

Abstract
To reduce peroxides, peroxiredoxins (Prx) require a key ‘peroxidatic’ cysteine, that in a substrate-ready fully folded (FF) conformation becomes oxidized to sulfenic acid, and then after a local unfolding (LU) of the active site, forms a disulfide bond with a second ‘resolving’ Cys. For Salmonella typhimurium alkyl hydroperoxide reductase C (StAhpC) and some other Prxs, the FF structure is only known for a peroxidatic Cys→Ser variant, which may not accurately represent the wild type enzyme. Here, we obtain the structure of authentic reduced wild type StAhpC by dithiothreitol treatment of disulfide form crystals that fortuitously accommodate both the LU and FF conformations. The unique environment of one molecule in the crystal reveals a thermodynamic linkage between the folding of the active site loop and C-terminal regions, and comparisons with the Ser-variant show structural and mobility differences from which we infer that the Cys→Ser mutation stabilizes the FF active site. A structure for the C165A variant (a resolving Cys to Ala mutant) in the same crystal form reveals that this mutation destabilizes the folding of the C-terminal region. These structures prove that subtle modifications to Prx structures can substantially influence enzymatic properties. We also present a simple thermodynamic framework for understanding the various mixtures of FF and LU conformations seen in these structures. Based on this framework, we rationalize how physiologically-relevant regulatory posttranslational modifications may modulate activity and propose a non-conventional strategy for designing selective Prx inhibitors.
Introduction
Peroxiredoxins (Prx) are a ubiquitous family of enzymes which reduce peroxides and peroxynitrites via a reactive cysteine.1 Members of the widespread Prx1 subfamily, mostly found to form doughnut-shaped decamers, are thought to be responsible for reducing over 90% of cytosolic and mitochondrial peroxides.1,2,3 The study of Prx1 enzymes also has implications for antibiotic drug designs as knock-out strains of certain microbial pathogens show them to be important for surviving peroxynitrite exposure4 (such as is generated by the host immune system5), for scavenging peroxide,6 for colonization,7 or even growth at oxygen levels at or above 4%.8,9 Additionally, controlled inactivation of some eukaryotic Prx1 subfamily members by a peroxide-driven hyperoxidation of the reactive Cys to a Cys-sulfinate was proposed to play a role in allowing peroxide-mediated signaling.10 The downstream oxidation of target proteins by locally accumulated hydrogen peroxide is increasingly recognized as important in many such signaling pathways11 with particularly well-studied examples being the inhibition of protein tyrosine phosphatases via oxidation of an active site cysteine.11,12,13,14,15 Physiologically relevant regulation of Prx1 enzymes is also thought to occur by phosphorylation,11, 13,16,17 lysine acetylation,18 glutathionylation,19 and proteolysis.11,20
Structural studies have shed much light on the mechanism of the Prx1 family of Prx enzymes (also called typical 2-Cys Prxs). For these enzymes, key residues are a peroxidatic Cys, termed CP, contained within an absolutely conserved PXXX(T/S)XXC motif, an additional conserved Arg, and a resolving Cys, termed CR, located near the C-terminus1,21 (Fig. 1a). The basic active unit involves a so-called “B-type” homodimer with two active sites, each involving the CP from one subunit and CR from the other (Fig 1a), with the association of five such dimers at “A-type” interfaces building the toroidal (α2)5 decamer (Fig 1b).22 The active site stabilizes the CP thiolate (pKa ∼6.0),23 as well as specifically binding and activating the peroxide substrate.1,24 In the catalytic cycle (Fig. 1c), an SN2 nucleophilic attack of the CP thiolate on the peroxide substrate forms a CP-sulfenic acid and water (or corresponding alcohol); the CP-sulfenic acid is then attacked by CR to form an interchain CP—CR disulfide (and another molecule of water) which is in turn reduced most commonly by a thioredoxin-type protein.1,21,24
Figure 1.
