin. Our findings suggest that these combination strategies targeting autophagic cell death may yield significant benefits for cancer patients, because lowering rapamycin concentration for cancer treatment minimizes its side effects in patients undergoing chemotherapy.
Keywords: autophagy, Beclin 1, chemotherapy, MDR, Rapamycin
INTRODUCTION
The efficacy of anticancer therapy in the clinic is often hampered by the acquisition of multidrug resistance (MDR) by cancer cells. This loss of efficacy is correlated predominantly with the overexpression of P-glycoproteins that actively efflux chemotherapeutic drugs (Gottesman, 2002). The development of compounds that block P-glycoprotein-mediated efflux has been the conventional approach used to overcome MDR (Wu et al., 2008). However, inhibitors of P-glycoproteins do not efficiently increase the sensitivity of chemotherapeutic drugs because the mechanisms leading to MDR are complex. In fact, many tumors have mutations that confer resistance to apoptosis (Mashima and Tsuruo, 2005), which is crucial to suppressing tumorigenesis. Therefore, alternative approaches have been suggested for exploiting non-apoptotic cell death modes for anticancer therapy in cases of MDR. One potential mechanism is the autophagic pathway (Moretti et al., 2007). Kim et al. (2008) found that autophagy increases the cytotoxicity of irradiation in apoptosis-deficient cells, demonstrating the ability of autophagy to overcome resistance to apoptosis in cancer cells.
Autophagy is a complex cellular process with a dual role (Sir and Ou, 2010). Enhanced cell death has been reported in the absence of gene products essential for autophagy, suggesting a role for this process in cell survival (Yue et al., 2003). Conversely, dysregulation of mammalian target of rapamycin (mTOR), a major negative regulator of autophagy, is frequently observed in cancer. Thus, mTOR inhibition has been proposed recently as a strategy for cancer therapy (Marinov et al., 2007). Clinical data suggest that mTOR inhibitors may induce tumor regression in some patients (Dancey, 2005; Panwalkar et al., 2004). Rapamycin (sirolimus), one of the main mTOR inhibitors used in clinical trials (Hartford and Ratain, 2007), has shown significant chemotherapeutic antitumor activity in various models (Kim et al., 2008; Weppler et al., 2007). Moreover, studies have established that rapamycin can downregulate and disable the P-glycoprotein pump (Pop et al., 2009) and enhance cellular drug uptake in cells overexpressing P-glycoprotein (Pawarode et al., 2007).
Owing to the relatively low cytotoxic activity of the mTOR inhibitors, many clinical trials have been performed with this class of compounds in combination with other anticancer agents to achieve better therapeutic outcomes (Meric-Bernstam and Gonzalez-Angulo, 2009). The present study evaluated the in vitro cytotoxic activity of rapamycin in combination with ectopic expression of Beclin-1, which is one component of a complex that includes the class III phosphotidylinositol-3-kinase that stimulates autophagy. Drug-resistant v-Ha-ras transformed NIH 3T3 (Ras-NIH 3T3/Mdr) cells were used to test the therapeutic efficacy and mechanism of action of combining Beclin-1 and rapamycin. We clearly showed the concentration-dependent effects of rapamycin on autopahgy induction and growth inhi-bition. Further we demonstrated that Beclin-1 ectopic expres-sion led to increased rapamycin sensitivity, indicating synergy in eliminating MDR-positive transformed cells.
MATERIALS AND METHODS
Antibodies and reagents
Rabbit polyclonal anti-MDR, anti-p21Cip1 and anti-p27Kip1 were obtained from Santa Cruz Biotechnology (USA). Apoptosis kit was purchased from Roche Molecular Biochemicals (Indianapolis, IN). Dulbecco’s modified Eagle’s medium (DMEM), fetal calf serum (FCS) and penicillin-streptomycin were purchased from GIBCO-Invitrogen (USA). Reagents for SDS-polyacrylamide gel electrophoresis were from Bio-Rad (USA). Rapamycin, Paclitaxel, monodansylcadaverine (MDC) and 3-methyl-adenine (3-MA) were obtained from Sigma (USA).
Mammalian cell culture, transient transfection and chemical treatment
Multidrug-resistant Ras-NIH 3T3/Mdr was derived from v-Ha-ras transformed NIH 3T3 cell line (Ras-NIH 3T3), which show morphologically transformed foci of cells with the characteristics of crisscrossed margins, piling up properties and invasiveness (Lee et al., 2009), by stepwise increased concentrations of paclitaxel. The Ras-NIH 3T3 cells and its drug-resistant counterpart were maintained at 37℃ in DMEM supplemented with 10% FCS, penicillin-streptomycin, and glutamine. Before experimental use, Ras-NIH 3T3/Mdr cells were maintained in a paclitaxel-free culture medium and subcultured at least three times. MEFs WT and Atg5-/- were kindly given by Dr. Young Sang Kim (Chungnam National University, Korea) with Dr. Noboru Mizushima’s (Tokyo Medical and Dental University, Japan) permission (Kuma et al., 2004). Where indicated the cells were transiently transfected with vector expressing the fulllength Beclin 1 (OriGene Technologies, Inc., USA) or pmCherry-Atg5 (Addgene, USA), using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. After 24 h, the transfected cells were serum-deprived overnight prior to treatment with chemical. For Atg5 knockdown, Atg5 siRNA and a non-targeting siRNA were from Santa Cruz Biotechnology (USA). RT-PCR primer for detecting Atg5 knockdown was also from Santa Cruz Biotechnology.
