

Abstract
Tissue inhibitor of metalloproteinase-1 (TIMP-1) plays various roles in cell growth in different cell types. However, few studies have focused on TIMP-1’s effect on fibroblast cells. In this study, we investigated the effects of TIMP-1 overexpression on NIH3T3 fibroblast proliferation and potential transduction signaling pathways involved. Overexpression of TIMP-1, by transfection of the pLenti6/ V5-DEST-TIMP-1 plasmid, significantly promoted NIH3T3 proliferation as determined by the BrdU array. Neither 5 nor 15 nM GM6001 (matrix metalloproteinase system inhibitor) affected NIH3T3 proliferation, but 45 nM GM6001 inhibited proliferation. TIMP-1 overexpression activated the p-Akt pathway, but not the p-ERK or p-p38 pathway. In TIMP-1-transfected cells, cyclinD1 was upregulated and p21CIP1 and p27KIP1 were downregulated, which promoted cell entry into the S and G2/M phases. The PI3-K inhibitor LY294002 abolished the TIMP-1-induced effects. Overex-pression of intracellular TIMP-1 stimulated NIH3T3 fibroblast proliferation in a matrix metalloproteinase (MMP)-indepen-dent manner by activating the p-Akt pathway and related cell cycle progression.
Keywords: Akt, cell cycle, NIH3T3 cells, proliferation, tissue inhibitor of metalloproteinase-1 (TIMP-1)
INTRODUCTION
As a major regulator of extracellular matrix (ECM) degradation and a cytokine, TIMP-1 not only influences the balance of ECM degradation, but also regulates cell growth. In recent years, it has been demonstrated that TIMP-1 has multiple effects on cell growth. On the one hand, TIMP-1 enhances cell survival by inhibiting apoptosis and stimulating cell proliferation. For example, TIMP-1 inhibits apoptosis in some cell lines (e.g., human breast epithelial cells, normal human granulocytes, rat mesangial cells, breast carcinoma cells, hepatic stellate cells, pancreatic islets B-cells) by modulating signaling pathways, such as the caspase pathway, PI3-K pathway, and MEPK pathway (Chromek et al., 2004; Han et al., 2001; Li et al., 1999; Lin et al., 2002; Liu et al., 2003; Park et al., 2003; Yoshiji et al., 2002). TIMP-1 also promotes cell proliferation in some cells (e.g., human aortic smooth muscle cells, primary melanoma cell lines, rabbit corneal epithelial cells) by regulating signaling pathways and the cell cycle (Akahane et al., 2004; Hoashi et al., 2001; Saika et al., 1998). However, TIMP-1 can also attenuate cell proliferation and promote apoptosis. For example, TIMP-1 overexpression inhibits proliferation of BEL-7402 (a hepatocellular carcinoma cell line) and pancreatic cancer (Bloomston et al., 2002; Guo et al., 2007) by signaling pathways such as stat. TIMP-1 also stimulates apoptosis in some cells, such as normal mammary epithelial cells and non-malignant MCF10A human breast epithelial cells, by cell cycle regulation (Fata et al., 1999; Taube et al., 2006). Thus, TIMP-1’s multiple functions on various cells may be dependent on cell-specific signal transduction pathways and cell cycle regulation.
Fibroblast cell proliferation is a major factor promoting tissue fibrosis, causing dysfunction in organs such as the kidney and liver. TIMP-1, an important cell growth regulatory cytokine, may play a crucial role in fibroblast cell proliferation. Several studies report the relationship between TIMP-1 and fibroblast cell proliferation. For example, Lovelock et al. showed that the TIMP family, including TIMP-1, stimulated cardiac fibroblast proliferation (Lovelock et al., 2005), but the mechanism involved in TIMP-1’s proliferative effect was unclear. In this study, we investigated TIMP-1’s effect on NIH3T3 proliferation and signaling pathways involved in the proliferative effects.
MATERIALS AND METHODS
Reagents
Anti-TIMP-1, anti-total-Akt, anti-p27KIP1; anti-cyclinD1, ant-p21CIP1, anti-MMP-1, and anti-actin antibodies were all from Santa Cruz Biotechnology (USA). Anti-phospho-Akt (ser-473) was from Promega Corporation (USA). A BrdU (5-bromo-2-deoxy-uridine) labeling and detection kit was purchased from Roche Applied Science (USA). LY294002 and GM6001 were purchased from Sigma-Aldrich Corp. (USA).
Cell culture
NIH3T3 (3 × 105) cells seeded in 25-cm2 flasks and 2000 cells seeded in 96-well culture plates were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal calf serum (FCS; 100 units/ml penicillin, 100 mg/ml streptomycin) in a 95% air / 5% CO2 incubator at 37℃ for 24 h before being prepared for the next procedure (i.e., transfection and chemical treatment).