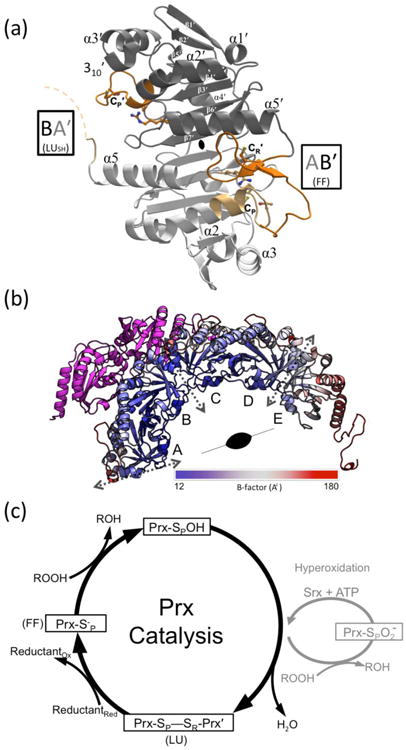
Structure and catalysis of StAhpC. (a) A “B-type” dimer unit of StAhpC with the key catalytic residues depicted as sticks, and select Prx fold core elements of seven β-strands and five helices22 labeled. Chains A (light grey/light orange) and B (dark grey/dark orange) from the WTDTT structure illustrate the FF AB′ active site and the LUSH BA′ active site (seen at ∼50% occupancy). The active site loop (residues 40-50) and C-terminal (residues 161-186) regions are highlighted (orange tones), as is the disordered BA′ C-terminal region (dashed curve). (b) The half-decamer observed in the StAhpC WTDTT asymmetric unit is colored according to mobility from low (blue) to high (red) as indicated. Decamer-building interfaces between B-type dimers (dotted grey two-headed arrows) and the crystallographic two-fold axis that generates the decamer are indicated. Also shown is the symmetry mate hindering folding of the chain A C-terminus (magenta). (c) The Prx catalytic cycle (black) and the hyperoxidation regulatory shunt active in some eukaryotes (grey) are shown. FF and LU conformations involved in catalysis are indicated.
In the enzyme form that reacts with peroxide, CP and CR are ∼14 Å apart and CR is buried, so that formation of the CP—CR disulfide requires a substantial local unfolding of both the active site loop (i.e. residues ∼40-50 which contains CP) of one chain and the C-terminus (containing CR) of its partner chain in the homodimer. Each active site of these enzymes, therefore, has at least two discrete conformations relevant to the catalytic cycle (Fig 1): (1) a fully folded conformation (FF), with a substrate-ready active site pocket, and (2) a locally unfolded conformation (LU) having the active site loop rearranged and the C-terminal residues beyond CR disordered.1,25 This FF↔LU transition is not only important for catalysis, but it has been shown to be the main factor governing the sensitivity of Prx1 subfamily enzymes to hyperoxidative inactivation.10 Some remaining points of uncertainty are, however, to what extent the unfolding of the active site loop and C-terminus are coordinated, how the conformational equilibrium is altered by modifications, and whether the catalytic states of CP-SH and CP-SOH are locked into a certain conformation or dynamically transitioning between them. NMR and crystallographic evidences imply that there is a preexisting dynamic equilibrium for the CP-SH form for a monomeric plant enzyme in the PrxQ subfamily,25,26 while one human Prx1 subfamily enzyme has recently been reported to specifically convert to the LU conformation when in the CP-SOH form.27
We address these questions here using Salmonella typhimurium alkyl hydroperoxide reductase C (StAhpC), one of the first discovered Prxs and a model system for studying Prx1 catalysis,28, 29 oligomerization,22 and regulation.10 Among the six StAhpC crystal structures reported, four exhibit the LU disulfide conformation (LUS-S) – with one wild type22 and four mutants29,23 – while only one, a CP→Ser (i.e. C46S) mutant, adopts the FF conformation.10 We speculated that the StAhpC C46S mutant structure may not accurately represent the properties of the reduced wild type enzyme, and thus sought to obtain a structure of the authentic wild-type FF active site. Here, we report that soaking the LUS-S form crystals in dithiothreitol (DTT) provides a structure of FF wild-type StAhpC (FFWT) in a crystal environment that can accommodate both the FF and LU conformations. In addition, we developed a protocol to crystallize the CR→Ala mutant in the same crystal form. This allows for a rare level of insight into the conformational, dynamic, and thermodynamic aspects of the FF↔LU equilibrium that are essential for Prx function, including how they are influenced by CP→Ser and CR→Ala mutations.
Experimental Procedures
Crystallography
C165A creation and purification of wild type and mutant AhpC proteins
The C165A mutation of StAhpC was created using the QuikChange site-directed mutagenesis kit (Stratagene) and validated by sequencing the entire gene. Wild type and mutant StAhpC were expressed from the pTHCm-ahpC vector23 in JW0598 (lacking ahpC) E. coli cells30 grown in Studier's ZYM-5052 auto-induction media.31 The purification procedure for all AhpC mutant proteins was essentially the same as described previously32 except that 10 mM β-mercaptoethanol (BME) was included in all buffers used during the purification of C165A in order to prevent hyperoxidation of the CP. After purification by phenyl sepharose and ion exchange chromatography, the C165A protein was concentrated and exchanged into 25 mM potassium phosphate, pH 7.0, 1 mM EDTA, 2 mM DTT. As seen from the results, apparently some BME remained present after the buffer exchange. The concentration of AhpC was determined by absorbance at 280 nm with ε = 24,300 M-1 cm-1.32
Crystallization of wild type StAhpC and C165A mutant
Initial crystallization was essentially as described by Wood et al.22 For wild type, optimal crystals were grown at 300 K in hanging drops formed by 4 μL of 14.3 mg/ml protein (in 25 mM phosphate-buffered saline (PBS), 1mM EDTA, pH 7.0) mixed with 1 μL of artificial mother liquor (AML) containing 1.4 M MgSO4 and 0.1 M MES at pH 6.5. Micro-seeding produced larger and better-diffracting crystals. Briefly, initial crystals were crushed in 100 μL of AML and vortexed, and a serial dilution of seed stock concentrations was created. Drops were seeded by dipping a 21-gauge needle into the seed stock and then streaking it across the new drop. Large, tapering column crystals on the order of ∼0.5 mm grew in 1-14 days. As expected, these crystals contained protein in the disulfide form, and for reduction, crystals were soaked for two minutes in freshly prepared AML containing 0.1 M DTT (Fig. S1). Some stress lines did appear on the crystals when this soak was performed.