Quantitation of autophagy
The cells were grown on chamber slides (Nunc), washed with PBS, and fixed in 10% formalin solution for 10 min. Autophagic vacuoles were labeled with MDC by incubating cells with 0.05 mM MDC in PBS at 37℃ for 10 min. After incubation, cells were washed four times with PBS and immediately analyzed by a ZeissAxio Scope.A1 epifluorescence microscope (Carl Zeiss MicroImaging, Inc., USA). Also, cells were transfected with pEGFP-LC3 (Addgene, USA) for 48 h followed by the treat-ment with chemicals. To quantify autophagy, fixed cells were classified as cells with predominantly diffuse GFP-LC3 fluore-scence or punctate GFP-LC3 pattern. The autophagic index was determined as the percentage of MDC-labeled or GFP-positive cells out of 200 cells from each treatment group.
Cell cycle assay
Ras-NIH 3T3 cells/Mdr cells were washed with PBS once, trypsinized, and collected by centrifugation at 400 × g for 5 min. The cells (106 cells per sample) were fixed with 70% ethanol, and stained with 50 μg/ml propidium iodide (Becton Dickinson) for 5 min. Cell cycle distribution was examined by measuring DNA content using a FACSCalibur-A flow cytometer and CellQuest Pro software (BD Biosciences, USA). A minimum of 104 cells per data point was examined.
Cell growth assay
The cell proliferation reagent WST-1 was used for the quantita-tive determination of cellular proliferation and activation (Roche Molecular Biochemicals, Germany). For proliferation assays, the cells were plated in quadruplicates into 96-well microliter plates (Costar, USA) at 5 × 103 cells/well and then treated with paclitaxel at 37℃ in a humidified 5% CO2/95% air incubator. After incubation for 2 or 3 days, the cells were incubated for additional 4 h in the presence of WST-1 labeling mixture (10 μl per well). The absorbance of the samples against a back-ground control (medium alone) as a blank was measured at 450 nm using a microliter plate (ELISA) reader (Molecular Devices, USA).
Clonogenic assay of cells in vitro
Ras-NIH 3T3/Mdr cells grown as monolayer cultures were harvested using a standard trypsinization procedure and were pipetted to produce a single cell suspension. Live cell counts were obtained by Trypan blue dye exclusion assay. Cells were plated at a low density (200 viable cells per well) in quadruplicate in 24-well culture plates without or in the presence of rapamycin. Seven days later, the cells were fixed with methanol and stained with 5% Giemsa solution, and colonies of more than 20 cells were scored.
Caspase-3 activity for apoptosis assay
Activation of caspase-3 was determined by detection of the chromophore p-nitroanilide (pNA) after cleavage from the labeled substrate DEVD-pNA (ApoAlert Caspase-3 Colorimetric Assay; Clontech Laboratories, USA). Briefly, an aliquot of cell suspension (2 × 106 cells/ml) after treatment with paclitaxel for 24 h was washed with PBS and centrifuged (400 × g, 10 min, 4℃). Cells were then lysed in 50 μl lysis buffer. After centrifuga-tion at 1000 × g, 3 min, 4℃, the supernatants were incubated with DEVD-pNA for 1 h at 37℃, and then optical density was measured at 405 nm. Caspase activity was defined as nmol pNA/h/mg of protein. The protein concentration was determined with a BCA protein assay reagent kit as described by the manufacturer (Pierce, USA).
Preparation of Cell Lysates and Immunoblot Analysis
Whole cell lysates were prepared as follows. Cells were washed twice with ice-cold phosphate-buffered saline (PBS), and harvested by scraping the cells into lysis buffer (20 mM Tris, pH 8.0, 150 mM NaCl, 1% Triton X-100, 2 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 20 μg/ml aprotinin, 10 μg/ml leu-peptin, 20 mM β-glycerophosphate and 2 mM sodium fluoride). Cell lysates were clarified by centrifugation at 15,000 × g for 10 min at 4℃, and lysate protein concentrations were determined with a BCA protein assay reagent kit. For immunoblotting, whole cell lysates were denatured in Laemmli sample buffer, and resolved by SDS-polyacrylamide gel. The proteins were transferred to nitrocellulose, and immunoblot analysis was performed using appropriate primary antibodies. Immune complexes on nitrocellulose were detected by the ECL-Plus chemiluminescent system (Amersham Pharmacia Biotech, USA). Fluorescent images were captured using KODAK Image Station 4000R (Carestream Health, Inc., USA). Bands were quantified using Kodak Molecular Imaging software, version 4.5.0 (Carestream Health, Inc.).
