

Abstract
The endocardium forms the inner lining of the heart tube, where it enables blood flow and also interacts with the myocardium during the formation of valves and trabeculae. Although a number of studies have identified regulators of the morphogenesis of the myocardium, relatively little is known about the molecules that control endocardial morphogenesis. Prior work has implicated the bHLH transcription factor Tal1 in endocardial tube formation: in zebrafish embryos lacking Tal1, endocardial cells form a disorganized mass within the ventricle and do not populate the atrium. Through blastomere transplantation, we find that tal1 plays a cell-autonomous role in regulating endocardial extension, suggesting that Tal1 activity influences the behavior of individual endocardial cells. The defects in endocardial behavior in tal1-deficient embryos originate during the earliest steps of endocardial morphogenesis: tal1-deficient endocardial cells fail to generate a cohesive monolayer at the midline and instead pack tightly together into a multi-layered aggregate. Moreover, the tight junction protein ZO-1 is mislocalized in the tal1-deficient endocardium, indicating a defect in intercellular junction formation. In addition, we find that the tal1-deficient endocardium fails to maintain its identity; over time, a progressively increasing number of tal1-deficient endocardial cells initiate myocardial gene expression. However, the onset of defects in intercellular junction formation precedes the onset of ectopic myocardial gene expression in the tal1-deficient endocardium. We therefore propose a model in which Tal1 has distinct roles in regulating the formation of endocardial intercellular junctions and maintaining endocardial identity.
Keywords: zebrafish, cardiac morphogenesis, endocardium, heart tube assembly, scl
INTRODUCTION
In its initial embryonic form, the vertebrate heart is a simple two-layered tube, in which an outer layer of muscular myocardium surrounds an inner layer of vascular endocardium. Both layers of the heart tube are essential for cardiac function: while the myocardium is responsible for the contractility that propels circulation, its endocardial lining provides continuity with the rest of the embryonic vessels. Furthermore, signaling between the endocardium and myocardium plays a fundamental role in controlling later steps of chamber growth, valve formation, and trabeculation (Chang et al., 2004; Delaughter et al., 2011; Stankunas et al., 2008; Wagner and Siddiqui, 2007). Therefore, the mechanisms controlling assembly of the two-layered heart tube are of great interest.
In all vertebrates, heart tube assembly begins with the migration of bilateral groups of endocardial and myocardial cells from the anterior lateral plate mesoderm (ALPM) to the embryonic midline (Evans et al., 2010). In amniotes, migrating endocardial cells seem to attach to the medial endoderm, where they will eventually merge into a central vessel (Aleksandrova et al., 2012; DeRuiter et al., 1992; Drake et al., 2006; Viragh et al., 1989). At the same time, cardiomyocytes meet at the midline to form the cardiac crescent, which then remodels into a myocardial tube that encases the endocardium (Abu-Issa and Kirby, 2008; DeRuiter et al., 1992; Moreno-Rodriguez et al., 2006).
Similar processes of myocardial and endocardial fusion take place in the zebrafish, where time-lapse analyses have been valuable in revealing the patterns of cell movement that underlie heart tube assembly (Baker et al., 2008; Bussmann et al., 2007; de Campos-Baptista et al., 2008; Fish et al., 2011; Holtzman et al., 2007; Proulx et al., 2010; Rohr et al., 2008; Smith et al., 2008). In zebrafish, bilateral populations of endocardial cells move toward the midline, where they assemble a sheet of tissue (Bussmann et al., 2007). The migrating cardiomyocytes form a ring surrounding and dorsal to the endocardial sheet, setting up the initial orientation of endocardium within the myocardium (Holtzman et al., 2007; Motoike et al., 2000; Stainier et al., 1993). Subsequently, endocardial and myocardial cells move toward the left side of the embryo and then extend to create the two-layered structure of the heart tube (Rohr et al., 2008).
The dynamic changes in tissue organization that occur during heart tube assembly raise a number of intriguing questions about the genetic regulation of this aspect of cardiac morphogenesis. For example, we do not yet fully understand how cardiac cells are recruited toward the midline, what configures the architecture of the endocardial sheet and myocardial ring, or which factors control the transition toward leftward movement and extension. In the case of the myocardium, several requirements for these processes have been revealed. Studies of zebrafish and mouse mutations that impair cardiac fusion have revealed that both the endoderm and the extracellular matrix are required for the initial migration of the myocardium and the formation of a central myocardial ring (Alexander et al., 1999; Arrington and Yost, 2009; Garavito-Aguilar et al., 2010; George et al., 1997; George et al., 1993; Kuo et al., 1997; Kupperman et al., 2000; Molkentin et al., 1997; Trinh and Stainier, 2004). In addition, Nodal and BMP signaling have been shown to be important for the leftward migration of myocardial cells (de Campos-Baptista et al., 2008; Lenhart et al., 2013; Rohr et al., 2008; Smith et al., 2008; Veerkamp et al., 2013).
Less is known about the mechanisms that regulate endocardial tube formation. Like the myocardium, the endocardium requires interactions with the endoderm to facilitate its movement toward the midline (Alexander et al., 1999; Holtzman et al., 2007; Sugi and Markwald, 1996, 2003). Additionally, Slit/Robo signaling and Vegf signaling have been shown to influence the medial migration of both the endocardium and the myocardium (Fish et al., 2011). However, the genes responsible for organizing the endocardial sheet and directing its transition as it lines the heart tube remain largely mysterious.
It is therefore interesting to investigate the functions of Tal1, a bHLH transcription factor that has been implicated in endocardial morphogenesis (Bussmann et al., 2007). Tal1 (also known as Scl) was originally characterized as a key player in the specification of hematopoietic lineages (Begley et al., 1989; Dooley et al., 2005; Juarez et al., 2005; Patterson et al., 2005; Porcher et al., 1996; Robb et al., 1995; Shivdasani et al., 1995). In addition, Tal1 is important for the formation of major embryonic blood vessels (Dooley et al., 2005; Patterson et al., 2005; Visvader et al., 1998). More recently, studies of a zebrafish tal1 mutant have revealed a phenotype in which endocardial cells aggregate in the ventricular portion of the heart tube and fail to populate the atrial portion (Bussmann et al., 2007). Thus, Tal1 has a potent influence on the formation of the endocardial tube, but the initial causes of the endocardial aggregation in tal1 mutants are not yet clear.
Here, we investigate the impact of tal1 on the cell behaviors that govern endocardial tube formation. Despite the well-known role of tal1 in hematopoietic specification, tal1 does not seem to be required for the specification of an appropriate number of endocardial cells. Instead, we find that tal1 plays a cell-autonomous role in regulating endocardial cell behavior. Furthermore, we find that the endocardial defects in tal1-deficient embryos originate with the disorganization of cell-cell contacts and intercellular junctions within the endocardial sheet at the midline. In addition, we find that tal1 is required for the maintenance of endocardial identity; loss of tal1 function leads to a progressive accumulation of ectopic myocardial gene expression in the endocardium. Since the onset of defects in intercellular junction formation precedes the accumulation of ectopic myocardial gene expression in tal1-deficient embryos, our work suggests that Tal1 has distinct roles in regulating the formation of endocardial intercellular junctions and maintaining endocardial identity.
MATERIALS AND METHODS
Zebrafish
We used the following established transgenic and mutant lines: Tg(kdrl:GRCFP) (Cross et al., 2003), Tg(fli1a:negfp)y7 (Roman et al., 2002), Tg(kdrl:HsHRAS-mCherry)s896 (Chi et al., 2008), Tg(−0.8myl7:DsRed.T4)sk74 (Garavito-Aguilar et al., 2010), and hans6 (Yelon et al., 2000). The Tg(myl7:H2A-mCherry) transgene was assembled using Gateway constructs to place the myl7 promoter upstream of the chimeric reporter H2A-mCherry (Kwan et al., 2007; Lin et al., 2012). Transgenic founders were established using standard techniques for Tol2-based transgenesis (Fisher et al., 2006), and were bred to isolate single stable integrants. We examined 4 independent integrants and found identical patterns of mCherry fluorescence in each case. Specifically, fluorescent nuclei were observed in a domain matching the fluorescent cells observed in Tg(myl7:EGFP)twu277 (Huang et al., 2003) embryos at all developmental stages examined between the 19 somite stage (19s) and 72 hours post-fertilization (hpf) (data not shown). We selected one of these representative transgenic lines, Tg(myl7:H2A-mCherry)sd12, to propagate for further work.
