

Abstract
Platelet-derived growth factors (PDGFs) are critical for development; their over-expression is associated with fibrogenesis. Full-length PDGF-C is secreted as an inactive dimer, requiring cleavage to allow receptor binding. Previous studies indicate that tissue-type plasminogen activator (tPA) is the specific protease that performs this cleavage; in vivo confirmation is lacking. We demonstrate that primary hepatocytes from tpa KO mice produce less cleaved active PDGF-CC than do wild type hepatocytes, suggesting that tPA is critical for in vitro activation of this growth factor. We developed mice that over-express full-length human PDGF-C in the liver; these mice develop progressive liver fibrosis. To test whether tPA is important for cleavage and activation of PDGF-C in vivo, we intercrossed PDGF-C transgenic (Tg) and tpa knock-out (KO) mice, anticipating that lack of tPA would result in decreased fibrosis due to lack of hPDGF-C cleavage. To measure levels of cleaved, dimerized PDGF-CC in sera, we developed an ELISA that specifically detects cleaved PDGF-CC. We report that the absence of tpa does not affect the phenotype of PDGF-C Tg mice. hPDGF-C Tg mice lacking tPA have high serum levels of cleaved growth factor, significant liver fibrosis, and gene expression alterations similar to those of PDGF-C Tg mice with intact tPA. Furthermore, urokinase plasminogen activator and plasminogen activator inhibitor-1 expression was increased in PDGF-C Tg; tpa KO mice. Our ELISA data suggest a difference between in vitro and in vivo activation of this growth factor, and our mouse model confirms that multiple proteases cleave and activate PDGF-C in vivo.
Keywords: platelet-derived growth factor, plasminogen activator, liver fibrosis
Introduction
Tissue fibrosis results from an imbalance in the metabolism of extracellular matrix proteins, and the plasminogen activation system is a critical regulator of this balance [1–3]. Tissue plasminogen activator (tPA) is a serine protease first discovered via its ability to cleave plasminogen to its active form, plasmin [1–4]. Given that tPA has a specific affinity for fibrin, its ability to activate plasminogen is more restricted to areas of thrombosis than those of other plasminogen activators. Mice deficient in tPA develop and reproduce normally and have a normal life span, though they display decreased thrombolytic potential and increased endotoxin-induced thrombosis [5]. In the liver, tPA appears to contribute to liver repair after injury [6], though uninjured tpa knock-out (KO) mice have histologically normal livers[5]. tPA, along with urokinase plasminogen activator (uPA) and plasmin, is additionally important in the degradation of collagen [4]. The balance of creation and degradation of collagen determines the composition of the extracellular matrix (ECM) of multiple organs, including the liver, and imbalance of these systems can lead to organ fibrosis. Increased activity of plasminogen activator inhibitor-1 (PAI-1), which inhibits tPA and uPA function, has been shown to lead to fibrosis, for example [1]. The ECM pf the liver contains latent growth factors, which upon cleavage after injury can lead to cell proliferation or inflammation, suggesting that the fibrinolytic pathway could have additional functions in the ECM of the liver, beyond collagen homeostasis.
The platelet-derived growth factor (PDGF) family has been extensively studied over the past 40 years, and these ligands are required for normal development [7]. PDGFs appear to be critical for adult physiologic functions, as their overexpression results in a number of pathological responses via mesenchymal cells, including liver fibrosis and cirrhosis [8–10]. PDGF-C and PDGF-D specifically are secreted as latent factors, from which the complement subcomponents Clr/Cls, Uegf, Bmp1 (CUB) domain must be cleaved to allow the active growth factor dimers to bind their receptors [11, 12]. Activated PDGF-CC binds to either PDGFRα homodimers or PDGFRα/β heterodimers to initiate intracellular signaling cascades, leading to diverse cellular processes such as proliferation and migration. Upon the initial discovery of PDGF-C, it was suggested that plasmin was the primary protease responsible for cleavage of the CUB domain from the growth factor domain, thus allowing downstream receptor activation [12]; a second group of investigators demonstrated that this cleavage is serum-dependent [11]. Later studies in fibroblasts demonstrated that tPA has the ability to cleave full-length PDGF-C to the active form [13–15]. Further, it has been suggested that activation of latent PDGF-C by tPA in ischemic stroke is responsible for the neurotoxic side effects of tPA in this setting; use of imatinib, a kinase inhibitor whose targets include PDGFRs, has been suggested for use in stroke patients to prevent this toxicity [15, 16]. Direct in vivo studies confirming the relationship between tPA and PDGF-C activation are lacking, however.