Many attempts to grow C2221 crystals of untreated C165A produced only a single crystal that grew after more than a month. Peroxide at 100 mM was added to some crystallization trials to attempt to produce homogeneous oxidized protein, and crystals grew much more readily. Analysis of the treated protein by mass spectrometry showed that the predominant redox states of the enzyme were CP-SO3- and a form with the molecular weight expected for a BME adduct that presumably was produced by residual BME from the purification reacting with transiently formed CP-SOH (Fig. S2). These crystals yielded a structure that was 100% LU but when soaked with DTT a portion of the enzyme shifted to the FF conformation. We inferred that the portion of the protein forming the BME-adduct was being reduced and shifting its conformation to FF, and the portion containing CP-SO3- was not being reduced and was remaining in the LU conformation. Though not conclusive, this observations implies that the CP-SO3- form of StAhpC behaves differently than two other Prxs for which this form was shown to be FF.33,34 In refining the protocol to produce maximal C165A-BME adduct with minimal CP-SO2-/SO3- formation, we settled on the addition of 10 μM BME and 20 μM peroxide based on trials of several concentrations that were analyzed with mass spectrometry (Fig S2). In our final protocol, crystals of C165A were obtained by seeding with crushed wild type LUS-S crystals in drops first treated with 10 μM BME for 30 minutes, with the subsequent addition of hydrogen peroxide to a final concentration of 20 μM. A DTT soak of these crystals was carried out as for wild type StAhpC. Here, we refer to the DTT-soaked structures of wild type and C165A as WTDTT and C165ADTT, respectively.
For freezing both wild type and C165A crystals, glycerol was added to the drop as a cryoprotectant to make a final concentration of ∼20%. The crystals dissolved if the glycerol was added too quickly, so the glycerol was placed beside the crystal drop and a small channel was created between them with a pipette tip and equilibrated for ∼2 min. The crystals were then scooped and frozen by plunging into liquid nitrogen.
Data collection
Data were collected at the Advanced Light Source (ALS) Lawrence Berkeley National Laboratory beam-lines 5.0.2. and 8.2.2. The data were indexed and integrated with Mosflm version 7.0.9.35 All crystals of StAhpC were found to be in the space group C2221 and exhibited similar unit cell dimensions as previous StAhpC structures, with a∼127 Å, b∼171 Å, c∼135 Å (see Table 1). The resolution cutoff criterion was based on the new statistic CC1/2 recently introduced by Karplus and Diederichs,36 which demonstrated that better models are obtained when resolution is extended to the CC1/2 = 0.1-0.2 range despite the fact that Rmeas. values become disturbingly large and <I/σ> unconventionally low. We have applied this new criteria in recent studies and discussed its value,25,37 and note a recent detailed study by Evans & Murshudov38 concluded that useful signal is contained in data out to the CC1/2 ∼0.2-4 range and “it seems sensible to set a generous limit so as not to exclude data containing real (if weak) information.” For our structures, we implemented a cutoff of CC1/2 ∼0.2 in the highest resolution shell (Table 1) and obtained improved maps. The effective resolution of a structure is difficult to define, but for comparison with past structures, we also note the resolution at which <I/σ> ∼2 (Table 1). Rfree flags were randomly assigned to five percent of the WTDTT dataset, and the same flags were imported to use for the C165ADTT dataset.