Morpholino injection
The tal1 E2I2 and E3I3 morpholinos (MOs) used in our studies were previously characterized and shown to be effective and specific; moreover, they phenocopy all aspects of the tal1 mutant phenotype (Bussmann et al., 2007; Juarez et al., 2005). We injected 12.5 ng of a 2:3 mix of E2I2 and E3I3 MOs into 1-cell stage embryos as previously described (Schoenebeck et al., 2007).
Transplantation
Blastomere transplantation was performed at the midblastula stage as previously described (Garavito-Aguilar et al., 2010). 75–100 cells were removed from Tg(kdrl:GRCFP) donor embryos and placed into the margin of either non-transgenic or Tg(kdrl:HsHRAS-mCherry) host embryos. For transplantation into non-transgenic hosts, rhodamine-dextran was injected into donors as a lineage tracer. We scored contribution to the endocardium at 24 hpf, and we checked chimeras again at 48 hpf to score contribution to individual chambers.
In situ hybridization and immunofluorescence
Whole mount in situ hybridization for fli1a (ZDB-GENE-980526-426) was performed using standard protocols (Yelon et al., 1999). For immunofluorescence, we employed MF20 supernatant (1:10; Developmental Studies Hybridoma Bank), chicken anti-GFP (1:1000; Abcam 13970), rabbit anti-GFP (1:500; Invitrogen A-11122), rabbit anti-DsRed (1:4000; Clontech 632496) rabbit anti-Fibronectin (1:100; Sigma F3648), mouse anti-β-catenin (1:500: Sigma C7207), and mouse anti-ZO-1 (1:200; Zymed 33-9100) as primary antibodies, followed by goat anti-mouse IgG2b TRITC, goat anti-rabbit FITC, goat anti-mouse Cy5 (Southern Biotech), goat anti-mouse Alexa 647, goat anti-chicken Alexa 488, and goat anti-rabbit Alexa 594 (Invitrogen) as secondary antibodies. We used a previously described protocol for whole mount immunofluorescence (Alexander et al., 1998). For cryosections, embryos were fixed overnight in 4% paraformaldehyde at 4°C, followed by cryoprotection, mounting, sectioning, and staining as performed previously (Garavito-Aguilar et al., 2010). Actin was visualized using rhodamine phalloidin (1:50; Invitrogen R415), which was incorporated into the secondary antibody stain.
Imaging and cell counting
Images were captured using Zeiss M2Bio and Axioplan microscopes outfitted with Zeiss Axiocam cameras and processed using Adobe Photoshop software. Confocal stacks were collected using Zeiss LSM510 and Leica SP5 microscopes and analyzed using Imaris software (Bitplane).
To determine the number of endocardial cells in wild-type and tal1-deficient hearts, we flattened embryos carrying Tg(fli1a:negfp) under a cover slip and counted the number of endothelial nuclei residing within the boundaries of the myocardium. We calculated the mean and standard deviation for each cell number data set and used a two-tailed, unpaired t-test to compare data sets.
To assess the percentage of endocardial cells expressing ectopic myocardial markers in tal1-deficient hearts, we compared the number of endocardial nuclei expressing both Tg(myl7:H2A-mCherry) and Tg(fli1a:negfp) to the number of endocardial nuclei expressing Tg(fli1a:negfp). We scored every tenth optical section from confocal stacks of individual hearts; between 3 and 10 sections were analyzed per heart.
RESULTS
The endocardial aggregate in tal1-deficient embryos is composed of a normal number of endocardial cells
In order to investigate the cellular mechanisms through which tal1 regulates endocardial morphogenesis, we have analyzed the phenotype of zebrafish embryos injected with anti-tal1 morpholinos (MOs). Embryos injected with these previously characterized MOs recapitulate the defects observed in tal1 mutant embryos (Bussmann et al., 2007; Juarez et al., 2005; Schoenebeck et al., 2007). In particular, MO-injected embryos clearly display the same endocardial defect seen in tal1 mutants: instead of forming an inner layer that lines the entire heart tube, the endocardium aggregates in a disorganized cluster in the ventricle and does not populate the atrium (Fig. 1). To analyze the origins of this defect, we employed MO-injected embryos (hereafter referred to as tal1-deficient embryos) in all of the experiments described below.
Figure 1. Endocardium fails to populate the atrium in tal1-deficient embryos.

(A–F) Lateral views of wild-type (WT) (A, C, E) and tal1-deficient hearts (B, D, F) in live embryos at 36 hpf; atrial end of the heart tube to the left. Three-dimensional reconstructions of confocal stacks depict myocardial cells expressing Tg(myl7:H2A-mCherry) (red; A, B, E, F) and endothelial cells, including the endocardium, expressing Tg(fli1a:negfp) (green; A–D). In wild-type embryos (A, C), the endocardium fully lines the myocardial tube, and extends past the atrium. In tal1-deficient embryos (B, D), the endocardium is clumped within the ventricle (n=151/158).
Our first step toward understanding the regulation of endocardial morphogenesis by tal1 was to examine the influence of tal1 on endocardial cell number. To determine whether the failure of the tal1-deficient endocardium to line the entire heart reflects a shortage of endocardial cells in tal1-deficient embryos, we counted endocardial cells just after heart tube elongation (25–30 hours post fertilization (hpf)) (Fig. S1). We found that the average number of cells in wild-type (59.1 ± 14.7; n=69) and tal1-deficient (58.6 ± 11.3; n=72) embryos was not significantly different at this stage (p >0.1). These results indicate that tal1-deficient embryos generate a normal number of endocardial cells and that their endocardial extension defects are not due to a deficiency in cell number.
tal1 acts cell autonomously to promote endocardial extension into the atrium
Since tal1 is expressed in the endocardium (Bussmann et al., 2007; Drake and Fleming, 2000; Elefanty et al., 1999; Zhang and Rodaway, 2007), we reasoned that it could act in a cell-autonomous fashion to control endocardial morphogenesis. Alternatively, tal1 could have an indirect impact on endocardial cell behavior, perhaps through an influence on the endocardial environment. To assess the cell-autonomy of tal1 function, we generated mosaic embryos through blastomere transplantation (Fig. 2).
Figure 2. tal1 promotes endocardial extension into the atrium in a cell-autonomous fashion.

(A–N) Examples of mosaic embryos generated through blastomere transplantation; lateral views, anterior to the left, at 48 hpf. Overlay of brightfield and fluorescent images indicates location of donor-derived cells.
(A–D) Examples of wild-type donor-derived cells expressing Tg(kdrl:GRCFP) (green) in wild-type non-transgenic hosts. (A, B) Wild-type donor-derived cells are located in the ventricular endocardium. (C, D) Wild-type donor-derived cells (arrow) are located in the atrial endocardium.
(E–H) Examples of tal1-deficient cells expressing Tg(kdrl:GRCFP) in wild-type non-transgenic hosts. (E, F) tal1-deficient donor-derived cells are located in the ventricular endocardium. (G, H) A single tal1-deficient donor-derived cell (arrow), weakly expressing Tg(kdrl:GRCFP), is located in the atrial endocardium. We found only 2 such examples among our mosaic embryos (Table 1).
(I–L) Examples of wild-type cells expressing Tg(kdrl:GRCFP) in tal1-deficient non-transgenic hosts. (I, J) Large number of wild-type donor-derived endocardial cells form a chain of cells through the ventricle (arrow), and extend to line the host atrium. (K, L) Small number of wild-type donor-derived endocardial cells form a small tube of endocardium within the host atrium.
(M, N) Example of wild-type cells expressing Tg(kdrl:GRCFP) in a tal1-deficient host expressing Tg(kdrl:HsHRAS-mCherry) (red). Wild-type donor-derived endocardial cells (green) populate the outflow tract and atrium, whereas tal1-deficient host endocardium (red) is clumped within the ventricle.
(O) Pie charts indicate the relative contributions of donor-derived cells to the ventricular endocardium (white), atrial endocardium (black), or ventricular and atrial endocardium (grey) within the mosaic embryos that exhibited donor-derived endocardium (Table 1).