Transfection method
Cells cultured in 25-cm2 flasks and 96-well culture plates were transfected with 2 μg plasmid (pLenti6/V5-DEST- mTIMP-1; gift from Prof. HanXiao) per ml culture medium using Lipofectin in fresh 10% FCS/DMEM medium for 24 h. Lenti6/V5-DEST was used as a control. When performing the Akt block array, cells were pretreated with 20 μM LY294002 for 30 min before cell transfection. Cells cultured in 25-cm2 flasks were prepared for protein isolation and flow cytometry; cells cultured in 96-well culture plates were prepared for BrdU assay. All experiments were repeated at least three times.
MMP inhibition array
Cells were cultured in 96-well culture plates in 10% FCS medium for 24 h and synchronized in 0.1% FCS DMEM for 24 h. Cells were treated with different GM6001 concentrations (5, 15, or 45 nM) for 24 h. Untreated cells were used as a control. The proliferation rates of cells were tested using the BrdU array. All experiments were repeated at least three times.
BrdU incorporation
Cells in 96-well plates were exposed to BrdU (10 μmol/L) for 1 h at 37℃, fixed in ethanol at -20℃ for 2 h, anti-BrdU working solution was added for 1 h at 37℃, and then anti-mouse-Ig-fluorescein was added for 30 min at 37℃. Cells were finally treated with 5 μg/ml Hoechst 33258 in phosphate-buffered saline (PBS) for 20 min and washed three times with PBS. Cells were examined by fluorescence microscopy.
Western blot
Cells that were cultured in 25-cm2 flasks were extracted with modified RIPA buffer (150 mM NaCl, 1% Nonidet P-40, 1 mM EDTA, 0.25% sodium deoxycholate, 50 mM Tris-HCl pH 7.4, 1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 mM PMSF). Protein concentration was determined by BCA protein assay. Protein (100 μg) was electrophoresed on a 12% SDS-polyacrylamide gel and transferred onto a nitrocellulose membrane (Millipore, USA).The nitrocellulose membrane was blocked with 1× Caseinin (Vector Laboratories, USA). The blot was incubated with the appropriate primary antibody and then the appropriate horseradish peroxidase-conjugated secondary antibodies. Antigen was detected using ECL regents (Santa Cruz Biotechnology, USA) according to the manufacturer’s instructions.
Gelatin zymography
Cells were lysed with lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Triton X-100) on ice for 30 min. The supernatant was collected after centrifugation. 10 mg of protein per lane was separated by 8% SDS-PAGE containing 1 mg/ml gelatin. After electrophoresis, the gel was washed at room temperature for 1 h in 2.5% Triton X-100, 50 mM Tris-HCl (pH 7.5), and then incubated at 37℃ overnight in incubation buffer (150 mM NaCl, 5 mM CaCl2, and 50 mM Tris-HCl pH 7.6, 1 μM ZnCl2). The gel was subsequently stained with 0.1% (w/v) Coomassie Brilliant Blue R-250 and destained in 30% (v/v) methanol and 10% (v/v) glacial acetic acid.
Detection of cell cycle phase by PI staining
Cells cultured in 25-cm2 flasks were harvested and fixed in 75% ethanol for 24 h at 4℃. The nuclei were stained with 20 mg/ml propidium iodide (PI) in 1% Triton-X100/PBS containing 200 mg/ml DNase-free RNase, and the DNA content was analyzed by flow cytometry using a FACS Calibur (BD, Research Trian-gle Park, USA) flow cytometer. The percentage of cells in each phase of the cell cycle was determined using the Modfit LT program (Verity Software House, USA).
Statistical analyses
Chi-squared analysis was used for comparisons of proliferation rates and cell phase distribution (%). The t-test and ANOVA analyses were conducted for two team comparisons and multiclass comparisons, respectively.
RESULTS
TIMP-1 plasmid transfection stimulated NIH3T3 proliferation independently of the MMP system
Initially, we examined TIMP-1 protein expression in cells transfected with the TIMP-1 plasmid (TIMP-1 group) by Western blotting. The results showed that the TIMP-1 protein was significantly increased in the TIMP-1 group compared with the control (Figs. 1A and 1B). Then, we detected the proliferation rate by a BrdU incorporation assay. There was a higher positive rate of BrdU in the TIMP-1 group than in controls (1.4-fold; Figs. 1C and 1D), illustrating that TIMP-1 intracellular overexpression could increase the cell proliferation rate.