Table 1. Data collection and refinement statistics.
| Data Collection | PDB code:4MA9 | PDB code: 4MAB |
|---|---|---|
| Structure | WTDTT | C165ADTT |
| Space group | C2221 | C2221 |
| Unit cell a, b, c (Å) | 126.81, 171.13, 135.34 | 127.23, 172.42, 136.21 |
| Resolution (Å) | 36.8-1.82 (1.92-1.82)a | 29.2-1.90 (2.00-1.90) |
| Completeness (%) | 96.7 (91.1) | 100.0 (100.0) |
| Unique reflections | 126642 (17246) | 117456 (17015) |
| Multiplicity | 13.0 (12.7) | 6.8 (6.4) |
| Rmeas (%) | 23.1b (408) | 23.8c (1048) |
| <I/σ> | 10.6 (0.6)d | 6.2 (0.2)e |
| CC1/2 | 1.00 (0.16) | 0.995 (0.20) |
| Refinement | ||
| Resolution range (Å) | 36.7-1.82 | 29.2-1.90 |
| R-factor (%) | 20.4 | 19.8 |
| R-free (%) | 24.0 | 23.9 |
| Molecules in AU | 5 | 5 |
| Protein residues | 907 | 909 |
| Water molecules | 518 | 232 |
| Total atoms | 14776 | 14454 |
| RMSD lengths (Å) | 0.012 | 0.016 |
| RMSD angles (°) | 1.3 | 1.6 |
| Ramachandran plotf | ||
| φ, ψ-Preferred (%) | 97.6 | 97.2 |
| φ, ψ-Allowed (%) | 2.4 | 2.7 |
| φ, ψ-Outliers (%) | 0.0 | 0.1g |
| B factors | 1 TLS group/monomer | 1 TLS group/monomer |
| <Main chain> (Å2) | 48 | 56 |
| <Side chains & waters> (Å2) | 59 | 64 |
Values in parentheses are for the highest resolution shell and preceding values are for all data.
Rmeas in the inner shell (40-5.76 Å) is 3.3%.
Rmeas in the inner shell (30-6.01 Å) is 3.8%.
<I/σ> of the inner shell is 58.1 and falls to ∼2 at 2.15 Å.
<I/σ> of the inner shell is 30.8 and falls to ∼2 at 2.30 Å.
Preferred, allowed, and outlier angles as assigned by Molprobity.66
V164 chain A has weak density but is near an allowed region.
Refinement of DTT-soaked structures
An initial model for WTDTT was constructed based on the StAhpC C46S crystal structure (PDB entry 1n8j of spacegroup P1)10 with active site residues 40-50 and 161-186 removed to produce maps that were unbiased for these regions. Chains K, L, O, P, and R of 1n8j were superimposed on the half-decamer of PDB entry 1yep, one of the previously solved locally-unfolded C2221 StAhpC structures,22 and this model was refined using BUSTER,39 producing initial R/Rfree values of 25.0/28.9. All chains were found to have had their disulfides reduced, and the active sites were modeled as FF or LUSH (LU with no disulfide formed) or LUC-term (only the C-terminal region unfolded), based on omit map density (Table 2). Coot40 was used to perform manual rebuilding, with main initial changes including the addition of solvent molecules and adjustment of some side-chain rotamers. No non-crystallographic-symmetry (NCS) restraints were used. End stage refinements were performed with PHENIX.41 Adding riding hydrogens reduced Rfree by ∼0.5 percent, and using TLS refinement with one group per protein chain, dropped Rfree by ∼3 percent. The occupancy of the chain B active site loop was estimated to be ∼0.5:0.5 FF:LU. We expect that remaining positive difference density in this region is due to solvent molecules that coordinate with Arg119 when the active site is LU, as was observed in the LUS-S structure.22 Based on evidence (see Fig. 2 legend), residual positive difference map peaks at sites that had been modeled as waters were replaced by potassium and chloride ions and six glycerol molecules. There remains one fairly large and oddly-shaped difference density feature at each decamer-building interface, which we were not able to interpret (Fig. S3). The WTDTT final R/Rfree was 20.4%/24.0%, and the final refinement statistics are reported in Table 1.
Table 2.
Conformations and their occupancies as seen in representative active sites of StAhpC structures.a
| FF | LUC-term | LUSH | LUS-S | |
|---|---|---|---|---|
| Crystal: active sites |

|
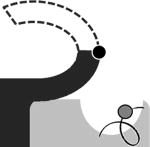
|
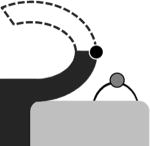
|
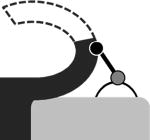
|
| WTDTT: AB′, CD′, DC′, EE′ | 100 | 0 | 0 | 0 |
| WTDTT: BA′ | 0 | 50 | 50 | 0 |
| C165ADTT: AB′, CD′, DC′, EE′ | 60 | 20 | 20 | 0 |
| C165ADTT: BA′ | 0 | 50 | 50 | 0 |
| C46S: all 20 | 100 | 0 | 0 | 0 |
| WTS-S: all 5 | 0 | 0 | 0 | 100 |
The conformation names used are all described in the text and the cartoon images provide a simple visual of the essential features of the conformation with the CP shown as a grey circle and CR as a black circle. The four crystal forms listed are also described in the text.