We have recently developed a mouse model in which liver fibrosis, angiogenesis, and tumor development are driven by hepatocyte-specific over-expression of human PDGF-C [17]. Elevated serum levels of cleaved, activated growth factor in these mice are associated with up-regulation of pro-fibrotic genes and liver fibrosis that progresses with age [17]. To maintain high levels of PDGF-CC, ongoing cleavage of full length PDGF-C by an in situ protease or proteases is required. The purpose of the current study was to determine whether tPA is indeed the protease responsible for activation of PDGF-C in the liver in an in vivo system. To this end, we intercrossed PDGF-C transgenic (Tg) mice with those deficient for tPA. Here we report that tPA appears important for PDGF-C cleavage in vitro, including in primary hepatocytes, but in the more complex in vivo environment cleavage and activation of the growth factor occurs normally in the absence of tPA. Moreover, PDGF-C Tg; tpa KO mice display a histologic phenotype identical to that of PDGF-C Tg mice with intact tPA. Our findings suggest that tPA is not the sole protease that is important for in vivo cleavage and activation of PDGF-C, and that there is significant redundancy amongst PDGF-C-activating proteases.
Materials and Methods
Enzyme-linked Immunosorbent Assay (ELISA) for Determination of PDGF-CC in Serum
We use the following nomenclature for PDGF-C protein: PDGF-C = non-cleaved, full-length protein including the growth factor domain and the CUB domain; PDGF-CC = cleaved, dimerized, activated growth factor domain. Levels of PDGF-CC in media or mouse sera were quantified using an antibody capture ELISA with two monoclonal antibodies specific for human PDGF-CC, as described previously [11]. Other ligands tested to validate the specificity of the ELISA included PDGF-AA, PDGF-AB, PDGF-BB, PDGF-CC and PDGF-DD (all from R&D), as well as full-length (FL) PDGF-C and PDGF-CC isolated from conditioned media derived from BHK cells overexpressing these two constructs (indicated as ‘z’ since they were developed at Zymogenetics) [11].
Culture of Primary Hepatocytes and Adenovirus Infection
Primary hepatocytes were isolated by collagenase perfusion and Percoll purification of wild type and tpa KO mice and cultured as previously described [18]. Primary hepatocytes were infected with adenoviruses over-expressing Lac-Z or full-length human PDGF-C [11] at an MOI of 2000; uninfected cells served as an additional control. Twenty-four hours after infection, infectious media were washed off and replaced with feeding media containing EGF (10ng/ml). Three days after plating, media were collected for detection of PDGF-CC using the PDGF-CC specific ELISA; and hepatocytes were harvested for RNA analysis as described below.
Animal studies
The generation and characterization of PDGF-C Tg mice has been previously described [17]. PDGF-C Tg mice were crossed with mice deficient for tPA (Plat tm1Mlg/J, Jackson Laboratories stock number 002508, referred to as tpa KO) to generate mice with the four genotypes used in study: wild type, tpa KO (lacking both tpa alleles), PDGF-C Tg, and PDGF-C Tg; tpa KO (expressing the PDGF-C transgene and lacking both tpa alleles). All mice were on the C57BL/6 background; mice heterozygous for tpa were initially kept and examined, but as we did not see any evidence of a gene dosage effect, tpa+/− were not used in our final analyses. Bromodeoxyuridine (BrdU, 50 μg/kg body weight) was injected intraperitoneally (IP) two hours prior to necropsy as described [17, 19]. At the indicated ages, mice were sacrificed by CO2 inhalation, sera collected by cardiac puncture, and livers and spleens harvested, weighed, and fixed or snap frozen for analysis. All animal studies were carried out under approved Institutional Animal Care and Use Committee protocols at the University of Washington.
Histology
Mouse livers were fixed in 10% neutral buffered formalin or Methacarn (60% methanol, 30% chloroform and 10% acetic acid: v/v/v) overnight, processed to paraffin blocks, sectioned and stained with hematoxylin and eosin (H&E), Picrosirius red, or trichrome using standard techniques. Immunostaining was performed on methacarn fixed sections as described [17, 19, 20] using a primary antibody specific for BrdU (Dako, Carpinteria, CA) and the mouse on mouse kit (Vector Labs). Nuclear incorporation of BrdU into both hepatocytes and non-parenchymal cells (NPCs) was used to measure cell proliferation; data are presented as the number of BrdU positive hepatocyte nuclei or NPCs present in thirty 400X fields (1.3 mm2; approximately 3000 hepatocytes). Morphometric analyses of liver fibrosis were performed on Picrosirius Red and Trichrome stained sections magnified to 40x using a method similar to described [21], using NIH Image J software.
RNA Isolation and Real-time PCR
Mouse livers or primary hepatocytes were harvested with TRIzol Reagent (Invitrogen) per the manufacturer’s protocol. 1μg of RNA template was used for reverse transcription by MMLV-RT (Applied Biosystems, Carlsbad, CA) per the manufacturer’s protocol. 50ng of the resultant cDNA were subjected to real-time PCR analysis using the following FAM labeled TaqMan primers (Applied Biosystems): 18s, Pdgfrα, Pdgfrβ, Col1a1, Col3, Col4a, Plat, Plau, and Serpine1; SYBR Green-labeled primers were used to detect expression of human 18S (Ambion, Life Technologies, Grand Island, NY) or PDGF-C (5′ ACAGAGGAGGTAAGATTA TACAGC 3′, 5′ AATTGTGGAGACAACAGGCACAGT 3′). A RotorGene 3000 thermocycler (Corbett Research, San Francisco, CA) was used for detection, and the ΔΔCt method was used to calculate fold change normalized to 18s or 18S [22].