Fig. 1. TIMP-1 plasmid transfection stimulates NIH3T3 cell proliferation independent of MMP system. “Con” refers to cells transfected with the control plasmid. “T” refers to cells transfected with the TIMP-1 plasmid. (A) TIMP-1 protein is over-expressed in the T group compared with the Con group, as evaluated by Western blotting. (B) The TIMP-1 protein expression in control group and TIMP-1 group are quantified by densitometry analysis. TIMP-1 band intensities are presented as the ratios to the β-actin levels. The band intensity in the Con group is arbitrarily set as 1.0, *p < 0.05 versus the Con group. (C) Representative images of BrdU staining in Con and T group are shown. Blue cell nuclei are considered as BrdU positive, Total cell nuclei are stained with Hoechst 33258. (D) The NIH3T3 proliferation rate is higher in the T group than in the Con group, as evaluated by BrdU incorporation assays. BrdU positive rate of Con is arbitrarily set as 1.0, *p < 0.05 versus the Con group. (E) TIMP-1 has little effect on the activity of MMP-2 when detected by gelatin zymography, but it could inhibit the expression of MMP-1 when detected by western blot. (F) There are no differences among N cells (normal group) and cells treated with 5 or 15 nM GM6001. There is a lower BrdU-positive rate in cells treated with 45 nM GM6001 com pared with the normal group. BrdU positive rate of N is arbitrarily set as 1.0, *p < 0.05 versus the N group.
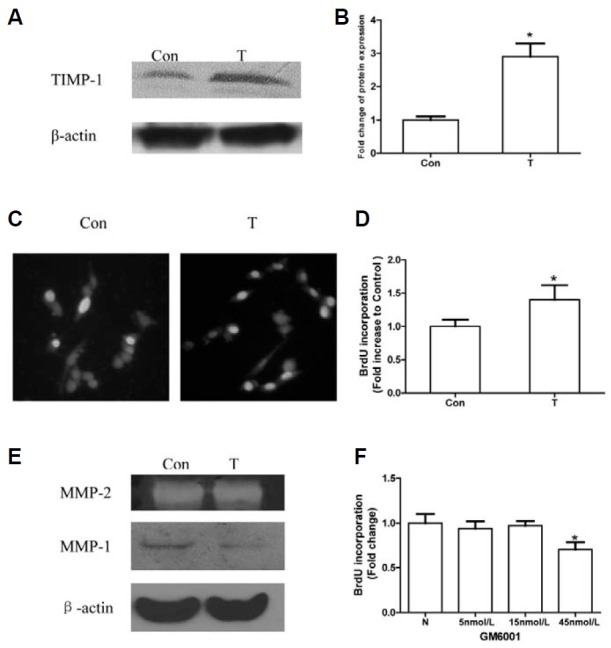
One of TIMP-1’s main functions is to inhibit the MMP system. Whether TIMP-1 promotes NIH3T3 proliferation by a MMP-independent mechanism needs clarification. First, we determined TIMP-1’s effect on MMP1 by Western blotting and MMP2 by gelatin zymography. The results showed that TIMP-1 had little effect on MMP2 activity, but inhibited MMP-1 expression (Fig. 1E). Then, we applied GM6001 to inhibit the MMP system and studied its role in NIH3T3 proliferation. GM6001 as an MMP inhibitor is often used in similar research (Akahane et al., 2004; Porter et al., 2004). We found that there was no difference in the BrdU-positive rate between the control group and the group treated with 5 or 15 nM GM6001, but cells treated with 45 nM GM6001 had a significantly lower BrdU positive rate than controls (Fig. 1F). The MMP inhibitor GM6001 had no positive effect on NIH3T3 proliferation, indicating that TIMP-1 promotes NIH3T3 proliferation by an MMP-independent mechanism.
TIMP-1 enhanced Akt phosphorylation, but had no effect on ERK and p38 phosphorylation
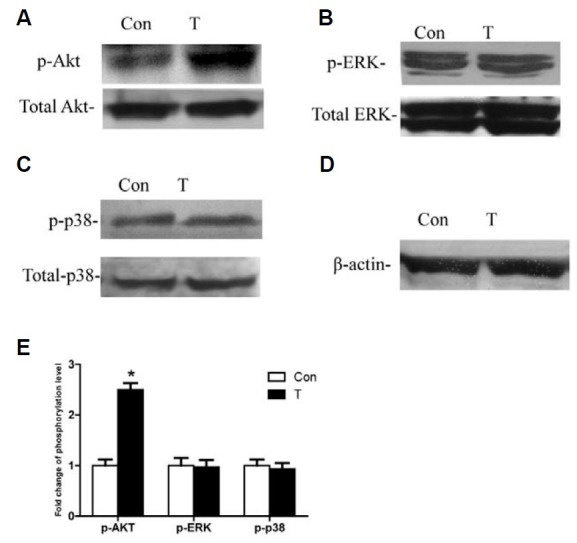
Signaling pathways played a crucial role in TIMP-1’s effect on cell proliferation (Fig. 2). Thus, we investigated TIMP-1’s effect on p-Akt, p-ERK, and p-p38. The results showed that the Akt phosphorylation level was higher in the TIMP-1 group at 24 h after transfection (Fig. 2A). However, there was no difference in p-ERK or p-p38 between the TIMP-1 group and controls when assayed by Western blot (Figs. 2B and 2C). This suggests that TIMP-1 overexpression activates the Akt signaling pathway, but not the MAPK (ERK, p38) signaling pathway.