First, we examined the ability of donor-derived cells to contribute to the endocardium, myocardium, and trunk endothelium in wild-type host embryos. We found that tal1-deficient donor cells contributed to the endocardium in 7% of our mosaic embryos, a frequency similar to that observed for wild-type donor cells (8.2%) (Table 1). This result suggests that tal1 is not required for endocardial specification, a conclusion consistent with our observation of normal endocardial cell number in tal1-deficient embryos (Fig. S1). Additionally, tal1-deficient donor cells contributed to the myocardium in 11.5% of mosaic embryos, a frequency comparable to that seen for wild-type donor cells (17.3%) (Table 1). However, the frequency of contribution of tal1-deficient donor cells to trunk endothelium (34.4% of mosaic embryos) was significantly reduced in comparison to the contribution made by wild-type donor cells (72.8%) (Table 1), consistent with the previously demonstrated role for tal1 in vessel formation (Dooley et al., 2005; Patterson et al., 2005; Visvader et al., 1998).
Table 1.
Transplantation experiments indicate a cell-autonomous role for tal1 during endocardial morphogenesis.
| Transplant type | Total endocardial contribution | Atrial endocardial contribution | Ventricular endocardial contribution | Endothelial contribution | Myocardial contribution |
|---|---|---|---|---|---|
| WT to WT | 15/184 (8.2%) | 10/15 (66.7%) | 9/15 (60.0%) | 134/184 (72.8%) | 37/214 (17.3%) |
| tal1 MO to WT | 21/299 (7.0%) | 2/21 (9.5%)* | 19/21 (90.5%) | 103/299 (34.4%)* | 52/451 (11.5%) |
| WT to tal1 MO | 9/238 (3.8%) | 8/9 (88.9%) | 2/9 (22.2%) | 185/238 (77.7%) |
Each fraction reports the number of embryos in which donor cells contributed to a particular tissue divided by the total number of mosaic embryos scored. The fractions representing atrial and ventricular endocardial contributions indicate the number of embryos in which donor cells contributed to the atrial or ventricular endocardium divided by the total number of embryos displaying donor contribution to the endocardium. In some cases, donor cells contributed to both the ventricular and atrial endocardium (Fig. 2O). Asterisks indicate statistically significant differences from control transplants (wild-type into wild-type), as determined by Fisher’s exact test (p < .05).
Next, we examined the relative contributions of donor cells to the ventricular and atrial endocardium. Wild-type donor cells contribute to the ventricular and atrial endocardium in wild-type hosts with approximately the same frequency (60% vs. 67.7% of embryos displaying wild-type donor-derived endocardial cells) (Table 1; Fig. 2A–D, O). In contrast, tal1-deficient donor-derived endocardial cells were nearly always found in the ventricle of wild-type hosts (90.5% of embryos with tal1-deficient donor-derived endocardial cells) (Table 1; Fig. 2E, F, O). We rarely observed tal1-deficient donor cells in the atrial endocardium of wild-type hosts (Table 1); furthermore, in each of the 2 embryos in which this was observed, we found only a single donor-derived atrial endocardial cell with weak expression of Tg(kdrl:GRCFP) (Fig. 2G, H). The inability of the wild-type host environment to facilitate the contribution of tal1-deficient cells to the atrial endocardium strongly suggests that tal1 plays a cell-autonomous role in promoting the extension of endocardial cells into the atrium.
In addition, we performed the converse transplantation experiment, placing wild-type blastomeres into tal1-deficient host embryos. Strikingly, in tal1-deficient embryos, which lack atrial endocardium, wild-type donor-derived endocardial cells were frequently able to populate the atrium (Table 1; Fig. 2I–O). Moreover, in hosts with large numbers of donor-derived endocardial cells, wild-type donor cells were able to fully line the atrium (Fig. 2I, J). In hosts with smaller numbers of donor-derived endocardial cells, wild-type donor cells formed small tubes of endocardium within the atrium (Fig. 2K, L). Interestingly, we observed that wild-type donor-derived endocardial cells tend to associate with each other and segregate away from tal1-deficient host endocardial cells, instead of mixing with them (Fig. 2M, N). The ability of wild-type donor cells to contribute to the atrial endocardium in tal1-deficient host embryos reinforced our conclusion that tal1 is required cell autonomously in order for endocardial cells to line the atrial chamber. In addition, our experiments suggest that tal1 affects endocardial cell behaviors, such as cell adhesion or movement, that impact the patterns of intercellular interaction.
Early endocardial architecture is disrupted in tal1-deficent embryos
To investigate the nature of the cell-intrinsic influence of tal1 on endocardial cell behavior, we proceeded to examine the origins of the endocardial morphogenesis defects in tal1-deficient embryos. As previously documented in tal1 mutants (Bussmann et al., 2007), loss of tal1 function does not seem to disrupt the initial migration of endocardial cells from the anterior lateral plate mesoderm (ALPM) to the embryonic midline (Fig. 3A, B). However, defects in tal1-deficient endocardial cell behavior become evident once the cells reach the midline. In wild-type embryos, endocardial cells that have arrived at the midline spread out along the anterior-posterior axis to create a sheet of tissue (Fig. 3A) (Bussmann et al., 2007). In contrast, tal1-deficient cells do not form a proper endocardial sheet: instead of spreading out along the anterior-posterior axis, tal1-deficient cells occupy a smaller area at the midline (Fig. 3B).
Figure 3. Defects in endocardial sheet formation appear after tal1-deficient endocardial cells arrive at the midline.

(A–F) Dorsal views of wild-type (A, C, E) and tal1-deficient embryos (B, D, F), anterior to the top, displaying the expression of fli1a at the 18 somite (18s) (A, B), 20s (C, D), and 22s (E, F) stages.
(A′-F′) Cartoons indicate regions of fli1a expression in the endocardium (grey and black) and cranial endothelium (white). fli1a expression in the branchial arches is visible in the images (A–F) but is not indicated in the cartoons (A′-F′).
(A, C, E) In wild-type embryos, an endocardial sheet spreads along the anterior-posterior axis by 18s (A). Increased expression of fli1a becomes apparent at the future apex (black areas in C′ and E′) of the endocardial tube as the endocardial sheet transforms into a tube extending toward the left side of the embryo (C, E).
(B, D, F) In tal1-deficient embryos, the expression of fli1a is maintained normally, consistent with previous reports (Patterson et al., 2005). Instead of spreading out into a sheet, the tal1-deficient endocardial cells aggregate at the midline (B) and subsequently fail to migrate toward the left (D, F).
Further examination of the cellular architecture of the endocardium revealed that tal1 influences the arrangement and shape of endocardial cells during sheet formation at the midline. In wild-type embryos, the endocardial sheet is a single cell layer thick; in addition, wild-type endocardial cells have a flattened, elongated appearance and only contact each other at their lateral edges (Fig. 4A, C, E, G, I). However, in tal1-deficient embryos, endocardial cells stack on top of one another to form a multi-layered aggregate (Fig. 4B, D, F, H, J). Additionally, tal1-deficient endocardial cells display a relatively rounded morphology and make contacts with each other on their apical and basal surfaces as well as on their lateral surfaces (Fig. 4B, D, F, H, J).
Figure 4. The architecture of the early endocardium is disrupted in tal1-deficient embryos.

(A–J) Transverse sections of wild-type (A, C, E, G, I) and tal1-deficient embryos (B, D, F, H, J), dorsal to the top, at 16s (A, B) and 18s (C–J).
Immunofluorescence detects localization of fibronectin (FN; green) (E–H) and β-catenin (red) (E, F, I, J); in addition, an anti-GFP antibody allows visualization of Tg(kdrl:GRCFP) expression (blue) in the endocardium (A–F). Together, these markers highlight the shapes and arrangement of the endocardial cells.
Endocardial cells form a single-layered sheet of cells at 16s (A), after they have migrated to the midline, and this sheet of cells is maintained at 18s (C). In tal1-deficient embryos, endocardial cells stack on top of each other, forming a multilayered cluster (B, D). The boxed areas in (C, D) are highlighted in (E–J).
The apical-basal polarity of wild-type endocardial cells (E) is indicated by basal deposition of FN (G) and apical enrichment of β-catenin (I). In tal1-deficient embryos (F), localization of FN (H) and β-catenin (J) highlight the dysmorphic endocardial architecture and aberrant cell shapes.
(E′–J′) Cartoons indicate regions of FN (green) and β-catenin (red) localization within the endocardium. Endocardial cell outlines are marked in black. The FN and β-catenin that are presumed to be located within the endoderm that overlies the endocardium are excluded from the cartoons.