RESULTS
The effect of rapamycin on cell viability and autophagy of drug-sensitive and drug-resistant cells
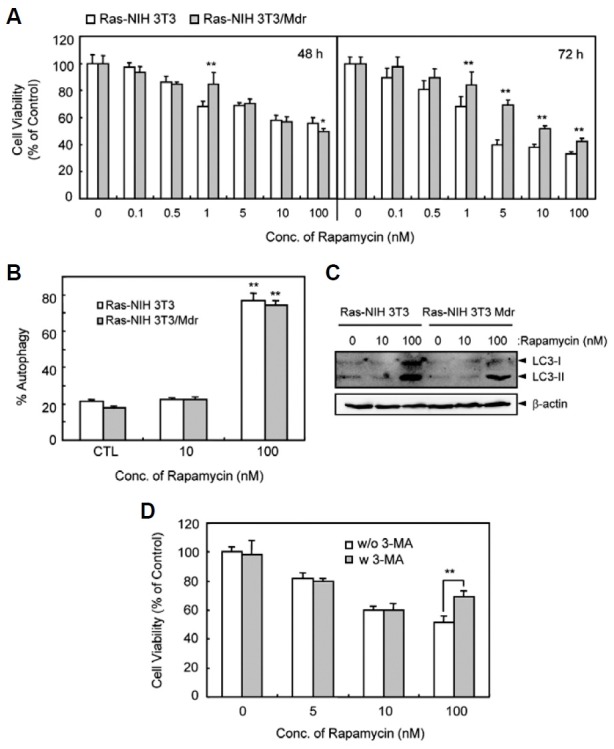
Rapamycin is a natural macrolide antibiotic that inhibits mTOR signaling and causes cell cycle arrest, and importantly, induces autophagy (Hartford and Ratain, 2007). We examined whether rapamycin affects cell death in Ras-NIH3T3 cells versus their drug-resistant counterpart. In this study, we created a new MDR-cell line that stably expresses the drug efflux pump P-glycoprotein that can be blocked by verapamil. As shown in Fig. 1A, the cell growth of the 2 cell lines was moderately inhibited by rapamycin in a dose-dependent manner. Ras-NIH3T3/Mdr cells were more resistant to rapamycin than Ras-NIH3T3 cells after a 72-h treatment. These results imply rapid recovery from cell death in Ras-NIH3T3/Mdr cells. Next, to determine the features of cell death induced by rapamycin in both cell lines, we examined the rapamycin’s effect on autophagy. As shown in Fig. 1B, rapamycin caused a marked increase in the number of GFP-LC3-positive autophagic vacuoles in both cell lines treated with 100 nM rapamycin. In addition, immunoblotting analysis also indicated conversion of nonautophagic LC3-I to autophagic LC3-II, one of the early hallmarks of cellular autophagy, in response to 100 nM rapamycin exposure (Fig. 1C). However, 10 nM rapamycin did not induce tautophagy, as measured by LC3 conversion and GFP-LC3 punctate dots. This indicates that growth inhibition induced by rapamycin at low concentrations does not depend completely on autophagy induction. Moreover, treatment with 3-methyladenine (3-MA), a specific inhibitor of autophagy, failed to rescue Ras-NIH3T3/ Mdr cells from cell death induced by 10 nM rapamycin (Fig. 1D). Conversely, growth inhibition mediated by treatment with 100 nM rapamycin was protected by 3-MA, whose protective effect may be entirely linked to its role in increasing autophagy.
Fig. 1. Effect of rapamycin on viability and autophagy of Ras-NIH 3T3 and Ras-NIH 3T3/Mdr cells. (A) Cytotoxic effect of rapamycin. Cells were treated with increasing concentrations of rapamycin ranging from 0.1 to 100 nM for 48 h (left) or for 72 h (right). Cell viability was then evaluated with WST-1 reagent. (B) After 48 h transfection with pEGFP-LC3, cells were incubated for 24 h at 37℃ with rapamycin and immediately analyzed by fluorescence microscopy. To quantify autophagic cells, we counted the number of autophagic cells among 200 GFP-positive cells. *P < 0.01, compared with vehicle control. The results presented are representative of at least three independent exper-iments. (C) The cells were treated with rapamycin as indicated. Electrophoretic mobility change of LC3 from non-autophagic (LC3-I) form to autophagic membrane recruited (LC3-II) form was determined by immunoblotting. (D) Ras- NIH 3T3/Mdr cells were treated with various doses of rapamycin for 24 h in the presence or absence of 1 mM 3-MA and then incubated in 96-well plates for 3 days. Cell growth inhibition was measured by the WST assay. In (A) and (D), the viability of cells treated with vehicle alone was regarded as 100%. Values represent the mean ± SD of quadrupli-cate determinants from one of three representative experiments. **P < 0.01 as determined by Dunnett’s t-test.
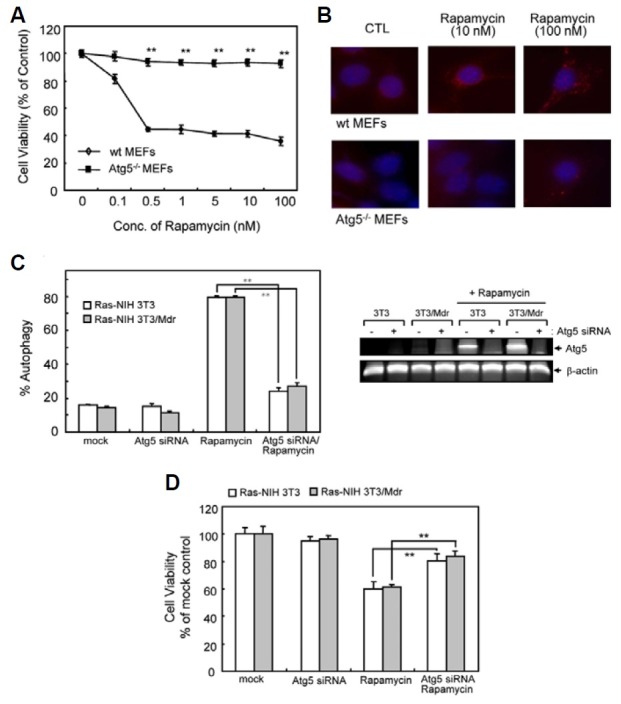
In order to test the hypothesis that autophagy induction plays a positive role in rapamycin-induced growth inhibition, we examined the rapamycin effect in Atg5-deficient mouse embryonic fibroblasts (Atg5-/- MEFs), which are defective in autophagy (Kuma et al., 2004). Atg5-/- MEFs were more resistant than wild-type cells to rapamycin-induced cell death (Fig. 2A) and did not undergo autophagy upon treatment with rapamycin (Fig. 2B). Conversely, treatment of wild-type MEFs with rapamycin induced autophagy, as evidenced by punctate patterns of LC3 immunoreactivity. In addition, we examined the effect of Atg5 knockdown by siRNA transfection in Ras-NIH3T3 cells and their drug-resistant counterpart. We confirmed the knockdown efficiency of the siRNA against endogenous Atg5 by RT-PCR analysis (Fig. 2C, right inset). The results presented in Fig. 2C showed that Atg5 knockdown by siRNA reduced the ability of rapamycin to induce autophagy in both cell lines. Further, Atg5 knockdown partially induced resistance toward rapamycin-induced growth inhibition (Fig. 2D). Taken together, these findings demonstrate that autophagy plays a prominent role in rapamycin-induced cancer cell death.