Figure 2.
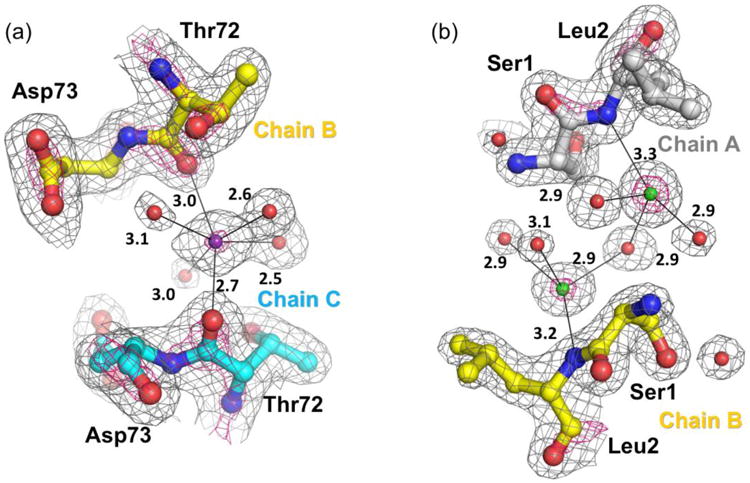
Evidence for bound ions at subunit interfaces in the WTDTT structure. (a) Representative of all decamer-building interfaces, a potassium (purple sphere) is coordinated by Thr72-O of chains B and C and four water sites (red spheres). The 2FO-FC density is contoured at 1.5 ρrms (grey mesh) and 5.0 ρrms (hot pink mesh) and coordinating interactions (black thin bonds; distances in Å) are shown. Evidences for the assignment are the strong electron density, the roughly octahedral coordination with distances near 2.8 Å as is uniquely expected for a potassium,67 and the presence of 25 mM potassium-phosphate in the protein buffer. If modeled as a water, strong difference density appears. This site is incorrectly modeled as a water in previous StAhpC structures. (b) Representative of all dimer-interfaces, chloride ions (green spheres) modeled near the positively-charged α-amino group of chains A and B. The 2FO-FC density is contoured at 2.0 ρrms (grey mesh) and 6.0 ρrms (hot pink mesh). When modeled as a water, strong difference density is observed. These ions are also present in the C165ADTT structure, but do not appear to be present in C46S StAhpC crystals.
Refinement for C165ADTT utilized the final WTDTT model as a starting model with active site residues 40-50 and 160-186 removed (initial R/Rfree = 24.4/26.4). As for WTDTT, the chain B active site loop was modeled as 50:50 LUSH/LUC-term. Residues 40-50 of chains A, C, D, and E were modeled only as fully-occupied FF despite residual low level density that could be due to some LU population; the C-terminal regions were modeled as partially occupied FF based on consistent 2FO-FC and FO-FC density for the expected FF chain path. Test refinements varying the occupancy in increments of 0.1 gave 0.6 as the occupancy at which the B-factors for this region were closest to those observed for the WTDTT structure. A residual difference peak near the position occupied by wild type Cys165-Sγ was left uninterpreted, and we suspect it is due to a partially-occupied sulfhydryl from DTT (present at ∼0.1 M) that binds at this site when the C-terminus is not folded. Both mass spectrometry on these crystals (Fig. S2), and reasonably clear electron density for Ala165 in chain A confirm the crystals are of the C165A mutant. Using the reference restraint option of Phenix41 the WTDTT structure was utilized to restrain the geometries of the weakly-occupied C-terminal regions. Riding hydrogens were added to the model and one TLS group per chain was implemented to reach final R/Rfree values of 19.9/23.9 (Table 1). The coordinates and structure factors for the WTDTT and C165ADTT structures have been deposited in the Protein Data Bank as PDB codes 4ma9 and 4mab.