Fig. 2. TIMP-1 enhances Akt phosphorylation, but had no effect on the ERK and p38 phos-phorylation. “Con” refers to cells transfected with the control plasmid. “T” refers to cells transfected with the TIMP-1 plasmid. (A) p-Akt is upregulated in the T group. (B) p-ERK is not changed significantly in the T group. (C) p-p38 is not changed significantly in the T group. (D) β-actin as internal control protein is detected. (E) Densitometry analysis is performed to compar-ing the level of p-Akt, p-ERK and p-p38 between T group and Con group, p-Akt, p-ERK, and p-p38 bands are presented as a ratio to the its respective total protein level. The band intensity in con group is arbitrarily given as 1.0, *p < 0.05 versus Con.
PI3-K inhibitor LY294002 abolished TIMP-1’s effect on proliferation by blocking the p-Akt pathway
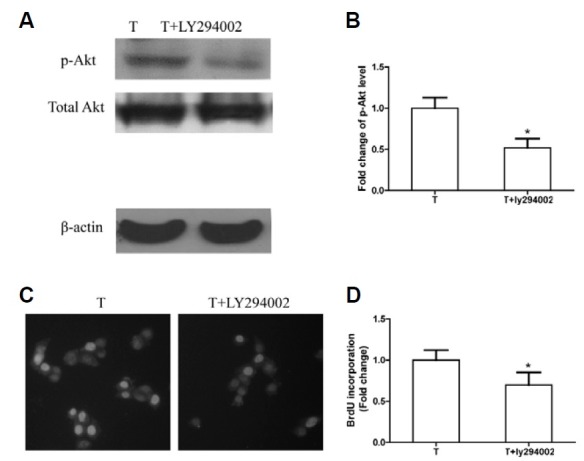
The role of p-Akt activation in TIMP-1’s proliferation promoting effect on NIH3T3 was examined by a blocking assay. A PI3-K inhibitor, LY294002, was applied to block p-Akt activation stimulated by TIMP-1. The results showed that, due to the inhibition of p-Akt activation by LY294002 (Figs. 3A and 3B), the positive BrdU rate was lower in the TIMP-1 group treated with LY294002 (T+ LY294002 group) than in the TIMP-1 group (Figs. 3C and 3D). Furthermore, there was no significant apoptosis when cells were treated with 20 μM LY294002 (data not shown), indicating that the p-Akt pathway inhibited by LY294002 did not trigger NIH3T3 apoptosis. These results illustrated that the Akt signaling pathway played a key role in TIMP-1’s effect on NIH3T3 proliferation and that p-Akt blockage abolished TIMP-1’s proliferative effects.
Fig. 3. The proliferative effects of TIMP-1 are terminated by the p-Akt inhibitor LY294002. “T” refers to cells transfected with the TIMP-1 plasmid. “T+LY294002” refers to the T group treated with LY294002. (A) LY294002 blocks the p-Akt upregulation as evaluated by Western blotting, β-actin is internal reference. (B) p-Akt levels in control group and TIMP-1 group are quantified by densitometry analysis. p-Akt band intensities are presented as the ratios to total Akt levels. The band intensity in the Con group is arbitrarily set as 1.0, *p < 0.05 versus the Con group. (C) Representative images of BrdU staining in T and T+LY294002 group are shown. Blue cell nuclei are considered as BrdU positive, Total cell nuclei are stained with Hoechst 33258. (D) The BrdU-positive rate is lower in the T+LY294002 group than in the T group, BrdU positive rate of Con is arbitrarily set as 1.0, *p < 0.05 versus the T group.
TIMP-1 affected cell cycle proteins including cyclinD1, p21CIP1, and p27KIP1 by activating the p-Akt pathway
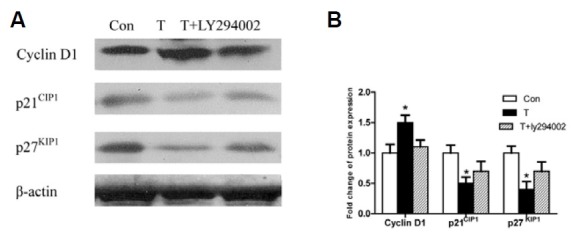
Cell cycle proteins are essential for the TIMP-1 proliferation promoting effect on NIH3T2 cells by activating the Akt signaling pathway. TIMP-1 overexpression upregulated cyclinD1 and downregulated p21CIP1 and p27KIP1. However, when LY294002 was applied to block p-Akt activation, TIMP-1’s effects on cell cycle proteins were abolished (Figs. 4A and 4B). These results illustrated that TIMP-1 affected cell cycle proteins by activating the p-Akt pathway, which promoted NIH3T3 proliferation.