Fig. 2. Effect of rapamycin on viability and autophagy of WT and Atg5-/- MEFs. (A) Autophagy-proficient (WT MEFs) and -deficient (Atg5-/- MEFs) cells were treated with increasing concentrations of rapamycin ranging from 0.1 to 100 nM for 72 h. Cell viability was evaluated with WST-1 reagent. The viability of cells treated with vehicle alone was regarded as 100%. Values represent the mean ± SD of quadruplicate determinants from one of three representative experiments. **P < 0.01 as determined by the Dunnett’s T-test as compared to drug-sensitive cells. (B) Autophagy induction was measured by immunofluorescent staining using anti-LC3 (Red). Blue = DAPI staining. (C) Ras-NIH 3T3/Mdr cells were transiently transfected with Atg5 siRNA or scrambled control siRNA for 24 h, together with GFP-LC3. Cells were subsequently washed and then treated with or without rapamycin (10 nM) for 24 h. Autophagy was quantified by the GFP-LC3 puncta. Right inset, RT-PCR showing knock-down of Atg5 mRNA expression by Atg5 siRNA. (D) Atg5 knockdown cells were incubated in the presence or absence of rapamycin in 96-well plates for 3 days. Cell growth inhibition was eva-luated with WST-1 reagent. In (C) and (D), the value of mock-transfected cells was regarded as 100%. Values represent the mean ± SD of quadruplicate determinants from one of three representative experiments. **P < 0.01 as determined by Dunnett’s t-test.
G1-phase arrest induced by rapamycin in drug-resistant cells
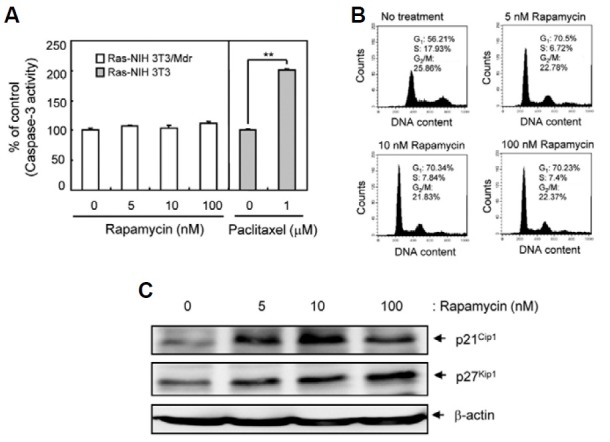
Next, we elucidated how rapamycin exerts its concentration-dependent cytotoxic effects. We determined whether rapamycin promoted cell cycle progression or apoptosis. Our experiments indicated no significant increase in apoptosis of Ras-NIH3T3/Mdr cells, as determined by caspase-3 activity (Fig. 3A). However, flow cytometric analysis revealed that rapamycin treatment increased the number of G1-phase cells and decreased S-phase cells (Fig. 3B). To examine the molecular mechanisms underlying the changes in cell cycle progression, we investigated the expression of the cyclin-dependent kinase inhibitors (CKIs) p21Cip1 and p27Kip1, which block G1- to S-phase transition. Western blot analysis demonstrated that rapamycin treatment enhanced CKI expression (Fig. 3C), suggesting that low concentrations of rapamycin elicit cell cycle arrest in Ras-NIH3T3/Mdr cells without a significant increase in autophagy.
Fig. 3. Detection of G1-phase arrest in Ras-NIH3T3/Mdr cells treated with rapamycin. (A) Pro-apoptotic activity of caspase-3 was determined by detection of the chromophore p-nitroanilide (pNA) after cleavage from the labeled substrate DEVD-pNA in rapamycin- or paclitaxel-treated (positive control) cells. Values represent the mean ± SD of duplicate determinants from one of three representative experiments. **P < 0.01 as determined by the Dunnett’s T-test. (B) Cell cycle progression was assessed by staining fixed cells with propidium iodide to estimate the percentage of cells in the G1 (2N DNA content), G2/M (4N DNA content), and S phases (2 to 4N DNA content). The percentage of cells in each phase of the cell cycle was quantitated using Cell-Quest Pro software and plotted. (C) Whole cell extracts were prepared 48 h post-rapamycin treatment. The expression of p21Cip1 and p27Kip1 were assessed by immuno-blotting. β-actin was assessed as a loading control. The results presented are representative of at least three independent experiments.