Statistical analyses
Statistical analysis was done by non-parametric analysis using the Mann-Whitney U t-test using GraphPad Prizm software (GraphPad for Science Inc, San Diego CA). Data are presented as average +/− S.E.M., with p<0.05 (*) considered statistically significant.
Results
Tpa KO hepatocytes have deficient cleavage of PDGF-C
To determine whether the profound phenotype of PDGF-C Tg mice was solely dependent on cleavage of PDGF-C by tPA, we first needed an assay that could specifically measure cleaved, active PDGF-CC. We thus developed an ELISA for cleaved, activated PDGF-CC that does not cross-react with other members of the PDGF family (Figure 1A). Using full-length (FL) and cleaved growth factor forms of the C ligand at varying concentrations, we next demonstrated that the ELISA is specific for the cleaved form, and does not cross-react with the full-length secreted protein (1B). Thus, PDGF-AA, PDGF-AB, PDGF-BB, PDGF-DD, and FL-PDGF-C are undetectable by this assay. Given prior in vitro data in fibroblasts suggesting that tPA is the protease responsible for cleavage of PDGF-C, prior to testing our Tg mouse model, we wondered whether primary hepatocytes isolated from wild type and tpa KO would have an equivalent ability to cleave PDGF-C. We infected these primary hepatocytes with adenoviral vectors over-expressing human PDGF-C, LacZ, or a scrambled control sequence. We subjected conditioned media from infected hepatocytes to our specific ELISA, and found that tpa KO hepatocytes had lower levels of cleaved active growth factor 3 days after infection with Ad-PDGF-C than did wild type hepatocytes (Figure 1C). The decrease in protein levels does not appear to be due to differences in adenoviral PDGF-C transduction in wild type and tpa KO hepatocytes, as similar levels of hPDGF-C mRNA were detected in hepatocytes of each genotype (Figure 1D)., These in vitro studies suggest that tPA does cleave PDGF-C in primary hepatocytes, corroborating previous observations that tPA cleaves PDGF-C in fibroblast cultures [14].
Figure 1. A PDGF-CC specific ELISA demonstrates in vitro dependence on tPA for PDGF-C cleavage.

We developed an ELISA specific for cleaved, active PDGF-CC; this ELISA does not recognize other PDGFs (A) or full length (FL) PDGF-C (B). PDGFs –AA, -AB, -BB, and -DD were not detectable by this assay. Ligands were purified from BHK over-expressing cells as previously described [11] and are indicated by “z”, or purchased from R&D. We isolated wild type and tpa KO primary hepatocytes and infected them with adenoviruses over-expressing PDGF-C. ELISA measurements of cleaved, activated PDGF-CC in media from infected wild type and tpa KO cells are shown (C). We achieved similarly high levels of PDGF-C expression in vitro (by qPCR and normalized to 18S, D). Ad-Lac-Z infected and uninfected cells are shown as controls. * indicates p < 0.05. Data are representative of two separate experiments, each done in triplicate.
PDGF-C Tg; t-PA KO mice are born at the expected frequency
Given the published evidence that tPA protease activity is critical for cleavage and activation of PDGF-C in vitro, and our findings in primary hepatocytes, we intercrossed tpa KO mice with mice over-expressing hPDGF-C in the liver to examine the interaction between these two proteins in vivo. Given that PDGF-C Tg mice with intact tPA develop progressive liver fibrosis [17], we reasoned that if the inactive full-length PDGF-C dimer could not be cleaved by tPA, this phenotype would not develop. We found that PDGF-C Tg; tpa KO mice develop and breed normally, and do not have an overt phenotype other than expected consequences of PDGF-C overexpression. Supplementary table 1 depicts the birth frequencies of the progeny resulting from our crosses, and demonstrates that PDGF-C Tg; tpa KO mice were born at the expected Mendelian ratio.
PDGF-C Tg; tpa KO mice develop hepatomegaly, splenomegaly, and cleave PDGF-C
PDGF-C over-expression drives enhanced proliferation of both hepatocytes and NPCs in our Tg model, leading to hepatomegaly [17, 19]. PDGF-C Tg mice also have splenomegaly, either due to parenchymal proliferation or portal hypertension [17]. Figure 2, panels A & B demonstrate that the increased size of these organs in PDGF-C Tg mice was still present in the absence of tPA. If tPA were necessary for activation of PDGF-C in vivo, we would expect this hyper-proliferative state to be abolished in Tg animals lacking tPA. There were no differences between liver and spleen size between wild type and tpa KO mice. Further, Figure 2, panels C & D demonstrate equivalent BrdU incorporation in hepatocytes and NPCs of 6-month-old animals over-expressing PDGF-C with or without tpa expression. We then performed the ELISA described in Figure 1 on sera from wild type, tpa KO, PDGF-C Tg, and PDGF-C Tg; tpa KO mice. Surprisingly, we found that PDGF-C Tg; tpa KO mice had nearly identical levels of PDGF-CC in serum as did PDGF-C Tg mice with intact tPA at one month (Figure 2E) and six months (2F) of age, suggesting that tPA is not necessary for activating PDGF-C in the liver. We hypothesize that in the complex in vivo environment, other proteases may compensate for lack of tPA than in the simplified cell culture environment, explaining the deficit in growth factor cleavage in tpa KO cells in vitro, while in vivo cleavage of PDGF-C is intact in the absence of tPA.