Fig. 4. Overexpression of TIMP-1 upregulates cyclinD1 and downregulates p21CIP1 and p27KIP1, but this effect can be abolished by LY294002. (A) CyclinD1, are highly upregulated while p21CIP1 and p27KIP1 are downer-gulated in the T group compared to Con group, but these effect of TIMP-1 are blocked by LY294002(T+LY294002 group). (B) Densitometry analysis of the intensities of these protein bands is presented as their ratios to the actin levels. The band intensity of the control group is arbitrarily set as 1.0, *p < 0.05 versus the Con group.
TIMP-1 induced cell cycle arrest across G1, but this could be terminated by LY294002
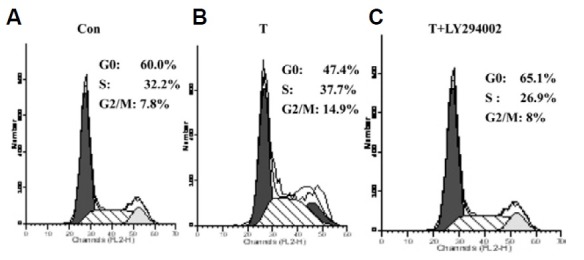
Finally, cell cycle distribution was examined to determine TIMP-1’s effects on NIH3T3 proliferation via the p-Akt pathway. In a flow cytometric DNA assay, TIMP-1 overexpression induced the cell cycle across the G1 phase and increased the S and G2/M phase in TIMP-1-transfected cells compared with controls (Figs. 5A and 5B). However, when the p-Akt inhibitor LY294002 was used, cell cycle phase changes stimulated by TIMP-1 were blocked (Fig. 5C). These results are consistent with TIMP-1 induced NIH3T3 proliferation via the p-Akt pathway.
Fig. 5. TIMP-1 induces NIH3T3 across G1 phase and increases the cells in the S and G2/M phase G0/G1 phase is decreased and S, G2/M phase are increased in the T group (A) compared with the Con group (B); but when LY294002 is applied, there is higher percentage of G0/G1 and lower S phase and G2/M phase in the T+LY294002 group than in the T group (C).
DISCUSSION
The present study showed that TIMP-1 stimulated NIH3T3 proliferation by activating the p-Akt pathway. As an important cytokine, TIMP-1 has multiple functions in fibroblast cells, including proliferation and extracellular matrix accumulation. In the present study, we confirmed that TIMP-1 overexpression promoted NIH3T3 fibroblast cell proliferation, similar to a previous report (Lovelock et al., 2005). We deduced that TIMP-1 bound membrane receptors, which influenced cell growth. Previous studies showed that TIMP-1 could bind to CD63 (Jung et al., 2006) or the proMMP-9/CD44 complex (Lambert et al., 2009), thereby activating survival pathways in other cells, such as breast cancer cells and erythroid cells. However, only CD44 expression on the surface of NIH3T3 cells has been reported (Zhu and Bourguignon, 1996). We assumed that TIMP-1 interacted with CD44, mediated cell signaling pathways, and affected NIH3T3 cell growth. However, additional studies are needed to address this. Moreover, we found that a higher concentration of Lipofectin could delay TIMP-1’s effect on cell proliferation through influencing cell growth in our previous study. Although there were differences in the results due to different conditions, both experiments showed that TIMP-1 overexpression, by plasmid transfection, could stimulate NIH3T3 proliferation
Some studies have shown that TIMP-1’s proliferative effect was dependent on its effects on the MMP system (Porter et al., 2004; 2005) Thus, we examined TIMP-1’s function on the MMP system and used GM6001 to help understand the role MMP inhibition on NIH3T3 proliferation. TIMP-1 inhibited MMP-1 expression, with less effect on MMP-2 activity, in NIH3T3 cells. TIMP-1 exerted this effect on MMP-1 by forming a TIMP-1/MMP-1 complex, whereas MMP-2 was primarily inhibited by TIMP-2 by forming a TIMP-2/MMP2 complex and was thus less affected by TIMP-1 due to its poor binding capacity with MMP-2 (Howard and Banda, 1991; Stetler-Stevenson et al., 1989). However, when GM6001, a MMP inhibitor, was used, we found that MMP-1 inhibition could not stimulate NIH3T3 proliferation, illustrating that TIMP-1 promoted NIH3T3 proliferation independent of the MMP system. Other studies have shown similar results. Hayakawa found that alkylated TIMP-1, which had no effect on inhibiting MMPs, still could stimulate cell growth in Raji cells, supporting the notion that TIMP-1 possessed novel cytokine-like activity (Hayakawa et al., 1994). Liu et al. (2005) found that neither the broad spectrum MMP inhibitor BB94, nor the synthetic peptide hydroxamate metalloproteinase inhibitor/tumor necrosis factor-protease inhibitor (TAPI) affected cell growth or death in MCF10A cells, suggesting that an MMP-independent TIMP-1 signaling pathway may exist. However, how TIMP-1 promotes proliferation independent of MMP in NIH3T3 cells needs further clarification.