Effect of combined ectopic Beclin-1 expression and rapamycin treatment on autophagy induction and cell growth
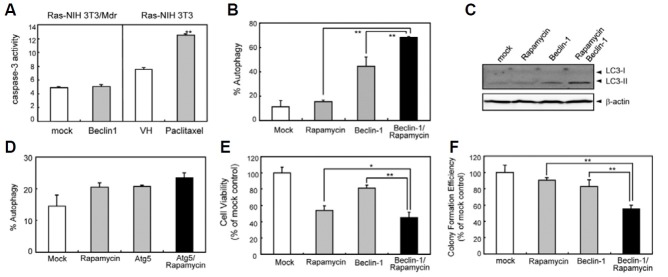
Increased Beclin-1 expression regulates autophagy tightly by initiating autophagosome formation (Cao and Klionsky, 2007). Ectopic expression of Beclin-1 failed to modulate apoptosis (Fig. 4A), while inducing a great increase in autophagosome numbers (Fig. 4B). Moreover, combination treatment synergistically increased the incidence of autophagy significantly compared to the individual treatments alone (Fig. 4B). Immunoblotting analysis also indicated the synergistic increase in conversion of nonautophagic LC3-I to autophagic LC3-II in response to combination treatment (Fig. 4C). We further investigated whether ectopic expression of Atg5, a necessary component of the autophagic machinery downstream of Beclin-1, also increases autophagy. Unexpectedly, autophagic induction of cells overexpressing Atg5 was virtually the same for mock-transfected cells (Fig. 4D), suggesting that Atg5 is not a rate-limiting factor in Beclin1-dependnt autophagic pathway. Next, studies were carried out to determine whether Beclin-1 ectopic expression enhanced the loss of cell survival observed in Ras-NIH3T3/Mdr cells treated with low concentrations of rapamycin. Beclin-1 expression sensitized Ras-NIH3T3/Mdr cells to rapamycin-induced growth inhibition and cell death (Fig. 4E). We also investigated the effect of rapamycin on Ras-NIH 3T3/Mdr cells in vitro clonogenic survival alone or in combination with Beclin-1 ectopic expression (Fig. 4F). Clonogenic assay is an in vitro cell survival assay based on the ability of a single cell to grow into a colony and can also be used to determine the effectiveness of other cytotoxic agents (Franken et al., 2006). When used as a single agent, rapamycin inhibited Ras-NIH 3T3/Mdr cell clonogenic survival. When rapamycin was used in combination with Beclin-1 ectopic expression, the effect of rapamycin on clonogenic survival and growth of Ras-NIH 3T3/Mdr cells was even greater.
Fig. 4. The effect of combined Beclin-1 and rapamycin treatment on autophagy induction and cell growth in Ras-NIH 3T3/Mdr cells. (A) The cells were transiently transfected for 48 h with either a vector expressing full-length Beclin-1 or an empty vector. Ras-NIH 3T3 cells treated with paclitaxel were used as a positive control for apoptosis. Analysis of caspase-3 activity was performed as described in Fig. 2. (B) To quantify the incidence of autophagy, Beclin1-trans-fetced Ras-NIH 3T3/Mdr cells treated with or without rapamycin (10 nM) for 24 h were stained with MDC and visualized by fluorescence microscopy. The autophagic index was calculated as the percentage of MDC-labeled cells out of 200 cells from each treatment group. (C) Beclin1-transfected cells were treated with rapamycin as indicated. Electrophoretic mobility change of LC3 from nonautophagic (LC3-I) form to autophagic membrane recruited (LC3-II) form was determined by immunoblotting. (D) The cells were transiently transfected with either a vector expressing mCherry-Atg5 fusion proteins or an empty vector, together with GFP-LC3 and allowed to express for 24 h. Cells were incubated in the presence or absence of rapamycin (10 nM) for 24 h. To quantify autophagic cells, we counted the number of autophagic cells among 200 GFP-positive cells. (E) Beclin-1-transfetced Ras-NIH 3T3/Mdr cells were incubated in the presence or absence of rapamycin in 96-well plates for 3 days. Cell growth was measured by the WST assay. The viability of cells treated with vehicle alone was regarded as 100%. (F) To identify the effect of rapamycin and Beclin1 when used as single agents or in combination on Ras-NIH 3T3/Mdr cells clonogenic survival, Beclin-1-transfected Ras-NIH 3T3/Mdr cells were incubated in the absence and presence of rapamycin for 7 days. The cells were fixed with methanol and stained with 5% Giemsa solution, and colonies of more than 20 cells were scored. Values represent the mean ± SD of quadruplicate determinants from one of three representative experiments. **P < 0.01; *P < 0.05 as determined by the Dunnett’s t-test.
Effect of combined treatment of Beclin-1/rapamycin on sensitivity to paclitaxel in drug-resistant cells
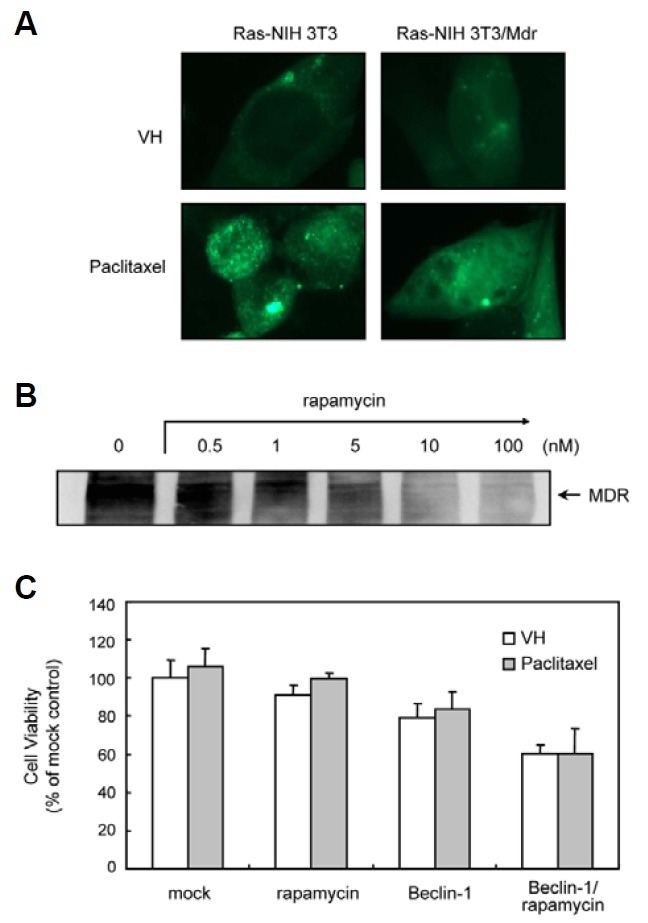