Figure 2. Organ size, liver BrdU Incorporation, and PDGF-C cleavage in PDGF-C Tg mice do not depend on tPA.

Weights of mouse whole liver (A) and spleen (B) relative to the body weight of the animal at six months of age are shown. Liver sections from six-month-old mice were stained with BrdU to assess cell proliferation in hepatocytes (C) and non-parenchymal cells (D). BrdU positive cells were counted in thirty 400X fields for a total of 3000 cells evaluated (n=6–12 per genotype). Serum levels of activated PDGF-CC were determined in 1-month (E) and 6-month-old (F) wild type, tpa KO, PDGF-C Tg, and PDGF-C Tg; tpa KO mice, demonstrating that lack of tPA does not affect the circulating levels of activated PDGF-CC (n=6–12 per genotype at each age.)
PDGF-C overexpression drives liver fibrosis with or without tPA
We previously showed that hepatic over-expression of PDGF-C results in pericellular and perivenular fibrosis by 6 weeks of age, accompanied by increased peri-sinusoidal α-smooth muscle actin and glial fibrillary acidic protein expression [17]. H&E staining of liver sections from PDGF-C Tg and PDGF-C Tg; tpa KO mice revealed a similar pattern of liver architecture abnormalities (data not shown). We then performed picrosirius red staining to specifically assess liver fibrosis in 3-month-old PDGF-C Tg mice with and without tPA. Minimal picrosirius red staining was seen in wild type littermates, an example of which is shown as a control (Figure 3A). Confirming previous work, we found that PDGF-C Tg mice with intact tpa have a moderate amount of fibrosis at this age (Figure 3B), and that PDGF-C Tg; tpa KO mice (Figure 3C) have fibrosis of qualitatively similar severity. To more precisely quantify fibrosis in PDGF-C Tg vs PDGF-C Tg; tpa KO mice, we additionally performed morphometric analyses on these picrosirius red stained sections, and found virtually identical amounts of fibrosis in mice of these two genotypes (Figure 3D). To confirm these results, we also performed trichrome staining at this same time point, and did not detect any significant fibrosis in wild type animals (Figure 3E), nor did we detect a difference in the amount of collagen deposition between PDGF-C Tg (Figure 3F) and PDGF-C Tg; tpa KO mice (Figure 3G, quantified in 3H). We thus conclude that PDGF-C Tg mice develop fibrosis independent of tPA. Since we also detect cleaved PDGF-CC in the absence of tPA, we hypothesize that there are other proteases acting on the full-length ligand to cleave and activate PDGF-CC.
Figure 3. PDGF-C-driven liver fibrosis does not require tPA.

Picroirius red staining was used to assess the development of liver fibrosis in wild type (A), PDGF-C Tg (B), and PDGF-C Tg; tpa KO (C) mice at 3 months of age. Scale bar indicates 100 microns. Morphometric analysis of Picrosirius red stained sections reveals equivalent amounts of fibrosis in PDGF-C Tg vs. PDGF-C Tg; tpa KO mice (D). n=3–4 mice/genotype. Trichrome staining in wild type (E), PDGF-C Tg (F), and PDGF-C Tg; tpa KO (G) mice confirmed similar levels of fibrosis with or without tpa (H). Scale bar indicates 50 microns.
Modulation of gene expression in PDGF-C Tg mice by tpa
The fibrosis seen in PDGF-C Tg livers is preceded by up-regulation of collagen gene expression. To determine whether tPA activity modulates collagen expression in PDGF-C Tg mice, we performed qPCR for the collagens Col1a1, Col3, and Col4 on RNA extracted from whole livers from 3-month-old wild type, tpa KO, PDGF-C Tg, and PDGF-C Tg; tpa KO mice. As expected given the amount of fibrosis in these animals, PDGF-C Tg mice had marked induction of all three genes versus wild type or tpa KO mice (Figure 4A–C). Interestingly, Col1a1 and Col4 expression were significantly higher in PDGF-C Tg; tpa KO livers when compared to PDGF-C Tg livers at this age, though there was no difference in Col3 expression with and without tPA. In our PDGF-C Tg mouse model, we have demonstrated that over-expression of PDGF-C leads to up-regulation of expression of its receptor components, PDGFR-α and -β (ref Figure 4D, E). Using qPCR on the samples listed above, we found that the absence of tPA did not affect expression of Pdgfrα, but slightly increased expression of Pdgfrβ, in PDGF-C Tg mice. These data suggest that tPA may subtly temper pro-fibrotic signaling in PDGF-C Tg mice, though we did not detect obvious differences in our histologic analyses (Figure 3).