Together, these results indicate that the endocardial morphogenesis defects in tal1-deficient embryos originate with errors in the formation of an endocardial sheet at the midline. Subsequently, tal1-deficient endocardial cells fail to undergo the normal process of leftward migration along the length of the extending heart tube; instead, tal1-deficient endocardial cells aggregate into a dense cluster that ultimately resides within the ventricle (Fig. 3C–F). Thus, the inability of tal1-deficient endocardial cells to populate the atrium suggests that the early architecture of the endocardial sheet is an important prerequisite for normal endocardial tube formation.
tal1 influences the distribution of endocardial intercellular junctions
The aberrant patterns of cell-cell contacts observed in tal1-deficient embryos led us to investigate whether tal1 influences the formation of intercellular junctions in the endocardium. We examined the subcellular localization of ZO-1, a core component of the tight junction complex (Fanning and Anderson, 2009). In the wild-type heart tube at 25 hpf, ZO-1 is found in discrete puncta at the apical vertices connecting endocardial cells (Fig. 5C). In tal1-deficient embryos, ZO-1 is dramatically mislocalized, with broad and irregular distribution around endocardial cell membranes (Fig. 5D), whereas myocardial localization of ZO-1 appears unaffected (Fig. 5A, B).
Figure 5. Endocardial intercellular junctions are disorganized following heart tube extension in tal1-deficient embryos.

(A–D) Immunofluorescence detects localization of ZO-1 (red) in wild-type (A, C) and tal1-deficient (B, D) hearts at 25 hpf; lateral views, ventricular end of the heart tube at the top. An anti-GFP antibody allows visualization of Tg(kdrl:GRCFP) expression (green) in the endocardium.
(A, B) Three-dimensional reconstructions of confocal stacks demonstrate that ZO-1 is located around the perimeter of the myocardial cells in both wild-type and tal1-deficient hearts. (C) Optical slice of a wild-type heart reveals that ZO-1 is localized in discrete puncta at points of endocardial cell-cell contact; in addition, wild-type endocardial cells exhibit a flattened and elongated morphology. (D) Optical slice of a tal1-deficient heart reveals that ZO-1 is mislocalized in large clusters around the perimeter of the endocardial cells; in addition, the tal1-deficient endocardial cells exhibit a rounded shape, rather than the normal elongated shape.
(E, F) Immunofluorescence detects ZO-1 (purple) in wild-type (E) and tal1-deficient (F) hearts at 25 hpf; lateral views, ventricular end of the heart tube at the top. Expression of the Tg(fli1a:negfp) (green) and Tg(myl7:H2A-mCherry) (red) transgenes reveals one endocardial cell with ectopic myocardial gene expression (arrow). Subcellular localization of ZO-1 is aberrant in the majority of tal1-deficient endocardial cells, nearly all of which do not display ectopic myocardial gene expression.
The irregular pattern of ZO-1 localization within the tal1-deficient endocardium could simply be a result of aberrant endocardial cell packing within the ventricle, rather than being an initial component of the morphogenesis defect in tal1-deficient embryos. To investigate whether tal1 influences tight junction establishment during endocardial sheet formation, we examined ZO-1 localization at 18s. In the wild-type endocardial sheet, ZO-1 is located in discrete puncta at apical-lateral cell-cell junctions (Fig. 6A, C, D, G), similar to its localization in the heart tube at 25 hpf. Even on occasions when wild-type endocardial cells were found on top of one another instead of in a side-by-side orientation within the forming sheet, ZO-1 was found in individual puncta at cell-cell vertices (Fig. S2). In contrast, ZO-1 is located in multiple irregular puncta along the endocardial cell boundaries of the multi-layered tal1-deficient endocardium (Fig. 6B, E, F, H). These observations suggest that tal1 is required during endocardial sheet formation to promote proper localization of intercellular junctions.
Figure 6. Endocardial intercellular junctions are disorganized during early endocardial morphogenesis in tal1-deficient embryos.

(A–H) Transverse sections of wild-type (A, C, D, G) and tal1-deficient embryos (B, E, F, H), dorsal to the top, at 18s. Immunofluorescence detects localization of ZO-1 (red) (C–H) and F-actin (blue) (D, F); in addition, an anti-GFP antibody allows visualization of Tg(kdrl:GRCFP) expression (green) in the endocardium (A, B, G, H). The boxed areas in (A, B) are highlighted in (C–H).
(A) Wild-type endocardium forms a single-layered sheet with ZO-1 localized to discrete puncta at cell-cell contacts (C, D, G). (B) tal1-deficient endocardium is multi-layered and disorganized, and ZO-1 is mislocalized in multiple puncta along the endocardial cell boundaries (E, F, H).
(G′, H′) Cartoons indicate regions of ZO-1 (red) and F-actin (blue) localization within the endocardium.
tal1 is required for maintenance of endocardial identity
Recent studies in mouse have indicated that Tal1 is required for the repression of myocardial differentiation within the endocardium (Van Handel et al., 2012). Along the same lines, our previous studies in zebrafish have demonstrated that tal1, together with the ETS family transcription factor gene etsrp, plays a critical role in inhibiting myocardial specification within the portion of the ALPM that normally gives rise to the endocardium (Schoenebeck et al., 2007). Together, these studies indicate the importance of Tal1 in repressing myocardial gene expression within the endocardial lineage. Therefore, we wondered whether a defect in endocardial identity assignment or maintenance could be responsible for the morphogenetic defects observed in the tal1-deficient endocardium.
In order to examine whether ectopic myocardial differentiation occurs within tal1-deficient endocardial cells, we employed the Tg(myl7:H2A-mCherry) transgene as a reporter of myocardial gene expression. At 60 hpf, we readily observed Tg(myl7:H2A-mCherry) expression within the tal1-deficient endocardium; these Tg(myl7:H2A-mCherry)-expressing cells also expressed Tg(fli1a:nEGFP) and were located within the endocardial aggregate inside the ventricle (Fig. 7C, D; n=26/29). In contrast, we never detected Tg(myl7:H2A-mCherry) expression within the wild-type endocardium (Fig. 7A, B; n=0/23). In addition, we used the MF20 antibody to visualize the localization of sarcomeric myosin heavy chain. Similar to our observations using Tg(myl7:H2A-mcherry), we observed ectopic localization of sarcomeric myosin heavy chain in the tal1-deficient endocardium (Fig. 7F), but not in the wild-type endocardium (Fig. 7E).
Figure 7. Myocardial genes are ectopically expressed in the tal1-deficient endocardium at 60 hpf.

(A–D) Three-dimensional reconstructions of confocal stacks (A, C) and single optical sections through the ventricle (B, D) depict wild-type (A, B) and tal1-deficient hearts (C, D) expressing Tg(myl7:H2A-mCherry) (red) and Tg(fli1a:negfp) (green) at 60 hpf, arterial pole at the top. Many tal1-deficient endocardial cells express both Tg(myl7:H2A-mCherry) and Tg(fli1a:negfp) (D), but wild-type endocardial cells express only Tg(fli1a:negfp) (B).
(E, F) Immunofluorescence with the monoclonal antibody MF20 detects sarcomeric myosin heavy chain localization (blue) in wild-type (E) and tal1-deficient hearts (F) expressing Tg(myl7:H2A-mCherry) (red) and Tg(fli1a:negfp) (green). Optical sections through the ventricle reveal ectopic sarcomeric myosin heavy chain within the tal1-deficient endocardium (F). In contrast, sarcomeric myosin heavy chain is not found in the wild-type endocardium (E); section shown depicts both the atrial endocardium (bottom) and outflow tract endocardium (top). (B′, D′, F′) Images display only the myocardial markers from the sections shown in (B, D, F).
Extension of these studies revealed that ectopic myocardial gene expression accumulates progressively over time in the tal1-deficient endocardium. At 24 hpf, we rarely detected Tg(myl7:H2A-mCherry) expression in tal1-deficient endocardium: on average, we found ectopic myocardial gene expression in 5% of tal1-deficient endocardial cells (Fig. 8A, B, I). As development proceeds, this percentage progressively increases. At 36 hpf, we detected Tg(myl7:H2A-mCherry) expression in 27% of tal1-deficient endocardial cells (Fig. 8C, D, I); at 48 hpf, this value had increased to 38% (Fig. 8E, F, I). By 60 hpf, we routinely encountered prominent expression of Tg(myl7:H2A-mCherry) in the majority of tal1-deficient endocardial cells (71%; Fig. 8G–I). Furthermore, we observed a decrease in the expression of Tg(fli1a:negfp) in the cells ectopically expressing Tg(myl7:H2A-mCherry) (Fig. 7B, D). Together, these data demonstrate the importance of tal1 function for maintenance of endocardial identity through repression of myocardial gene expression.