Our unpublished data show that autophagy and apoptosis act as cooperatively to induce cell death in Ras-NIH3T3 cells treated with paclitaxel. The effects of paclitaxel on autophagy induction in Ras-NIH3T3/Mdr cells are shown in Fig. 5A. The cellular localization of the autophagy marker LC3 was evaluated using transiently transfected cells expressing GFP-LC3. Paclitaxel caused an accumulation of GFP-LC3 in Ras-NIH3T3 cells, resulting in a punctate fluorescent pattern that indicates a redistribution of LC3 to autophagic structures. In contrast, Ras-NIH3T3/Mdr cells showed diffuse staining. Previous studies have suggested that rapamycin can downregulate and disable the P-glycoprotein drug efflux pump, thereby increasing the cytotoxicity of chemotherapeutic agents in multidrug-resistant cells (Arceci et al., 1992; Pop et al., 2009). Therefore, we tested whether rapamycin enhances the susceptibility to paclitaxel treatment via P-glycoprotein downregulation. Ras-NIH3T3/Mdr cells were pretreated with rapamycin for 24 h, followed by induction of apoptosis with paclitaxel for 24 h. Despite the downregulation of P-glycoprotein (Fig. 5B), rapamycin failed to over-come the effects of paclitaxel, as demonstrated with a cytoto-xicity assay (Fig. 5C). We obtained similar results with Beclin-1 overexpression and/or rapamycin treatment. These findings suggest that the cytotoxic effect of rapamycin appears to be P-glycoprotein-independent.
Fig. 5. The effect of rapamycin on P-glycoprotein in Ras-NIH 3T3/ Mdr cells. (A) Cells were transfected for 48 h with pEGFP-LC3 and then incubated with paclitaxel for 24 h at 37℃. GFP-LC3 distribution was analyzed by fluorescence microscopy. The results presented are representative of at least three independent experiments. (B) The expression of P-glycoprotein was assessed by immunoblotting analysis. Whole cell extracts were prepared 24 h post-rapa-mycin treatment (0.5-100 nM). The expression of P-glycoprotein was assessed by immunoblotting. The results presented are representative of at least three independent experiments (C) Beclin-1-transfected or mock cells were pre-treated with rapamycin for 24 h or vehicle and subsequently treated with or without 1 μM paclitaxel for 2 days. Cell growth was measured by the WST assay. The viability of cells treated with vehicle alone was regarded as 100%. Values represent the mean ± SD of quadruplicate determinants from one of three representative experiments.
DISCUSSION
Recent studies have suggested that induction of autophagy may be an effective therapeutic approach for apoptosis-resistant cancer cells (Ullman et al., 2008). In the present study, we demonstrated that the mTOR inhibitor rapamycin induced autophagy in Ras-NIH3T3/Mdr cells and that this biological process mediated the growth inhibitory effect of rapamycin. Studies of Atg5-deficient MEF cells confirm that autophagy plays a prominent role in rapamycin-induced cancer cell death. However, our results demonstrate that rapamycin-induced cell death is concentration-dependent. The sensitivity of Ras-NIH3T3/Mdr cells to rapamycin is not related to the extent of autophagy induction by rapamycin. Autophagy induction was observed only with rapamycin concentrations of 100 nM and greater. However, growth inhibition without autophagy induction was seen at lower rapamycin concentrations (10 nM and less), indicating that rapamycin-induced cell death is independent of autophagy induction. Mechanistic studies showed that inhibition of the mTOR pathway by low concentrations of rapamycin induced G1-phase cell cycle arrest by upregulation of p27Kip1 and p21Cip1 expression in Ras-NIH3T3/Mdr cells. In support of this finding, Kawamata et al. (1998) reported G1-phase arrest of cancer cells by rapamycin via modulation of cell cycle regulatory proteins. Thus, low rapamycin concentrations may inhibit Ras-NIH3T3/Mdr cell proliferation by slowing G1- to S-phase transition or by arresting cells in the G1 phase of the cell cycle. These studies suggest that rapamycin-induced cell death may result from 2 different mechanisms. At lower rapamycin concentrations (≤ 10 nM), cell death may occur after G1 arrest via an autophagy-independent pathway, whereas at higher rapamycin concentrations (≥ 100 nM), cell death may occur through an autophagy-dependent pathway.