Figure 4. Differential gene expression in PDGF-C Tg vs PDGF-C Tg;tpa KO mice.

qPCR was used to assess expression of Col1a1 (A), Col3 (B), Col4 (C), Pdgfrα (D), and Pdgfrβ (E) in wild type, tpa KO, PDGF-C Tg, and PDGF-C Tg; tpa KO mice at three months of age. Gene expression data were normalized to that of 18s, and graphed as fold change relative to expression in wild type mice. * indicates p < 0.05. n=3–6 per genotype.
Both tPA and uPA have the ability to convert plasminogen to its activated form, plasmin, while PAI-1 inhibits the activity of both of these proteases [3]. Both proteases have anti-fibrotic properties as well, and we wondered whether expression of other components of this pathway could compensate for the lack of tPA activity in PDGF-C Tg; tpa KO mice. To evaluate how this pathway could be affected by over-expression of PDGF-C and/or loss of tpa in the liver, we measured expression of these genes in 3-month-old mice of our four genotypes by qPCR. We found a marked up-regulation of the gene for tPA (Plat) in PDGF-C Tg mice compared to wild type littermates, and additionally confirmed the complete absence of the gene for Plat expression in tpa KO and PDGF-C Tg; tpa KO mice (Figure 5A). Interestingly, both the gene for uPA (Plau) and the gene for PAI1 (Serpine1) were up-regulated in PDGF-C Tg mice, and expression of both of these genes was further significantly enhanced by the absence of tpa (Figure 5B, C). These data suggest that uPA could be compensating for loss of tPA activity in PDGF-C Tg; tpa KO mice, and that the massive up-regulation of tPA and uPA in PDGF-C Tg mice is associated with a concomitant increase expression of their endogenous inhibitor, PAI-1. The latter fact may explain the absence of a significant difference in ECM deposition in PDGF-C Tg vs PDGF-C Tg; tpa KO mice (Figure 3) despite the observed differences in expression of genes for collagens and PDGFRs (Figure 4).
Figure 5. Genes of the plasminogen cascade are increased in PDGF-C Tg;tpa KO mice.
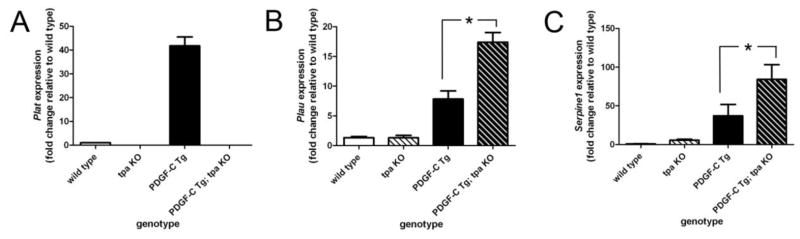
qPCR was used to assess expression of the genes for tPA (Plat, A), uPA (Plau, B), and PAI-1 (Serpine1, C) in three-month-old wild type, tpa KO, PDGF-C Tg, and PDGF-C Tg; tpa KO mice. Gene expression data were normalized to that of 18s, and graphed as fold change relative to expression in wild type mice. * indicates p < 0.05. n=3–6 per genotype.
Discussion
The main finding of the present study is that tPA is not essential for cleavage and activation of PDGF-C in vivo. While we demonstrated functional activation of PDGF-C by tPA in primary hepatocytes infected with adenoviruses over-expressing the inactive growth factor (Figure 1), corroborating prior work in vitro [13], these findings were not replicated in our Tg mouse model. In fact, mice that over-express PDGF-C in the liver in the absence of tPA were phenotypically similar to those transgenic for PDGF-C alone (Figures 2–3). These data suggest that another protease (or proteases) cleaves and activates PDGF-C in physiologic and pathologic conditions in the liver. Previously published work suggested that tPA is critical in activating PDGF-C to enhance blood brain barrier permeability [13, 15, 16]. At this time it is unclear whether the difference between our results and those of Su et al relate to fundamental differences between the brain and the liver, or variability in methods for measuring PDGF-CC and its activity in vivo.
We observed an interaction between tPA and PDGF-C in modulating collagen gene expression (Figure 4) in PDGF-C Tg mice. These data are consistent with those of multiple other investigators, which demonstrate that the fibrinolytic pathway modulates the development of fibrosis in multiple organs, and the liver in particular. For instance, Hsiao et al induced liver fibrosis in wild type and tpa KO mice with carbon tetrachloride, and found that tpa KO mice had increased α-smooth muscle expression and more severe fibrosis than did wild type mice. Contrary to these findings, Higazi et al performed similar experiments in upa and tpa KO mice, and found that both strains of KO animals had decreased fibrosis after chronic carbon tetrachloride treatment [23]. In humans, circulating levels of the receptor for uPA (uPAR) correlated with increasing stage of liver fibrosis in patients with hepatitis C virus [24], and another group found that both uPA and soluble uPAR levels were elevated in patients with cirrhosis, independent of etiology. Moreover, soluble uPAR levels correlate with liver function and systemic inflammation and have prognostic value for certain patients, particularly those with alcoholic liver disease [25, 26]. The mechanisms by which plasminogen activators may modulate fibrosis include activation of matrix metalloproteinases [27], which degrade the ECM, and activation of protective growth factors such as hepatocyte growth factor (HGF). Some authors have proposed the use of plasminogen activators as anti-fibrotic therapy, though clinical data are lacking. Regardless, despite the elevation in collagen gene expression in PDGF-C Tg;tpa KO mice vs. Tg mice, we could not detect a histologic difference in fibrosis (Figure 3), perhaps due to the associated up-regulation of upa.