Figure 8. Ectopic myocardial gene expression progressively increases within the tal1-deficient endocardium.
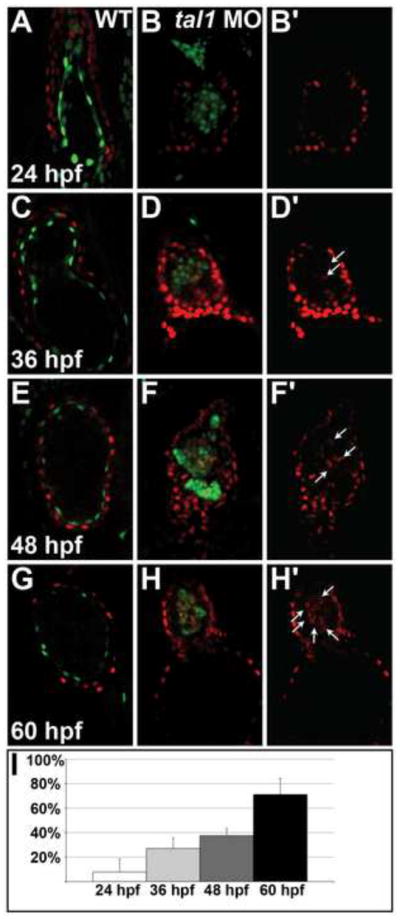
(A–H) Optical sections from confocal stacks of wild-type (A, C, E, G) and tal1-deficient (B, D, F, H) hearts expressing Tg(myl7:H2A-mCherry) (red) and Tg(fli1a:negfp) (green) at 24 hpf (A, B), 36 hpf (C, D), 48 hpf (E, F) and 60 hpf (G, H), arterial pole at the top. These representative hearts demonstrate the progressive increase in the percentage of endocardial cells expressing Tg(myl7:H2A-mCherry) that occurs between 24 and 60 hpf in tal1-deficient embryos (B, D, F, H). In contrast, the wild-type endocardium never expresses Tg(myl7:H2A-mCherry) (A, C, E, G).
(B′, D′, F′, H′) Images display only the myocardial marker from the sections shown in (B, D, F, H). Arrows indicate examples of endocardial cells ectopically expressing Tg(myl7:H2A-mCherry).
(I) Bar graph depicts the average percentage of tal1-deficient endocardial cells expressing Tg(myl7:H2A-mCherry) at each stage examined. Error bars indicate standard deviation (n=4–8 embryos per stage).
Finally, we considered whether the initiation of ectopic myocardial differentiation could underlie the abnormal endocardial cell behaviors seen in tal1-deficient embryos. Notably, at 24 hpf, the majority of the endocardial cells in tal1-deficient embryos displayed defects in ZO-1 localization, while very few of these cells displayed ectopic myocardial gene expression (Fig. 5E, F).
Additionally, the onset of aberrant intercellular contacts and ZO-1 localization (17–18 hpf, Figs. 4 and 6) precedes the onset of the appearance of ectopic Tg(myl7:H2A-mCherry) expression in the tal1-deficient endocardium (24 hpf; Fig. 8A, B, I). Furthermore, we found that hand2, a bHLH transcription factor gene essential for robust myocardial differentiation (Schoenebeck et al., 2007; Yelon et al., 2000), is not required for the aggregation of endocardial cells in tal1-deficient embryos (Fig. S3). We have previously shown that hand2 (han) mutants display a dramatic reduction in the number of myocardial cells as well as a minor defect in the formation of the posterior portion of the endocardial sheet (Garavito-Aguilar et al., 2010; Schoenebeck et al., 2007; Yelon et al., 2000). Strikingly, we observed that embryos deficient for both han and tal1 exhibit multi-layered endocardial aggregates similar to those seen in tal1-deficient embryos, whereas the endocardium in han mutants forms a single-layered sheet with relatively normal spacing between endocardial cells (Fig. S3). These data indicate that the aggregation of the tal1-deficient endocardium does not require hand2 function and suggest that this aggregation is not a consequence of ectopic myocardial differentiation. Together, these experiments suggest that tal1 could play independent roles in regulating endocardial morphogenesis and inhibiting myocardial gene expression.
DISCUSSION
Taken together, our studies of tal1-deficient embryos illuminate several intriguing elements of endocardial morphogenesis. Our data reveal a cell-autonomous requirement for Tal1 function during endocardial tube extension and point to a cell-intrinsic influence of Tal1 on the regulation of endocardial cell-cell contacts. In addition, our results provide the first demonstration that Tal1 function has a significant impact on the localization of endocardial intercellular junctions within the endocardial sheet at the embryonic midline. Moreover, our work highlights the importance of endocardial sheet architecture as a key prerequisite for proper endocardial tube formation.
Although prior studies in zebrafish have tracked the trajectories of endocardial cells while they migrate to the midline, the mechanisms controlling endocardial cell behavior during tube formation are not well understood (Bussmann et al., 2007; Proulx et al., 2010; Veerkamp et al., 2013). It is therefore particularly interesting that the endocardial tube defects in tal1-deficient embryos originate with inappropriate cell-cell contacts within the endocardial sheet at the midline. Without Tal1 function, endocardial cells migrate to the midline normally but then do not spread out into a single layer of flattened, endothelium-like tissue; instead, they create a cluster of irregularly stacked, cuboidal cells. Importantly, the failure of tal1-deficient endocardial cells to form a normally elongated tube appears to be a consequence of their early disorganization; once clustered together, the tal1-deficient endocardial cells exhibit no apparent inclination toward populating the atrium. At the same time, the relatively normal length of the tal1-deficient myocardial tube suggests that, even though endocardium-myocardium interactions are important for several aspects of heart development (Chang et al., 2004; de la Pompa and Epstein, 2012; Garavito-Aguilar et al., 2010; Holtzman et al., 2007; Stankunas et al., 2008; Totong et al., 2011; Wagner and Siddiqui, 2007), the process of myocardial tube elongation is largely independent from endocardial tube elongation. The derailment of endocardial tube formation by the defective architecture of the midline sheet strongly suggests that the sheet is an important intermediate structure during endocardial morphogenesis. We propose that formation of the endocardial sheet represents a fundamental transition in cell behavior, as endocardial cells shift from migrating as individuals to moving as a cohesive group, and that this transition is crucial for the elongation of the endocardial tube.
Regulation of intercellular junction formation is likely to contribute to the coordination of endocardial sheet architecture, and our data provide striking evidence for the influence of Tal1 on the organization of endocardial cell-cell junctions. In the tal1-deficient endocardial sheet, junctions form at inappropriate subcellular locations and cells aggregate in abnormal configurations. Our mosaic studies suggest the possibility that Tal1 regulates cell-intrinsic aspects of junction component localization; however, it is also feasible that the irregular distribution of junctions in the tal1-deficient endocardium reflects a secondary consequence of misregulation of another cell behavior such as migration or establishment of polarity. Additionally, although our data document the impact of Tal1 on the distribution of the tight junction component ZO-1, we speculate that additional components of intercellular junctions are likely to be disorganized in tal1-deficient embryos, including components of the adherens junctions, especially since adherens junctions have been shown to regulate tight junction establishment in cultured epithelial cells (Deleuze et al., 2007; Fanning and Anderson, 2009; Hartsock and Nelson, 2008; Taddei et al., 2008). In future studies, it will be valuable to acquire a deeper understanding of the molecular mechanisms underlying endocardial cell-cell interactions and to evaluate whether or not Tal1 has a direct influence on transcription of any of the key players.