mTOR-specific inhibitors such as rapamycin analogs have been established as promising agents for treating malignant tumors (Faivre et al., 2006). However, the efficacy of mTOR inhibitors alone in cancer treatment is only modest. These inhibitors may not completely inhibit mTOR function, because they do not act as mTOR kinase inhibitors, but instead interfere with the function of only the mTOR/raptor (regulatory-associated protein of mTOR) complex (Guertin and Sabatini, 2005). Thus, we evaluated the effects of combining Beclin-1 expression with the anticancer activity of rapamycin. A recent study has demonstrated that deletion of the Beclin-1 gene plays a causal role in promoting oncogenesis. In fact, the Beclin-1 gene is commonly deleted in brain tumors (Miracco and Cosci, 2007) and hepatocellular carcinomas (Daniel et al., 2007). Furthermore, transfection of a colon cancer cell line with beclin-1 resulted in cell growth inhibition (Koneri et al., 2007). In the present study, we found that rapamycin in combination with Beclin-1 ectopic expression acted synergistically and more effectively against Ras-NIH3T3/Mdr cells by inducing autophagy. This result is consistent with previous reports which suggest that Beclin-1 overexpression enhances chemosensitivity to anticancer drugs (Sun et al., 2010). However, the mechanisms of the synergistic activity of rapamycin and ectopic expression of Beclin-1 remain unknown. At concentrations lower than the critical concentration required for autophagy induction, rapamycin treatment resulted in enhanced expression of p27Kip1 and, particularly p21Cip1, which inhibits cell growth by acting on the G1-phase of the cell cycle. Several reports have indicated that growth inhibition and autophagy are associated with the upregulation of p21Cip1 and p27Kip1 expression (Komata et al., 2003; Mazzanti et al., 2009). Thus, it is likely that inhibition of mTOR by rapamycin at low concentrations is not sufficient to initiate autophagosome formation, which is required for autophagy, and that Beclin-1 over-expression triggers additional processes downstream of mTOR during G1-phase arrest. These findings suggest that further activation of the autophagic pathway is a potential therapeutic strategy to sensitize cancer cells to rapamycin. However, overexpression of Atg5, a component of the autophagosomal machinery downstream of Beclin1, did not affect autophagy induction, although there is a report that enfor-ced expression of Atg5 sensitized tumour cells to anticancer drug treatment both in vitro and in vivo (Yousefi et al., 2006). Consistent with our results, other laboratories also reported the lack of effect of Atg5 overexpression in the Atg5+/+ cells (Hamacher-Brady et al., 2006; King et al., 2008). Thus, this result implies that the autophagic machinery downstream of Beclin1-dependent induction was not a rate-limiting factor. On the other hand, rapamycin has been documented to modulate the transport function of P-glycoprotein, thereby acting as an agent that reverses the multidrugresistance phenotype (Arceci et al., 1992; Pawarode et al., 2007; Pop et al., 2009). Our results show that, although rapamycin inhibits P-glycoprotein expression in Ras-NIH3T3/Mdr cells, rapamycin has no effect on susceptibility to chemotherapeutic agents such as paclitaxel.
In summary, our in vitro findings demonstrate that the combination of Beclin-1 expression and rapamycin at low concentrations was superior to single-agent therapy in a drug-resistant cell line. Although several studies have been published on chemotherapy with rapamycin alone or with a gene therapy approach delivering Beclin-1 for cancer treatment, the present study is the first report to demonstrate that targeting the autophagic pathway in combination with cell cycle arrest may be an effective therapeutic strategy for eliminating MDR-positive cancer cells. Studies examining the concentration-dependent effects of mTOR inhibitors may provide significant insights into the development of the best drug combination regimens to achieve optimal anticancer activity.
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology (2009-0064464).
References
- 1.Arceci R.J., Stieglitz K., Bierer B.E. Immunosuppressants FK506 and rapamycin function as reversal agents of the multidrug resistance phenotype. Blood. (1992);80:1528–1536. [PubMed] [Google Scholar]
- 2.Cao Y., Klionsky D.J. Physiological functions of Atg6/ Beclin 1: a unique autophagy-related protein. Cell Res. (2007);17:839–849. doi: 10.1038/cr.2007.78. [DOI] [PubMed] [Google Scholar]
- 3.Dancey J.E. Inhibitors of the mammalian target of rapamycin. Expert Opin. Investig. Drugs. (2005);14:313–328. doi: 10.1517/13543784.14.3.313. [DOI] [PubMed] [Google Scholar]
- 4.Daniel F., Legrand A., Pessayre D. Beclin1 mRNA strongly correlates with Bcl-XL mRNA expression in human hepatocellular carcinoma. Cancer Invest. (2007);25:226–231. doi: 10.1080/07357900701206323. [DOI] [PubMed] [Google Scholar]
- 5.Faivre S., Kroemer G., Raymond E. Current development of mTOR inhibitors as anticancer agents. Nat. Rev. Drug Discov. (2006);5:671–688. doi: 10.1038/nrd2062. [DOI] [PubMed] [Google Scholar]
- 6.Franken N.A., Rodermond H.M., Stap J., Haveman J., van Bree C. Clongenic assay of cells in vitro. Nat. Protoc. (2006);1:2315–2319. doi: 10.1038/nprot.2006.339. [DOI] [PubMed] [Google Scholar]
- 7.Gottesman M.M. Mechanisms of cancer drug resistanace. Annu. Rev. Med. (2002);53:615–627. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- 8.Guertin D.A., Sabatini D.M. An expanding role for mTOR in cancer. Trends Mol. Med. (2005);11:353–361. doi: 10.1016/j.molmed.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 9.Hamacher-Brady A., Brady N.R., Gottlieb R.A. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J. Biol. Chem. (2006);281:29776–29787. doi: 10.1074/jbc.M603783200. [DOI] [PubMed] [Google Scholar]