Although previous studies indicated that tPA was the primary protease responsible for PDGF-C cleavage [14, 15], other studies suggest redundancy in this proteolytic activity [11, 12]. For instance in MCF7 cells, a breast cancer cell line, uPA and matriptase have been identified as proteases that can generate PDGF-CC, in addition to tPA [28]. Another group studied activation of PDGF-C in the vitreous humor of patients with proliferative vitreoretinopathy, and identified plasmin as the primary PDGF-C-cleaving protease [29], similar to the findings of the group of investigators who originally described PDGF-C [12]. It is likely that multiple proteases are able to cleave and activate PDGF-C, and that they are able to compensate for one another when necessary. All of these cleavage events seem to require Lys225 and/or Arg231, 234, which are located in the hinge region between the growth factor domain and the CUB domain [30], [28], [11]. Interestingly, Eriksson’s group recently demonstrated that uPA appears to activate PDGF-D, and that uPA and its receptor uPAR regulate the spatial distribution of this growth factor [31].
Mice deficient for tPA were initially developed and characterized in the 1990s [5], and have normal development, life span, and baseline histology of all organs, though their thrombolytic potential is lower than that of wild type mice. Interestingly upa KO mice develop spontaneous fibrin deposits, whereas tpa KO mice do not, and tpa KO mice do not have compensatory increased baseline activity of other proteases (e.g. uPA). We found that in tpa KO mice with the added stress of PDGF-CC induced liver fibrosis, however, there was significant compensatory up-regulation of upa (Figure 5), as well as Serpine1, the gene encoding PAI1, the primary physiologic inhibitor of tPA and uPA. These data are in concert with the notion that these two plasminogen activators are also vital to the body’s defenses against fibrosis, and suggest that the compensatory up-regulation of upa in PDGF-C Tg; tpa KO mice prevents more severe liver fibrosis in these animals. We suspect that a PDGF-C Tg; upa KO mouse would have more severe fibrosis, due to the phenotype of upa KO mice compared to tpa KO mice [5].
Conclusions
PDGF-C Tg; tpa KO mice have a phenotype nearly identical to that of PDGF-C Tg mice, thus tPA is not necessary for cleavage and activation of PDGF-C in the liver. Our findings suggest that multiple proteases are able to cleave and activate PDGF-C, not exclusively tPA, and call into question previous work on the interplay of these two proteins in health and disease.
Supplementary Material
tPA has been proposed as the primary protease that activates PDGF-C.
We crossed PDGF-C Tg mice with tPA KO mice to evaluate in vivo importance of tPA.
PDGF-C Tg; tPA KO mice had high levels of cleaved PDGF-CC by specific ELISA.
PDGF-C Tg; tPA KO mice develop fibrosis equivalent to PDGF-C Tg mice.
tPA is not critical for in vivo cleavage of PDGF-C in the liver.
Acknowledgments
This work was supported by NIH grants CA-23226 and CA-74131 (to N. Fausto) and CA-127228 (to JS Campbell), the Herbert S. Coe Foundation, American College of Surgeons Louis C. Argenta Fellowship, the American Surgical Association Foundation Fellowship (to KJ Riehle), and the Howard Hughes Medical Institute program in Molecular Medicine and NIH-HL007312 (to BJ Hayes). We thank James Surapisitchat for technical assistance.