In addition to its importance in organizing the morphology of the endocardial sheet, Tal1 is important for the maintenance of the endocardial differentiation program. Previously, we have shown that tal1 and estrp contribute to ALPM patterning by repressing cardiac progenitor specification and promoting vascular and hematopoietic specification (Schoenebeck et al., 2007). However, our prior work did not evaluate how each of these genes contributes to the progression of endocardial differentiation. Our current data show that, as development progresses, the tal1-deficient endocardium fails to maintain its identity and gradually accumulates myocardial characteristics. These results reinforce the conclusions reached from recent studies that revealed ectopic myocardial differentiation in the endocardium of Tal1 mutant mice (Van Handel et al., 2012). The failure of tal1-deficient embryos to maintain endocardial identity may reflect the early role of tal1 in repressing cardiac specification in the ALPM or may be due to a distinct, later role of tal1. Recent work has also demonstrated the importance of etsrp for promoting endocardial differentiation and repressing myocardial differentiation (Palencia-Desai et al., 2011; Rasmussen et al., 2011). However, etsrp may act at an earlier stage than tal1, since loss of etsrp function seems to inhibit the initiation, rather than the maintenance, of endocardial gene expression (Palencia-Desai et al., 2011; Rasmussen et al., 2011).
The failure of endocardial cells to maintain their identity in tal1-deficient embryos raises the possibility that their cell behavior problems are merely secondary to the misexpression of myocardial genes in the endocardium. Since we observe temporal separation between the sudden, early onset of defects in junction formation and the gradual, later appearance of ectopic myocardial differentiation, we propose a model in which Tal1 has distinct roles in regulating the formation of endocardial intercellular junctions and maintaining endocardial identity. However, we also acknowledge the possibility that the identity transformation in tal1-deficient endocardium begins prior to the expression of the myocardial markers that we have examined and that the earlier expression of particular myocardial genes, such as myocardium-specific adhesion molecules, could interfere with endocardial sheet formation. Evaluation of this model will require future elucidation of the precise kinetics of ectopic myocardial gene expression in the tal1-deficient endocardium.
Overall, our studies provide the first integration of two disparate and previously noted roles of Tal1 -- the role of Tal1 in promoting endocardial tube elongation (Bussmann et al., 2007) and its role in repressing myocardial differentiation within the endocardium (Schoenebeck et al., 2007; Van Handel et al., 2012) -- and provide higher resolution of the temporal dynamics of each of these Tal1 functions. The synthesis of our data suggests the possibility that Tal1 plays independent roles in endocardial junction organization and endocardial identity preservation but also leaves open the possibility that these functions are interconnected. Future studies focused on the identification of relevant Tal1 targets will be essential to determine whether each of the roles of Tal1 are executed by distinct downstream pathways.
Supplementary Material
Frontal views of wild-type (A, B) and tal1-deficient hearts (C, D) at 30 hpf; ventricular end of the heart tube at the top. The transgene Tg(fli1a:negfp)(green) was used to count the number of endocardial cells residing within the boundaries of the myocardium, which was labeled by MF20 immunofluorescence (red).
{kind=link}
(A–D) Transverse section of wild-type endocardium, dorsal to the top, at 18s. Immunofluorescence detects localization of ZO-1 (red) (B–D) and F-actin (blue) (C); in addition, an anti-GFP antibody allows visualization of Tg(kdrl:GRCFP) expression (green) in the endocardium (A, D). The boxed area in (A) is highlighted in (B–D). Even when wild-type cells are stacked on top of each other, ZO-1 is properly localized to individual vertices at cell-cell junctions (B–D), rather than being found in large patches as in tal1-deficient embryos (Fig. 6E, F, H).
(E) Cartoon indicates regions of ZO-1 (red) and F-actin (blue) localization within the endocardium.
{kind=link}
(A–D) Three-dimensional reconstructions of confocal stacks depict dorsal views of wild-type (A), han mutant (B), tal1-deficient(C) and han mutant; tal1-deficient (D) embryos expressing Tg(fli1a:negfp) (green) at 20s.
(A′–D′) Reconstructions of optical sections displaying transverse slices through the endocardium of the embryos shown in (A–D). Arrowheads mark progenitors of the lateral dorsal aortae.
The endocardium forms a single-layered sheet in wild-type (A, A′) and han mutant (B, B′) embryos. In contrast, the endocardium in embryos that are deficient for both han and tal1 (D, D′) forms a multi-layered aggregate similar to that seen in tal1-deficient embryos (C, C′).
{kind=link}
HIGHLIGHTS.
tal1 acts cell autonomously to regulate endocardial morphogenesis.
Early endocardial architecture is disrupted in tal1-deficient embryos.
tal1 influences the distribution of endocardial intercellular junctions.
tal1 is required for maintenance of endocardial identity.
Acknowledgments
We thank L. Pandolfo and C. McDaniel for expert zebrafish care, N. Chi, S. Evans, H. Mikkola, and B. Van Handel for helpful input, and members of the Yelon lab for constructive discussions. Work in the Yelon lab is supported by grants from the National Institutes of Health, American Heart Association, and March of Dimes. J.B. is supported by an American Heart Association Postdoctoral Fellowship (12POST11660038), J.S. was supported by a NIH NRSA Postdoctoral Fellowship (F32 HL094048), and Z.V.G.A. was supported by an American Heart Association Predoctoral Fellowship (0815795D).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abu-Issa R, Kirby ML. Patterning of the heart field in the chick. Dev Biol. 2008;319:223–233. doi: 10.1016/j.ydbio.2008.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleksandrova A, Czirok A, Szabo A, Filla MB, Hossain MJ, Whelan PF, Lansford R, Rongish BJ. Convective tissue movements play a major role in avian endocardial morphogenesis. Dev Biol. 2012;363:348–361. doi: 10.1016/j.ydbio.2011.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander J, Rothenberg M, Henry GL, Stainier DY. casanova plays an early and essential role in endoderm formation in zebrafish. Dev Biol. 1999;215:343–357. doi: 10.1006/dbio.1999.9441. [DOI] [PubMed] [Google Scholar]
- Alexander J, Stainier DY, Yelon D. Screening mosaic F1 females for mutations affecting zebrafish heart induction and patterning. Dev Genet. 1998;22:288–299. doi: 10.1002/(SICI)1520-6408(1998)22:3<288::AID-DVG10>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Arrington CB, Yost HJ. Extra-embryonic syndecan 2 regulates organ primordia migration and fibrillogenesis throughout the zebrafish embryo. Development. 2009;136:3143–3152. doi: 10.1242/dev.031492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker K, Holtzman NG, Burdine RD. Direct and indirect roles for Nodal signaling in two axis conversions during asymmetric morphogenesis of the zebrafish heart. Proc Natl Acad Sci U S A. 2008;105:13924–13929. doi: 10.1073/pnas.0802159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begley CG, Aplan PD, Denning SM, Haynes BF, Waldmann TA, Kirsch IR. The gene SCL is expressed during early hematopoiesis and encodes a differentiation-related DNA-binding motif. Proc Natl Acad Sci U S A. 1989;86:10128–10132. doi: 10.1073/pnas.86.24.10128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussmann J, Bakkers J, Schulte-Merker S. Early endocardial morphogenesis requires Scl/Tal1. PLoS Genet. 2007;3:e140. doi: 10.1371/journal.pgen.0030140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CP, Neilson JR, Bayle JH, Gestwicki JE, Kuo A, Stankunas K, Graef IA, Crabtree GR. A field of myocardial-endocardial NFAT signaling underlies heart valve morphogenesis. Cell. 2004;118:649–663. doi: 10.1016/j.cell.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Chi NC, Shaw RM, De Val S, Kang G, Jan LY, Black BL, Stainier DY. Foxn4 directly regulates tbx2b expression and atrioventricular canal formation. Genes Dev. 2008;22:734–739. doi: 10.1101/gad.1629408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross LM, Cook MA, Lin S, Chen JN, Rubinstein AL. Rapid analysis of angiogenesis drugs in a live fluorescent zebrafish assay. Arterioscler Thromb Vasc Biol. 2003;23:911–912. doi: 10.1161/01.ATV.0000068685.72914.7E. [DOI] [PubMed] [Google Scholar]