Mass spectrometry analysis
Crystal samples were prepared from crystals harvested into a 10 μL drop of degassed DI water, manually crushed, and kept from further chemical reactions by flash-freezing in liquid nitrogen prior to analysis. Proteins were detected from 800-2000 m/z using a LTQ-FT Ultra mass spectrometer (Thermo, San Jose, CA, US) in LTQ mode, with a Finnigan Ion Max API source set up for electrospray ionization in positive ion mode. All collections used the following conditions: spray voltage 5 kV, capillary temperature 200° C, capillary voltage 40 V, and tube lens 240 V. Proteins from samples described above were adsorbed onto a C4 Ziptip, washed with water and eluted in-line to the mass spectrometer with solvent as previously described.42,43 The solvent of 50% acetonitrile, 50% water and 0.1% formic acid was delivered at 20 μL/min. Maximum entropy deconvolution and integration of the mass spectrum was performed to generate zero charge parent masses from the mass spectra using the program MOP (Multiple Overlaying Pictures, Spectrum Square Associates, Inc) and Matlab.42 Spectra of crystals were noisy, but clearly showed that WTDTT was predominantly monomeric (indicating reduction of the intermolecular disulfide, Fig. S1) and C165ADTT was mostly in the thiol/thiolate state, though there were minor peaks near the expected positions of CP-SOH, CP-SO2-, and CP-SO3- (Fig. S2). The time course assay for C165A was performed on reduced protein at 43.5 μM in 100-fold diluted PBS buffer,44 pH 7.0, with the addition of peroxide to make the final concentration 1 mM. Samples were taken at 0, 1, 2, 3, 5, 10 and 20 minutes and analyzed as specified above.
Results and Discussion
Overall structure
Recognizing that the existing StAhpC wild type LUS-S structure22 had few crystal contacts involving residues that change positions in the FF↔LU transition (Fig 1b), we conceived of generating the FF conformation by soaking the LUS-S crystals in DTT. Fresh crystals of the disulfide form of wild type StAhpC22 survived the DTT soak with diffraction power and unit cell virtually unchanged, allowing the structure to be determined at 1.82 Å resolution using recently proposed36 more generous resolution cutoff criteria (Table 1). We identified bound potassium and chloride ions (Fig. 2), which may relate to how ionic strength can increase decamerization, FF stability, and catalytic activity,29 as has been reported for another AhpC.45 In this crystal form a half-decamer of five chains (labeled A-E) are in the asymmetric unit, with the AB, CD, and EEsym (symmetry mate of E) pairs being the functional homodimer units (Fig. 1b).22,29 For the wild-type DTT-treated structure (WTDTT), disulfide reduction did occur, as the active site loop (residues 40-50) for chains A, C, D, E, and the C-termini of their partner chains (B, D, C, and Esym, respectively) adopt the FF conformation, and the CP and CR thiolates/thiols are visible in their expected positions ∼14 Å apart (Fig. 3a,b). Mass spectrometry of dissolved WTDTT crystals confirmed the presence of the reduced monomeric form (Fig. S1). Interestingly, the active site loop of chain B exhibits a ∼50:50 mix of the FF/LU chain paths (Fig. 3c), and its partner in the dimer, chain A, has its C-terminus LU (only modeled through residue 163) even though no CP— CR disulfide is present (Fig 1a). This C-terminal LU conformation can be explained in that a crystal packing interaction blocks the position it would fill in the FF conformation (Fig 1b).
Figure 3.
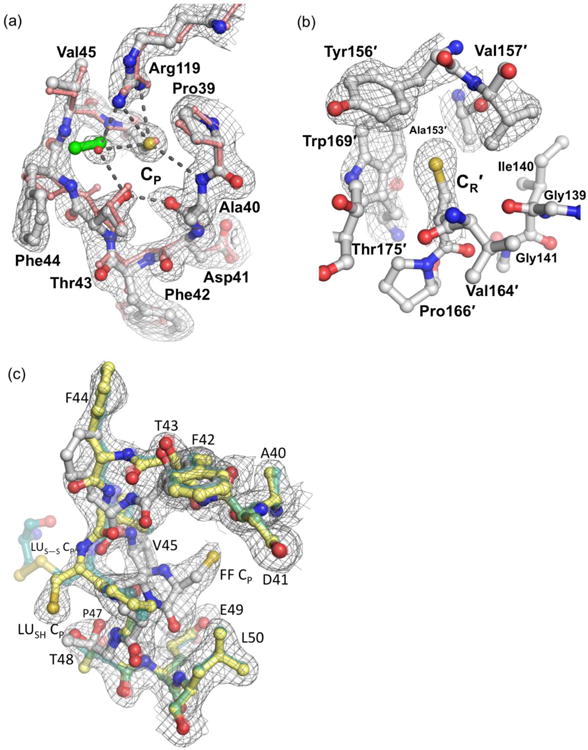
The WTDTT CP and CR environments in FF chains. (a) The FF active site loop of wild type StAhpC chain A (white) in 2FO-FC electron density (grey mesh contoured at 1.5 ρrms) matches the conformation of peroxide-bound ApTpx (pink protein and green peroxide; PDB code 3a2v). Selected polar interactions are indicated by dashed lines. In StAhpC, a bound water (red sphere) overlays with the position of the ApTpx hydrogen peroxide substrate. (b) The FF CR environment in chain B (white) is shown with 2FO-FC electron density (contoured at 1.0 ρrms). The FF CR is buried in a pocket formed by Ile140, Tyr156′, Val157′, Trp169′, and Thr175′ where it is sheltered from reacting with cellular oxidants and electrophiles. (c) The active site loop of chain B shown adopting both the FF path (solid white) and the LUSH path (solid yellow) with similar levels of 2FO-FC electron density (countered at 1.0 ρrms). The LUSH conformation is similar to that of LUS-S (transparent forest green; PDB code 1yep), but the CP thiol adopts a different position.