Abbreviations
- PDGF
platelet-derived growth factor
- tPA
tissue-type plasminogen activator
- Tg
transgenic
- KO
knock-out
- uPA
urokinase plasminogen activator
- ECM
extracellular matrix
- PAI-1
plasminogen activator inhibitor-1
- IP
intraperitoneally
- CUB
complement subcomponents Clr/Cls, Uegf, Bmp1
- BrdU
Bromodeoxyuridine
- H&E
hematoxylin & eosin
- NPC
non-parenchymal cell
- HCC
hepatocellular carcinoma
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ghosh AK, Vaughan DE. PAI-1 in tissue fibrosis. Journal of cellular physiology. 2012;227:493–507. doi: 10.1002/jcp.22783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zorio E, Gilabert-Estelles J, Espana F, Ramon LA, Cosin R, Estelles A. Fibrinolysis: the key to new pathogenetic mechanisms. Current medicinal chemistry. 2008;15:923–929. doi: 10.2174/092986708783955455. [DOI] [PubMed] [Google Scholar]
- 3.Irigoyen JP, Munoz-Canoves P, Montero L, Koziczak M, Nagamine Y. The plasminogen activator system: biology and regulation. Cellular and molecular life sciences: CMLS. 1999;56:104–132. doi: 10.1007/PL00000615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kang LI, Mars WM. Fibrinolytic factors in liver fibrosis. Current pharmaceutical biotechnology. 2011;12:1441–1446. doi: 10.2174/138920111798281036. [DOI] [PubMed] [Google Scholar]
- 5.Carmeliet P, Schoonjans L, Kieckens L, Ream B, Degen J, Bronson R, De Vos R, van den Oord JJ, Collen D, Mulligan RC. Physiological consequences of loss of plasminogen activator gene function in mice. Nature. 1994;368:419–424. doi: 10.1038/368419a0. [DOI] [PubMed] [Google Scholar]
- 6.Bezerra JA, Currier AR, Melin-Aldana H, Sabla G, Bugge TH, Kombrinck KW, Degen JL. Plasminogen activators direct reorganization of the liver lobule after acute injury. Am J Pathol. 2001;158:921–929. doi: 10.1016/S0002-9440(10)64039-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fredriksson L, Li H, Eriksson U. The PDGF family: four gene products form five dimeric isoforms. Cytokine Growth Factor Rev. 2004;15:197–204. doi: 10.1016/j.cytogfr.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 8.Olson LE, Soriano P. Increased PDGFRalpha activation disrupts connective tissue development and drives systemic fibrosis. Developmental cell. 2009;16:303–313. doi: 10.1016/j.devcel.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonner JA, Buchsbaum DJ, Rogers BE, Grizzle WE, Trummell HQ, Curiel DT, Fiveash JB, Ove R, Raisch KP. Adenoviral vector-mediated augmentation of epidermal growth factor receptor (EGFr) enhances the radiosensitization properties of anti-EGFr treatment in prostate cancer cells. International journal of radiation oncology, biology, physics. 2004;58:950–958. doi: 10.1016/j.ijrobp.2003.09.095. [DOI] [PubMed] [Google Scholar]
- 10.Wynn TA. Cellular and molecular mechanisms of fibrosis. The Journal of pathology. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gilbertson DG, Duff ME, West JW, Kelly JD, Sheppard PO, Hofstrand PD, Gao Z, Shoemaker K, Bukowski TR, Moore M, Feldhaus AL, Humes JM, Palmer TE, Hart CE. Platelet-derived growth factor C (PDGF-C), a novel growth factor that binds to PDGF alpha and beta receptor. J Biol Chem. 2001;276:27406–27414. doi: 10.1074/jbc.M101056200. [DOI] [PubMed] [Google Scholar]
- 12.Li X, Ponten A, Aase K, Karlsson L, Abramsson A, Uutela M, Backstrom G, Hellstrom M, Bostrom H, Li H, Soriano P, Betsholtz C, Heldin CH, Alitalo K, Ostman A, Eriksson U. PDGF-C is a new protease-activated ligand for the PDGF alpha-receptor. Nature cell biology. 2000;2:302–309. doi: 10.1038/35010579. [DOI] [PubMed] [Google Scholar]
- 13.Franke K, Curth K, Lenart J, Knochenhauer D, Kietzmann T. Enhanced plasminogen activator inhibitor-1 expression in transgenic mice with hepatocyte-specific overexpression of superoxide dismutase or glutathione peroxidase. Antioxid Redox Signal. 2004;6:721–728. doi: 10.1089/1523086041361613. [DOI] [PubMed] [Google Scholar]
- 14.Fredriksson L, Li H, Fieber C, Li X, Eriksson U. Tissue plasminogen activator is a potent activator of PDGF-CC. EMBO J. 2004;23:3793–3802. doi: 10.1038/sj.emboj.7600397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Su EJ, Fredriksson L, Geyer M, Folestad E, Cale J, Andrae J, Gao Y, Pietras K, Mann K, Yepes M, Strickland DK, Betsholtz C, Eriksson U, Lawrence DA. Activation of PDGF-CC by tissue plasminogen activator impairs blood-brain barrier integrity during ischemic stroke. Nat Med. 2008;14:731–737. doi: 10.1038/nm1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Su EJ, Fredriksson L, Schielke GP, Eriksson U, Lawrence DA. Tissue plasminogen activator-mediated PDGF signaling and neurovascular coupling in stroke. J Thromb Haemost. 