TIMP-1 regulates cell growth by activating several signaling pathways, such as Akt, MAPK, and RAS (Lambert et al., 2003; Porter et al., 2004; Wang et al., 2002). In this study, we found that the p-Akt pathway, but not the ERK or p38 pathway, was activated by TIMP-1 in NIH3T3. Akt activation was required for NIH3T3 proliferation. For example, the PI3-K-Akt pathway mediated Wnt3a-induced growth and proliferation of NIH3T3 cells (Kim et al., 2007); p-Akt inhibition suppressed NIH3T3 proliferation (Donapaty et al., 2006). Thus, p-Akt activation by TIMP-1 may play an important role in NIH3T3 proliferation. More work was performed to determine whether the Akt signaling pathway affected the cell cycle in TIMP-1’s proliferative effects.
In the current experiment, TIMP-1 upregulated cyclinD1 and downregulated p21CIP1 and p27KIP1, eventually promoting cell entry into the S phase and the G2/M phase. The PI3-K inhibitor LY294002 abolished these TIMP-1 effects. These results confirmed that the p-Akt pathway acts as key mediator in TIMP-1’s effects on the cell cycle. p-Akt, activated by TIMP-1 overexpression, triggered the p-Akt pathway, which upregulated cyclinD1 and downregulated p21CIP1 and p27KIP1 (Collado et al., 2000; Mirza et al., 2004). These cell cyclins promoted NIH3T3 entry into S phase and G2/M phase. Upregulated cyclinD1 stimulated cell entry into the S and G2/M phase via inhibition of pRB function, by activating pRB phosphorylation (Baldin et al., 1993; Sherr, 2000). Downregulation of p21CIP1 led to cell cycle progression across the G0/G1 phase by decreasing dephosphorylation of pRb and its lesser effect on inhibition of CDK activity and downregulation of p27KIP1 increased pRB phosphorylation by decreasing p27KIP1/cyclinE/CDK2 complex and promoted cell entry into S and G2/M phase (Tchernev and Orfanos, 2007). However, once activation of p-Akt was blocked, cell cycle regulation mediated by Akt was abolished, resulting in the loss of TIMP-1-induced NIH3T3 proliferation. These results confirmed that cell cycle regulation by p-Akt mediated TIMP-1’s proliferative effects in NIH3T3 cells.
In this study, we demonstrated that TIMP-1 overexpression stimulated NIH3T3 proliferation in a MMP-independent manner; the PI3-K/p-Akt pathway, but not the ERK or p38 MAPK pathway, was involved in TIMP-1’s proliferative effect. These results may help in exploring TIMP-1’s effects on fibroblast cell proliferation so that we may control fibroblast cell proliferation by mediating TIMP-1 and its related cell signaling pathways in the future.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation (No. 30630033, No. 8100077) and National Basic Research Program of China (No. 2007CB507400).
References
- 1.Akahane T., Akahane M., Shah A., Thorgeirsson U.P. TIMP-1 stimulates proliferation of human aortic smooth muscle cells and Ras effector pathways. Biochem. Biophys. Res. Commun. (2004);324:440–445. doi: 10.1016/j.bbrc.2004.09.063. [DOI] [PubMed] [Google Scholar]
- 2.Baldin V., Lukas J., Marcote M.J., Pagano M., Draetta G. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev. (1993);7:812–821. doi: 10.1101/gad.7.5.812. [DOI] [PubMed] [Google Scholar]
- 3.Bloomston M., Shafii A., Zervos E.E., Rosemurgy A.S. TIMP-1 overexpression in pancreatic cancer attenuates tumor growth, decreases implantation and metastasis, and inhibits angiogenesis. J. Surg. Res. (2002);102:39–44. doi: 10.1006/jsre.2001.6318. [DOI] [PubMed] [Google Scholar]
- 4.Chromek M., Tullus K., Lundahl J., Brauner A. Tissue inhibitor of metalloproteinase 1 activates normal human granulocytes, protects them from apoptosis, and blocks their transmigration during inflammation. Infect. Immun. (2004);72:82–88. doi: 10.1128/IAI.72.1.82-88.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collado M., Medema R.H., Garcia-Cao I., Dubuisson M.L., Barradas M., Glassford J., Rivas C., Burgering B.M., Serrano M., Lam E.W. Inhibition of the phosphoinositide 3-kinase pathway induces a senescence-like arrest mediated by p27Kip1. J. Biol. Chem. (2000);275:21960–21968. doi: 10.1074/jbc.M000759200. [DOI] [PubMed] [Google Scholar]