- de Campos-Baptista MI, Holtzman NG, Yelon D, Schier AF. Nodal signaling promotes the speed and directional movement of cardiomyocytes in zebrafish. Dev Dyn. 2008;237:3624–3633. doi: 10.1002/dvdy.21777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Pompa JL, Epstein JA. Coordinating tissue interactions: Notch signaling in cardiac development and disease. Dev Cell. 2012;22:244–254. doi: 10.1016/j.devcel.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaughter DM, Saint-Jean L, Baldwin HS, Barnett JV. What chick and mouse models have taught us about the role of the endocardium in congenital heart disease. Birth Defects Res A Clin Mol Teratol. 2011;91:511–525. doi: 10.1002/bdra.20809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleuze V, Chalhoub E, El-Hajj R, Dohet C, Le Clech M, Couraud PO, Huber P, Mathieu D. TAL-1/SCL and its partners E47 and LMO2 up-regulate VE-cadherin expression in endothelial cells. Mol Cell Biol. 2007;27:2687–2697. doi: 10.1128/MCB.00493-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRuiter MC, Poelmann RE, VanderPlas-de Vries I, Mentink MM, Gittenberger-de Groot AC. The development of the myocardium and endocardium in mouse embryos. Fusion of two heart tubes? Anat Embryol (Berl) 1992;185:461–473. doi: 10.1007/BF00174084. [DOI] [PubMed] [Google Scholar]
- Dooley KA, Davidson AJ, Zon LI. Zebrafish scl functions independently in hematopoietic and endothelial development. Dev Biol. 2005;277:522–536. doi: 10.1016/j.ydbio.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Drake CJ, Fleming PA. Vasculogenesis in the day 6.5 to 9.5 mouse embryo. Blood. 2000;95:1671–1679. [PubMed] [Google Scholar]
- Drake CJ, Wessels A, Trusk T, Little CD. Elevated vascular endothelial cell growth factor affects mesocardial morphogenesis and inhibits normal heart bending. Dev Dyn. 2006;235:10–18. doi: 10.1002/dvdy.20539. [DOI] [PubMed] [Google Scholar]
- Elefanty AG, Begley CG, Hartley L, Papaevangeliou B, Robb L. SCL expression in the mouse embryo detected with a targeted lacZ reporter gene demonstrates its localization to hematopoietic, vascular, and neural tissues. Blood. 1999;94:3754–3763. [PubMed] [Google Scholar]
- Evans SM, Yelon D, Conlon FL, Kirby ML. Myocardial lineage development. Circ Res. 2010;107:1428–1444. doi: 10.1161/CIRCRESAHA.110.227405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanning AS, Anderson JM. Zonula occludens-1 and -2 are cytosolic scaffolds that regulate the assembly of cellular junctions. Ann N Y Acad Sci. 2009;1165:113–120. doi: 10.1111/j.1749-6632.2009.04440.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish JE, Wythe JD, Xiao T, Bruneau BG, Stainier DY, Srivastava D, Woo S. A Slit/miR-218/Robo regulatory loop is required during heart tube formation in zebrafish. Development. 2011;138:1409–1419. doi: 10.1242/dev.060046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher S, Grice EA, Vinton RM, Bessling SL, Urasaki A, Kawakami K, McCallion AS. Evaluating the biological relevance of putative enhancers using Tol2 transposon-mediated transgenesis in zebrafish. Nat Protoc. 2006;1:1297–1305. doi: 10.1038/nprot.2006.230. [DOI] [PubMed] [Google Scholar]
- Garavito-Aguilar ZV, Riley HE, Yelon D. Hand2 ensures an appropriate environment for cardiac fusion by limiting Fibronectin function. Development. 2010;137:3215–3220. doi: 10.1242/dev.052225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George EL, Baldwin HS, Hynes RO. Fibronectins are essential for heart and blood vessel morphogenesis but are dispensable for initial specification of precursor cells. Blood. 1997;90:3073–3081. [PubMed] [Google Scholar]
- George EL, Georges-Labouesse EN, Patel-King RS, Rayburn H, Hynes RO. Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development. 1993;119:1079–1091. doi: 10.1242/dev.119.4.1079. [DOI] [PubMed] [Google Scholar]
- Hartsock A, Nelson WJ. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta. 2008;1778:660–669. doi: 10.1016/j.bbamem.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman NG, Schoenebeck JJ, Tsai HJ, Yelon D. Endocardium is necessary for cardiomyocyte movement during heart tube assembly. Development. 2007;134:2379–2386. doi: 10.1242/dev.02857. [DOI] [PubMed] [Google Scholar]
- Huang CJ, Tu CT, Hsiao CD, Hsieh FJ, Tsai HJ. Germ-line transmission of a myocardium-specific GFP transgene reveals critical regulatory elements in the cardiac myosin light chain 2 promoter of zebrafish. Dev Dyn. 2003;228:30–40. doi: 10.1002/dvdy.10356. [DOI] [PubMed] [Google Scholar]
- Juarez MA, Su F, Chun S, Kiel MJ, Lyons SE. Distinct roles for SCL in erythroid specification and maturation in zebrafish. J Biol Chem. 2005;280:41636–41644. doi: 10.1074/jbc.M507998200. [DOI] [PubMed] [Google Scholar]
- Kuo CT, Morrisey EE, Anandappa R, Sigrist K, Lu MM, Parmacek MS, Soudais C, Leiden JM. GATA4 transcription factor is required for ventral morphogenesis and heart tube formation. Genes Dev. 1997;11:1048–1060. doi: 10.1101/gad.11.8.1048. [DOI] [PubMed] [Google Scholar]
- Kupperman E, An S, Osborne N, Waldron S, Stainier DY. A sphingosine-1-phosphate receptor regulates cell migration during vertebrate heart development. Nature. 2000;406:192–195. doi: 10.1038/35018092. [DOI] [PubMed] [Google Scholar]
- Kwan KM, Fujimoto E, Grabher C, Mangum BD, Hardy ME, Campbell DS, Parant JM, Yost HJ, Kanki JP, Chien CB. The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev Dyn. 2007;236:3088–3099. doi: 10.1002/dvdy.21343. [DOI] [PubMed] [Google Scholar]
- Lenhart KF, Holtzman NG, Williams JR, Burdine RD. Integration of Nodal and BMP Signals in the Heart Requires FoxH1 to Create Left-Right Differences in Cell Migration Rates That Direct Cardiac Asymmetry. PLoS Genet. 2013;9:e1003109. doi: 10.1371/journal.pgen.1003109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YF, Swinburne I, Yelon D. Multiple influences of blood flow on cardiomyocyte hypertrophy in the embryonic zebrafish heart. Dev Biol. 2012;362:242–253. doi: 10.1016/j.ydbio.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molkentin JD, Lin Q, Duncan SA, Olson EN. Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis. Genes Dev. 1997;11:1061–1072. doi: 10.1101/gad.11.8.1061. [DOI] [PubMed] [Google Scholar]
- Moreno-Rodriguez RA, Krug EL, Reyes L, Villavicencio L, Mjaatvedt CH, Markwald RR. Bidirectional fusion of the heart-forming fields in the developing chick embryo. Dev Dyn. 2006;235:191–202. doi: 10.1002/dvdy.20601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motoike T, Loughna S, Perens E, Roman BL, Liao W, Chau TC, Richardson CD, Kawate T, Kuno J, Weinstein BM, Stainier DY, Sato TN. Universal GFP reporter for the study of vascular development. Genesis. 2000;28:75–81. doi: 10.1002/1526-968x(200010)28:2<75::aid-gene50>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Palencia-Desai S, Kohli V, Kang J, Chi NC, Black BL, Sumanas S. Vascular endothelial and endocardial progenitors differentiate as cardiomyocytes in the absence of Etsrp/Etv2 function. Development. 2011;138:4721–4732. doi: 10.1242/dev.064998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson LJ, Gering M, Patient R. Scl is required for dorsal aorta as well as blood formation in zebrafish embryos. Blood. 2005;105:3502–3511. doi: 10.1182/blood-2004-09-3547. [DOI] [PubMed] [Google Scholar]