Thus, in this crystal, three new conformations containing reduced CP and CR residues are observed (Table 2). One, as targeted, is the FF conformation adopted by the wild type enzyme that is seen four times in the asymmetric unit and which we refer to as “FFWT” (Fig. 3a and 3b). The others are both ∼50% occupied and pair an LU C-terminus with either an LU active site loop without a disulfide (“LUSH”), or with an FF active site loop (“LUC-term”)(Fig. 3c). For clarity, since each complete active site combines residues from two chains, we will use here a nomenclature that first defines the chain contributing CP and then the chain contributing CR, and using a prime (′) to denote residues from the chain contributing CR. Thus, the four ∼100% FFWT active sites are the AB′, CD′, DC′, EE′sym active sites, and the 50:50 mix of the LUSH and LUC-term conformations occurs in the BA′ active site. An advantage of this crystal form is it provides a controlled environment in which StAhpC structures are each seen in two basic contexts — one with unconstrained C-termini and active site loop regions (active sites AB′, CD′, DC′, and EE′sym) and one with a C-terminus constrained to be unfolded and unconstrained active site loop (active site BA′). A summary of the conformations seen in StAhpC structures compared in this paper are given in Table 2.
Linkage of the active site loop and C-terminal conformations
Asymmetry of interactions linking the C-terminal and active site loop regions
As noted above, for the BA′ active site, the A-chain C-terminal region is blocked from adopting the FF conformation by a crystal contact. Our observance of the LUC-term conformation in this active site proves that the active site loop can be FF even when the C-terminal region is LU. In addition, because the BA′ active site loop was ∼50% LU (as opposed to dominantly FF as for the other active sites) implies a thermodynamic linkage in which the absence of a folded C-terminal region destabilizes the FF active site loop.
Comparing the FFWT and LUSH structures in the region of the active site (Fig. 4a) shows that unfolding of the C-terminus could destabilize the active site loop in three ways. First is the loss of hydrophobic packing interactions between Leu176′, Leu182′, and Ile186′ side chains and the first turn of helix α2. Second is a less direct effect mediated by residues 137-142 which in the FF conformation make a short β-sheet with C-terminal residues 163′-166′ (Fig. 4a). When 163′-166′ move away in the LU transition, the 137-142 segment shifts ∼1.5 Å, pulling with it Glu49 which is H-bonded with 142-NH (Fig. 5). This shifting of the Glu49 and Glu138 carboxylates contribute to a disordering of Arg119 and the weakening of its interactions with the FF CP side chain (Fig. 5). Third, a contribution not relevant for the crystalline decamer but expected to occur in solution, is that the loss of the packing from the side chains of Leu180′ and Val183′ with a hydrophobic niche (residues 15-20) at the decamer-building interface (Fig. 4a) would destabilize the decamer and therefore, as has been noted before,22 destabilize the active site loop.
Figure 4.
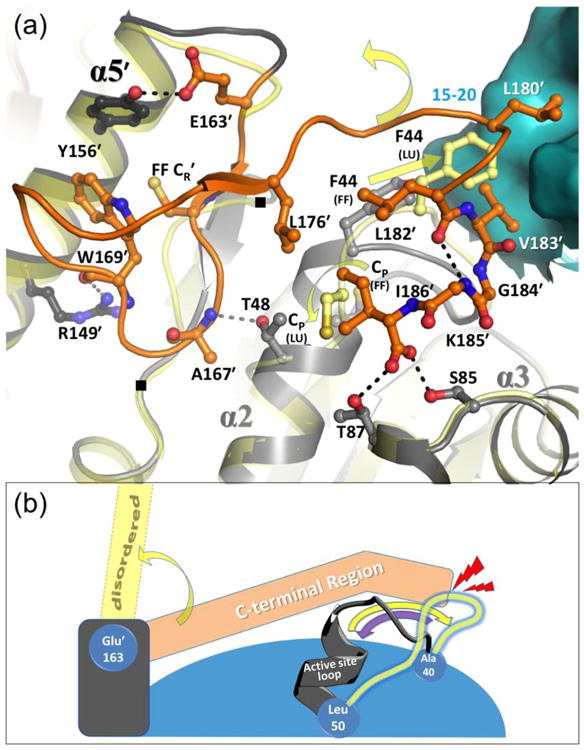
Packing interactions and coordination of unfolding of the FF C-terminal and active site loop regions. (a) The FF active site is shown (active site loop in grey; C-terminal region in dark orange; and the neighboring surface across the decamer-building interface in cyan) along with select interacting residues (labeled and shown as sticks) and stabilizing hydrogen bonds (dashed lines). Positions of residues 137 and 141 are also noted with (■). To provide context, the LUSH conformation (transparent yellow) is shown and yellow arrows indicate movements of the CP residue and Phe44. The C-terminal region has minimal regular secondary structure (residues Val164′–Cys165′ forms a short β-strand and Leu180′-Gly184′ is a short 310-helix), and it interacts with helix α5′ of the same chain (two H-bonds shown), across the decamer-building interface, and with the FF active site loop. The C-terminal residue Ile186′ side chain packs with Pro47, and C-terminal α-carboxylate H-bonds with the side chains of Ser85 and Thr87. (b) Cartoon scheme emphasizing that C-terminal region unfolding destabilizes but does not disrupt the active site loop, whereas active site loop unfolding does disrupt the folding of the C-terminal region. As in panel (a), FF conformations of the two regions are shown in grey and orange, yellow arrows indicate transitions to LU, and LU positions are depicted in transparent yellow. Shown also are approximate hinge points at Ala40, Leu50, and Glu163′ (blue cogs), and the collision of the LU active site loop with the FF C-terminal region (red lightning). The purple arrow emphasizes that the active site loop is not blocked from folding. The decamer-building interactions are not shown in this scheme.