- 10.Hartford C.M., Ratain M.J. Rapamycin: something old, something new, sometimes borrowed and now renewed. Clin. Pharmacol. Ther. (2007);82:381–388. doi: 10.1038/sj.clpt.6100317. [DOI] [PubMed] [Google Scholar]
- 11.Kawamata S., Sakaida H., Hori T., Maeda M., Uchiyama T. The upregulation of p27Kip1 by rapamycin results in G1 arrest in exponentially growing T-cell lines. Blood. (1998);91:561–569. [PubMed] [Google Scholar]
- 12.Kim K.W., Hwang M., Moretti L., Jaboin J.J., Cha Y.I., Lu B. Autophagy upregulation by inhibitors of caspase-3 and mTOR enhances radiotherapy in a mouse model of lung cancer. Autophagy. (2008);4:659–668. doi: 10.4161/auto.6058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.King M.A., Hands S., Hafiz F., Mizushima N., Tolsovsky A.M., Wyttenbach A. Rapamycin inhibits polyglutamine aggregation independently of autophagy by reducing protein synthesis. Mol. Pharmacol. (2008);73:1052–1063. doi: 10.1124/mol.107.043398. [DOI] [PubMed] [Google Scholar]
- 14.Komata T., Kanzawa T., Takeuchi H., Germano I.M., Schreiber M., Kondo Y., Kondo S. Antitumour effect of cyclin-dependent kinase inhibitors (p16(INK4A), p18(INK4C), p19(INK4D), p21(WAF1/CIP1) and p27(KIP1)) on malignant glioma cells. Br. J. Cancer. (2003);88:1277–1280. doi: 10.1038/sj.bjc.6600862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koneri K., Goi T., Hirono Y. Beclin1 gene inhibits tumor growth in colon cancer cell lines. Anticancer Res. (2007);27:1453–1457. [PubMed] [Google Scholar]
- 16.Kuma A., Hatano M., Matsui M., Yamamoto A., Nakaya H., Yo-shimori T., Ohsumi Y., Tokuhisa T., Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. (2004);432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 17.Lee M., Ahn J.-H., Eum K.-H. The differences in bio-logical properties between parental and v-Ha-ras transformed NIH3T3 cells. Cancer Res. Treat. (2009);41:93–99. doi: 10.4143/crt.2009.41.2.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marinov M., Fischer B., Arcaro A. Targeting mTOR signaling in lung cancer. Crit. Rev. Oncol. Hematol. (2007);63:172–182. doi: 10.1016/j.critrevonc.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 19.Mashima T., Tsuruo T. Defects of the apoptotic pathway as therapeutic target against cancer. Drug Resist. Updat. (2005);8:339–343. doi: 10.1016/j.drup.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 20.Mazzanti R., Platini F., Bottini C., Fantappie O., Solazzo M., Tessitore L. Down-regulation of the HGF/MET autocrine loop induced by celecoxib and mediated by P-gp in MDR-positive human hepatocellular carcinoma cell line. Biochem. Pharmacol. (2009);78:21–32. doi: 10.1016/j.bcp.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 21.Meric-Bernstam F., Gonzalez-Angulo A.M. Targeting the mTOR signaling network for cancer therapy. J. Clin. Oncol. (2009);27:2278–2287. doi: 10.1200/JCO.2008.20.0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miracco C., Cosci E. Protein and mRNA expression of autophagy gene Beclin1 in human brain tumours. Int. J. Oncol. (2007);30:429–436. [PubMed] [Google Scholar]
- 23.Moretti L., Yang E.S., Kim K.W., Lu B. Autophagy signaling in cancer and its potential as novel target to improve anticancer therapy. Drug Resist. Updat. (2007);10:135–143. doi: 10.1016/j.drup.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 24.Panwalkar A., Verstovsek S., Giles F.J. Mammalian target of rapamycin inhibition as therapy for hematologic malignancies. Cancer. (2004);100:657–666. doi: 10.1002/cncr.20026. [DOI] [PubMed] [Google Scholar]
- 25.Pawarode A., Shukla S., Minderman H., Fricke S.M., Pinder E.M., O’Loughlin K.L., Ambudkar S.V., Baer M.R. Differential effects of the immunosuppressive agents cyclosporin A, tacrolimus and sirolimus on drug transport by multidrug resistance proteins. Cancer Chemother. Pharmacol. (2007);60:179–188. doi: 10.1007/s00280-006-0357-8. [DOI] [PubMed] [Google Scholar]
- 26.Pop I.V., Pop L.M., Ghetie M.A., Vitetta E.S. Targeting mammalian target of rapamycin to both downregulate and disable the P-glycoprotein pump in multidrug-resistant B-cell lymphoma cell lines. Leuk. Lymphoma. (2009);50:1155–1162. doi: 10.1080/10428190903046722. [DOI] [PubMed] [Google Scholar]
- 27.Sir D., Ou J.H. Autophagy in viral replication and pathogenesis. Mol. Cells. (2010);29:1–7. doi: 10.1007/s10059-010-0014-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun Y., Liu J.H., Jin L., Lin S.M., Yang Y., Sui Y.X., Shi H. Over-expression of the Beclin1 gene upregulates chemosensitivity to anti-cancer drugs by enhancing therapy-induced apoptosis in cervix squamous carcinoma CaSki cells. Cancer Lett. (2010);294:204–210. doi: 10.1016/j.canlet.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 29.Ullman E., Fan Y., Stawowczyk M., Chen H.M., Yue Z., Zong W.X. Autophagy promotes necrosis in apoptosis-deficient cells in response to ER stress. Cell Death Differ. (2008);15:422–425. doi: 10.1038/sj.cdd.4402234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weppler S.A., Krause M., Zyromska A., Lambin P., Baumann M., Wouters B.G. Response of U87 glioma xenografts treated with concurrent rapamycin and fractionated radiotherapy: possible role for thrombosis. Radiother. Oncol. (2007);82:96–104. doi: 10.1016/j.radonc.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 31.Wu C.P., Calcagno A.M., Ambudkar S.V. Reversal of ABC drug transporter-mediated multidrug resistance in cancer cells: evaluation of current strategies. Curr. Mol. Pharmacol. (2008);1:93–105. doi: 10.2174/1874467210801020093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yousefi S., Perozzo R., Schmid I., Ziemiecki A., Schaffner T., Scapozza L., Brunner T., Simon H.U. Calpainmediated cleavage of Arg5 switches autophagy to apoptosis. Nat. Cell Biol. (2006);8:1124–1132. doi: 10.1038/ncb1482. [DOI] [PubMed] [Google Scholar]
- 33.Yue Z., Jin S., Yang C., Levine A.J., Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA. (2003);100:15077–15082. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]