2009;7(Suppl 1):155–158. doi: 10.1111/j.1538-7836.2009.03402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Campbell JS, Hughes SD, Gilbertson DG, Palmer TE, Holdren MS, Haran AC, Odell MM, Bauer RL, Ren HP, Haugen HS, Yeh MM, Fausto N. Platelet-derived growth factor C induces liver fibrosis, steatosis, and hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2005;102:3389–3394. doi: 10.1073/pnas.0409722102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Riehle KJ, Campbell JS, McMahan RS, Johnson MM, Beyer RP, Bammler TK, Fausto N. Regulation of liver regeneration and hepatocarcinogenesis by suppressor of cytokine signaling 3. The Journal of experimental medicine. 2008;205:91–103. doi: 10.1084/jem.20070820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Campbell JS, Johnson MM, Bauer RL, Hudkins KL, Gilbertson DG, Riehle KJ, Yeh MM, Alpers CE, Fausto N. Targeting stromal cells for the treatment of platelet-derived growth factor C-induced hepatocellular carcinogenesis. Differentiation. 2007;75:843–852. doi: 10.1111/j.1432-0436.2007.00235.x. [DOI] [PubMed] [Google Scholar]
- 20.Hudkins KL, Gilbertson DG, Carling M, Taneda S, Hughes SD, Holdren MS, Palmer TE, Topouzis S, Haran AC, Feldhaus AL, Alpers CE. Exogenous PDGF-D is a potent mesangial cell mitogen and causes a severe mesangial proliferative glomerulopathy. Journal of the American Society of Nephrology: JASN. 2004;15:286–298. doi: 10.1097/01.asn.0000108522.79652.63. [DOI] [PubMed] [Google Scholar]
- 21.Gately S, Twardowski P, Stack MS, Cundiff DL, Grella D, Castellino FJ, Enghild J, Kwaan HC, Lee F, Kramer RA, Volpert O, Bouck N, Soff GA. The mechanism of cancer-mediated conversion of plasminogen to the angiogenesis inhibitor angiostatin. Proc Natl Acad Sci U S A. 1997;94:10868–10872. doi: 10.1073/pnas.94.20.10868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vaquero J, Campbell JS, Haque J, McMahan RS, Riehle KJ, Bauer RL, Fausto N. Toll-like receptor 4 and myeloid differentiation factor 88 provide mechanistic insights into the cause and effects of interleukin-6 activation in mouse liver regeneration. Hepatology. 2011;54:597–608. doi: 10.1002/hep.24420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Torimura T, Ueno T, Kin M, Taniguchi E, Nakamura T, Inoue K, Sakata R, Hashimoto O, Sakamoto M, Ohira H, Kumashiro R, Sata M, Yano H, Kojiro M, Veitonmaki N, Cao Y. Gene transfer of kringle 1–5 suppresses tumor development and improves prognosis of mice with hepatocellular carcinoma. Gastroenterology. 2006;130:1301–1310. doi: 10.1053/j.gastro.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 24.Schmitz V, Wang L, Barajas M, Gomar C, Prieto J, Qian C. Treatment of colorectal and hepatocellular carcinomas by adenoviral mediated gene transfer of endostatin and angiostatin-like molecule in mice. Gut. 2004;53:561–567. doi: 10.1136/gut.2003.019307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zimmermann HW, Koch A, Seidler S, Trautwein C, Tacke F. Circulating soluble urokinase plasminogen activator is elevated in patients with chronic liver disease, discriminates stage and aetiology of cirrhosis and predicts prognosis. Liver international: official journal of the International Association for the Study of the Liver. 2012;32:500–509. doi: 10.1111/j.1478-3231.2011.02665.x. [DOI] [PubMed] [Google Scholar]
- 26.Zimmermann HW, Reuken PA, Koch A, Bartneck M, Adams DH, Trautwein C, Stallmach A, Tacke F, Bruns T. Soluble urokinase plasminogen activator receptor is compartmentally regulated in decompensated cirrhosis and indicates immune activation and short-term mortality. Journal of internal medicine. 2013 doi: 10.1111/joim.12054. [DOI] [PubMed] [Google Scholar]
- 27.Lee SR, Guo SZ, Scannevin RH, Magliaro BC, Rhodes KJ, Wang X, Lo EH. Induction of matrix metalloproteinase, cytokines and chemokines in rat cortical astrocytes exposed to plasminogen activators. Neuroscience letters. 2007;417:1–5. doi: 10.1016/j.neulet.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 28.Hurst NJ, Jr, Najy AJ, Ustach CV, Movilla L, Kim HR. Platelet-derived growth factor-C (PDGF-C) activation by serine proteases: implications for breast cancer progression. Biochem J. 2012;441:909–918. doi: 10.1042/BJ20111020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lei H, Velez G, Hovland P, Hirose T, Kazlauskas A. Plasmin is the major protease responsible for processing PDGF-C in the vitreous of patients with proliferative vitreoretinopathy. Investigative ophthalmology & visual science. 2008;49:42–48. doi: 10.1167/iovs.07-0776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fredriksson L, Ehnman M, Fieber C, Eriksson U. Structural requirements for activation of latent platelet-derived growth factor CC by tissue plasminogen activator. J Biol Chem. 2005;280:26856–26862. doi: 10.1074/jbc.M503388200. [DOI] [PubMed] [Google Scholar]
- 31.Ehnman M, Li H, Fredriksson L, Pietras K, Eriksson U. The uPA/uPAR system regulates the bioavailability of PDGF-DD: implications for tumour growth. Oncogene. 2009;28:534–544. doi: 10.1038/onc.2008.410. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.