- 6.Donapaty S., Louis S., Horvath E., Kun J., Sebti S.M., Malafa M.P. RRR-alpha-tocopherol succinate down-regulates oncogenic Ras signaling. Mol. Cancer Ther. (2006);5:309–316. doi: 10.1158/1535-7163.MCT-05-0330. [DOI] [PubMed] [Google Scholar]
- 7.Fata J.E., Leco K.J., Moorehead R.A., Martin D.C., Khokha R. Timp-1 is important for epithelial proliferation and branching morphogenesis during mouse mammary development. Dev. Biol. (1999);211:238–254. doi: 10.1006/dbio.1999.9313. [DOI] [PubMed] [Google Scholar]
- 8.Guo S.Y., Shen X., Yang J., Yuan J., Yang R.L., Mao K., Zhao D.H., Li C.J. TIMP-1 mediates the inhibitory effect of interleukin-6 on the proliferation of a hepatocarcinoma cell line in a STAT3-dependent manner. Braz. J. Med. Biol. Res. (2007);40:621–631. doi: 10.1590/s0100-879x2007000500004. [DOI] [PubMed] [Google Scholar]
- 9.Han X., Sun Y., Scott S., Bleich D. Tissue inhibitor of metalloproteinase-1 prevents cytokine-mediated dysfunction and cytotoxicity in pancreatic islets and beta-cells. Diabetes. (2001);50:1047–1055. doi: 10.2337/diabetes.50.5.1047. [DOI] [PubMed] [Google Scholar]
- 10.Hayakawa T., Yamashita K., Ohuchi E., Shinagawa A. Cell growth-promoting activity of tissue inhibitor of metalloproteinases-2 (TIMP-2). J. Cell Sci. (1994);107:2373–2379. doi: 10.1242/jcs.107.9.2373. [DOI] [PubMed] [Google Scholar]
- 11.Hoashi T., Kadono T., Kikuchi K., Etoh T., Tamaki K. Differential growth regulation in human melanoma cell lines by TIMP-1 and TIMP-2. Biochem. Biophys. Res. Commun. (2001);288:371–379. doi: 10.1006/bbrc.2001.5789. [DOI] [PubMed] [Google Scholar]
- 12.Howard E.W., Banda M.J. Binding of tissue inhibitor of metalloproteinases 2 to two distinct sites on human 72-kDa gelatinase. Identification of a stabilization site. J. Biol. Chem. (1991);266:17972–17977. [PubMed] [Google Scholar]
- 13.Jung K.K., Liu X.W., Chirco R., Fridman R., Kim H.R. Identification of CD63 as a tissue inhibitor of metalloproteinase-1 interacting cell surface protein. EMBO J. (2006);25:3934–3942. doi: 10.1038/sj.emboj.7601281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim S.E., Lee W.J., Choi K.Y. The PI3 kinase-Akt pathway mediates Wnt3a-induced proliferation. Cell. Signal. (2007);19:511–518. doi: 10.1016/j.cellsig.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 15.Lambert E., Boudot C., Kadri Z., Soula-Rothhut M., Sowa M.L., Mayeux P., Hornebeck W., Haye B., Petitfrere E. Tissue inhibitor of metalloproteinases-1 signalling pathway leading to erythroid cell survival. Biochem. J. (2003);372:767–774. doi: 10.1042/BJ20030187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lambert E., Bridoux L., Devy J., Dasse E., Sowa M.L., Duca L., Hornebeck W., Martiny L., Petitfrere-Charpentier E. TIMP-1 binding to proMMP-9/CD44 complex localized at the cell surface promotes erythroid cell survival. Int. J. Biochem. Cell Biol. (2009);41:1102–1115. doi: 10.1016/j.biocel.2008.10.017. [DOI] [PubMed] [Google Scholar]
- 17.Li G., Fridman R., Kim H.R. Tissue inhibitor of metalloproteinase-1 inhibits apoptosis of human breast epithelial cells. Cancer Res. (1999);59:6267–6275. [PubMed] [Google Scholar]
- 18.Lin H., Chen X., Wang J., Yu Z. Inhibition of apoptosis in rat mesangial cells by tissue inhibitor of metalloproteinase-1. Kidney Int. (2002);62:60–69. doi: 10.1046/j.1523-1755.2002.00403.x. [DOI] [PubMed] [Google Scholar]
- 19.Liu X.W., Bernardo M.M., Fridman R., Kim H.R. Tissue inhibitor of metalloproteinase-1 protects human breast epithelial cells against intrinsic apoptotic cell death via the focal adhesion kinase/phosphatidylinositol 3-kinase and MAPK signaling pathway. J. Biol. Chem. (2003);278:40364–40372. doi: 10.1074/jbc.M302999200. [DOI] [PubMed] [Google Scholar]