- Porcher C, Swat W, Rockwell K, Fujiwara Y, Alt FW, Orkin SH. The T cell leukemia oncoprotein SCL/tal-1 is essential for development of all hematopoietic lineages. Cell. 1996;86:47–57. doi: 10.1016/s0092-8674(00)80076-8. [DOI] [PubMed] [Google Scholar]
- Proulx K, Lu A, Sumanas S. Cranial vasculature in zebrafish forms by angioblast cluster-derived angiogenesis. Dev Biol. 2010;348:34–46. doi: 10.1016/j.ydbio.2010.08.036. [DOI] [PubMed] [Google Scholar]
- Rasmussen TL, Kweon J, Diekmann MA, Belema-Bedada F, Song Q, Bowlin K, Shi X, Ferdous A, Li T, Kyba M, Metzger JM, Koyano-Nakagawa N, Garry DJ. ER71 directs mesodermal fate decisions during embryogenesis. Development. 2011;138:4801–4812. doi: 10.1242/dev.070912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robb L, Lyons I, Li R, Hartley L, Kontgen F, Harvey RP, Metcalf D, Begley CG. Absence of yolk sac hematopoiesis from mice with a targeted disruption of the scl gene. Proc Natl Acad Sci U S A. 1995;92:7075–7079. doi: 10.1073/pnas.92.15.7075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohr S, Otten C, Abdelilah-Seyfried S. Asymmetric involution of the myocardial field drives heart tube formation in zebrafish. Circ Res. 2008;102:e12–19. doi: 10.1161/CIRCRESAHA.107.165241. [DOI] [PubMed] [Google Scholar]
- Roman BL, Pham VN, Lawson ND, Kulik M, Childs S, Lekven AC, Garrity DM, Moon RT, Fishman MC, Lechleider RJ, Weinstein BM. Disruption of acvrl1 increases endothelial cell number in zebrafish cranial vessels. Development. 2002;129:3009–3019. doi: 10.1242/dev.129.12.3009. [DOI] [PubMed] [Google Scholar]
- Schoenebeck JJ, Keegan BR, Yelon D. Vessel and blood specification override cardiac potential in anterior mesoderm. Dev Cell. 2007;13:254–267. doi: 10.1016/j.devcel.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivdasani RA, Mayer EL, Orkin SH. Absence of blood formation in mice lacking the T-cell leukaemia oncoprotein tal-1/SCL. Nature. 1995;373:432–434. doi: 10.1038/373432a0. [DOI] [PubMed] [Google Scholar]
- Smith KA, Chocron S, von der Hardt S, de Pater E, Soufan A, Bussmann J, Schulte-Merker S, Hammerschmidt M, Bakkers J. Rotation and asymmetric development of the zebrafish heart requires directed migration of cardiac progenitor cells. Dev Cell. 2008;14:287–297. doi: 10.1016/j.devcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- Stainier DY, Lee RK, Fishman MC. Cardiovascular development in the zebrafish. I. Myocardial fate map and heart tube formation. Development. 1993;119:31–40. doi: 10.1242/dev.119.1.31. [DOI] [PubMed] [Google Scholar]
- Stankunas K, Hang CT, Tsun ZY, Chen H, Lee NV, Wu JI, Shang C, Bayle JH, Shou W, Iruela-Arispe ML, Chang CP. Endocardial Brg1 represses ADAMTS1 to maintain the microenvironment for myocardial morphogenesis. Dev Cell. 2008;14:298–311. doi: 10.1016/j.devcel.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugi Y, Markwald RR. Formation and early morphogenesis of endocardial endothelial precursor cells and the role of endoderm. Dev Biol. 1996;175:66–83. doi: 10.1006/dbio.1996.0096. [DOI] [PubMed] [Google Scholar]
- Sugi Y, Markwald RR. Endodermal growth factors promote endocardial precursor cell formation from precardiac mesoderm. Dev Biol. 2003;263:35–49. doi: 10.1016/s0012-1606(03)00433-0. [DOI] [PubMed] [Google Scholar]
- Taddei A, Giampietro C, Conti A, Orsenigo F, Breviario F, Pirazzoli V, Potente M, Daly C, Dimmeler S, Dejana E. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat Cell Biol. 2008;10:923–934. doi: 10.1038/ncb1752. [DOI] [PubMed] [Google Scholar]
- Totong R, Schell T, Lescroart F, Ryckebusch L, Lin YF, Zygmunt T, Herwig L, Krudewig A, Gershoony D, Belting HG, Affolter M, Torres-Vazquez J, Yelon D. The novel transmembrane protein Tmem2 is essential for coordination of myocardial and endocardial morphogenesis. Development. 2011;138:4199–4205. doi: 10.1242/dev.064261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh LA, Stainier DY. Fibronectin regulates epithelial organization during myocardial migration in zebrafish. Dev Cell. 2004;6:371–382. doi: 10.1016/s1534-5807(04)00063-2. [DOI] [PubMed] [Google Scholar]
- Van Handel B, Montel-Hagen A, Sasidharan R, Nakano H, Ferrari R, Boogerd CJ, Schredelseker J, Wang Y, Hunter S, Org T, Zhou J, Li X, Pellegrini M, Chen JN, Orkin SH, Kurdistani SK, Evans SM, Nakano A, Mikkola HK. Scl represses cardiomyogenesis in prospective hemogenic endothelium and endocardium. Cell. 2012;150:590–605. doi: 10.1016/j.cell.2012.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veerkamp J, Rudolph F, Cseresnyes Z, Priller F, Otten C, Renz M, Schaefer L, Abdelilah-Seyfried S. Unilateral dampening of bmp activity by nodal generates cardiac left-right asymmetry. Dev Cell. 2013;24:660–667. doi: 10.1016/j.devcel.2013.01.026. [DOI] [PubMed] [Google Scholar]
- Viragh S, Szabo E, Challice CE. Formation of the primitive myo- and endocardial tubes in the chicken embryo. J Mol Cell Cardiol. 1989;21:123–137. doi: 10.1016/0022-2828(89)90856-0. [DOI] [PubMed] [Google Scholar]
- Visvader JE, Fujiwara Y, Orkin SH. Unsuspected role for the T-cell leukemia protein SCL/tal-1 in vascular development. Genes Dev. 1998;12:473–479. doi: 10.1101/gad.12.4.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner M, Siddiqui MA. Signal transduction in early heart development (II): ventricular chamber specification, trabeculation, and heart valve formation. Exp Biol Med (Maywood) 2007;232:866–880. [PubMed] [Google Scholar]
- Yelon D, Horne SA, Stainier DY. Restricted expression of cardiac myosin genes reveals regulated aspects of heart tube assembly in zebrafish. Dev Biol. 1999;214:23–37. doi: 10.1006/dbio.1999.9406. [DOI] [PubMed] [Google Scholar]
- Yelon D, Ticho B, Halpern ME, Ruvinsky I, Ho RK, Silver LM, Stainier DY. The bHLH transcription factor hand2 plays parallel roles in zebrafish heart and pectoral fin development. Development. 2000;127:2573–2582. doi: 10.1242/dev.127.12.2573. [DOI] [PubMed] [Google Scholar]
- Zhang XY, Rodaway AR. SCL-GFP transgenic zebrafish: in vivo imaging of blood and endothelial development and identification of the initial site of definitive hematopoiesis. Dev Biol. 2007;307:179–194. doi: 10.1016/j.ydbio.2007.04.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Frontal views of wild-type (A, B) and tal1-deficient hearts (C, D) at 30 hpf; ventricular end of the heart tube at the top. The transgene Tg(fli1a:negfp)(green) was used to count the number of endocardial cells residing within the boundaries of the myocardium, which was labeled by MF20 immunofluorescence (red).
(A–D) Transverse section of wild-type endocardium, dorsal to the top, at 18s. Immunofluorescence detects localization of ZO-1 (red) (B–D) and F-actin (blue) (C); in addition, an anti-GFP antibody allows visualization of Tg(kdrl:GRCFP) expression (green) in the endocardium (A, D). The boxed area in (A) is highlighted in (B–D). Even when wild-type cells are stacked on top of each other, ZO-1 is properly localized to individual vertices at cell-cell junctions (B–D), rather than being found in large patches as in tal1-deficient embryos (Fig. 6E, F, H).
(E) Cartoon indicates regions of ZO-1 (red) and F-actin (blue) localization within the endocardium.
(A–D) Three-dimensional reconstructions of confocal stacks depict dorsal views of wild-type (A), han mutant (B), tal1-deficient(C) and han mutant; tal1-deficient (D) embryos expressing Tg(fli1a:negfp) (green) at 20s.
(A′–D′) Reconstructions of optical sections displaying transverse slices through the endocardium of the embryos shown in (A–D). Arrowheads mark progenitors of the lateral dorsal aortae.
The endocardium forms a single-layered sheet in wild-type (A, A′) and han mutant (B, B′) embryos. In contrast, the endocardium in embryos that are deficient for both han and tal1 (D, D′) forms a multi-layered aggregate similar to that seen in tal1-deficient embryos (C, C′).