- 20.Liu X.W., Taube M.E., Jung K.K., Dong Z., Lee Y.J., Roshy S., Sloane B.F., Fridman R., Kim H.R. Tissue inhibitor of metalloproteinase-1 protects human breast epithelial cells from extrinsic cell death: a potential oncogenic activity of tissue inhibitor of metalloproteinase-1. Cancer Res. (2005);65:898–906. [PubMed] [Google Scholar]
- 21.Lovelock J.D., Baker A.H., Gao F., Dong J.F., Bergeron A.L., McPheat W., Sivasubramanian N., Mann D.L. Heterogeneous effects of tissue inhibitors of matrix metalloproteinases on cardiac fibroblasts. Am. J. Physiol. Heart Circ. Physiol. (2005);288:H461–468. doi: 10.1152/ajpheart.00402.2004. [DOI] [PubMed] [Google Scholar]
- 22.Mirza A.M., Gysin S., Malek N., Nakayama K., Roberts J.M., McMahon M. Cooperative regulation of the cell division cycle by the protein kinases RAF and Akt. Mol. Cell. Biol. (2004);24:10868–10881. doi: 10.1128/MCB.24.24.10868-10881.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park S.Y., Song C.Y., Kim B.C., Hong H.K., Lee H.S. Angiotensin II mediates LDL-induced superoxide generation in mesangial cells. Am. J. Physiol. Renal. Physiol. (2003);285:F909–915. doi: 10.1152/ajprenal.00160.2003. [DOI] [PubMed] [Google Scholar]
- 24.Porter J.F., Shen S., Denhardt D.T. Tissue inhibitor of metalloproteinase-1 stimulates proliferation of human cancer cells by inhibiting a metalloproteinase. Br. J. Cancer. (2004);90:463–470. doi: 10.1038/sj.bjc.6601533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Porter J.F., Sharma S., Wilson D.L., Kappil M.A., Hart R.P., Denhardt D.T. Tissue inhibitor of metalloproteinases-1 stimulates gene expression in MDA-MB-435 human breast cancer cells by means of its ability to inhibit metalloproteinases. Breast Cancer Res. Treat. (2005);94:185–193. doi: 10.1007/s10549-005-7728-4. [DOI] [PubMed] [Google Scholar]
- 26.Saika S., Kawashima Y., Okada Y., Tanaka S.I., Yamanaka O., Ohnishi Y., Ooshima A. Recombinant TIMP-1 and -2 enhance the proliferation of rabbit corneal epithelial cells in vitro and the spreading of rabbit corneal epithelium in situ. Curr. Eye Res. (1998);17:47–52. doi: 10.1076/ceyr.17.1.47.5247. [DOI] [PubMed] [Google Scholar]
- 27.Sherr C.J. The Pezcoller lecture: cancer cell cycles revisited. Cancer Res. (2000);60:3689–3695. [PubMed] [Google Scholar]
- 28.Stetler-Stevenson W.G., Krutzsch H.C., Liotta L.A. Tissue inhibitor of metalloproteinase (TIMP-2). A new member of the metalloproteinase inhibitor family. J. Biol. Chem. (1989);264:17374–17378. [PubMed] [Google Scholar]
- 29.Taube M.E., Liu X.W., Fridman R., Kim H.R. TIMP-1 regulation of cell cycle in human breast epithelial cells via stabilization of p27(KIP1) protein. Oncogene. (2006);25:3041–3048. doi: 10.1038/sj.onc.1209336. [DOI] [PubMed] [Google Scholar]
- 30.Tchernev G., Orfanos C.E. Downregulation of cell cycle modulators p21, p27, p53, Rb and proapoptotic Bcl-2-related proteins Bax and Bak in cutaneous melanoma is associated with worse patient prognosis: preliminary findings. J. Cutan. Pathol. (2007);34:247–256. doi: 10.1111/j.1600-0560.2006.00700.x. [DOI] [PubMed] [Google Scholar]
- 31.Wang T., Yamashita K., Iwata K., Hayakawa T. Both tissue inhibitors of metalloproteinases-1 (TIMP-1) and TIMP-2 activate Ras but through different pathways. Biochem. Biophys. Res. Commun. (2002);296:201–205. doi: 10.1016/s0006-291x(02)00741-6. [DOI] [PubMed] [Google Scholar]
- 32.Yoshiji H., Kuriyama S., Yoshii J., Ikenaka Y., Noguchi R., Nakatani T., Tsujinoue H., Yanase K., Namisaki T., Imazu H., et al. Tissue inhibitor of metalloproteinases-1 attenuates spontaneous liver fibrosis resolution in the transgenic mouse. Hepatology. (2002);36:850–860. doi: 10.1053/jhep.2002.35625. [DOI] [PubMed] [Google Scholar]
- 33.Zhu D., Bourguignon L. Overexpression of CD44 in pl85(neu)-transfected NIH3T3 cells promotes an up-regulation of hyaluronic acid-mediated membrane-cytoskeleton interaction and cell adhesion. Oncogene. (1996);12:2309–2314. [PubMed] [Google Scholar]