Figure 5.
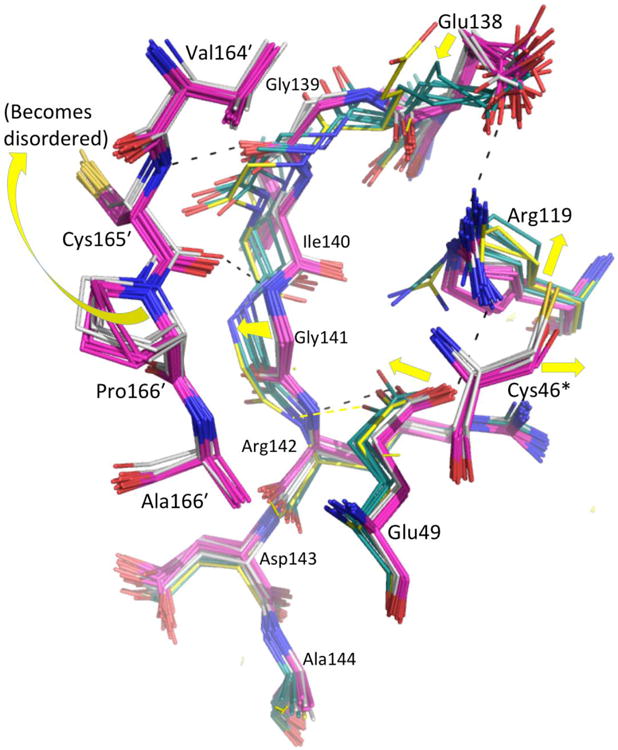
Role of the 137-142 segment in the FF↔LU transition. Twenty-four FF dimers (C46S, pink; wild type, white) and 6 LU dimers (LUS-S, forest green; LUSH, yellow) are shown, with yellow arrows highlighting FF→LU shifts. For clarity, only the FF position of CP (C46 or S46, denoted with an asterisk) is shown. Select H-bonds shown link residues 139-141 in a β-sheet with the FF C-terminal region and via the Glu138 and Glu49 side chains, to the catalytic Arg119.
In these ways, unfolding of the C-terminus destabilizes the FF active site loop, but does not force its unfolding. In contrast, the unfolding of the active site loop will not just destabilize the C-terminal segment, but will actually force it to unfold (Fig. 4b). This asymmetric linkage occurs because the LU positions of the active site loop backbone physically collide with the FF positions of Leu176′, Leu182′, and Ile186′.
Active site loop and C-terminal region B-factor patterns provide additional evidence of linkage
For the AB′, CD′, DC′, and EE′sym active sites in this crystal form, both LUS-S (as grown) and FF (after reduction by DTT) conformations can be adopted, proving that the mobility of these active sites are not hindered by the crystal packing. Therefore, additional evidence of a physical linkage between the active site loop and C-terminal conformations can be gleaned from their B-factors, which show that a correlation exists between their dynamic properties, with more ordered active site loops (lower B-factors) paired with more ordered C-termini (Fig. 6 inset). The detailed B-factor patterns of the chains, controlled for the crystal environment, further illustrates this linkage. Interestingly, all five regions associated with the FF↔LU transition are the high B-factor peaks, and of these regions three – the active site loop, the C-terminus, and residues 85-87 which H-bond to the Ile186′ α-carboxylate – become even more disordered in the transition from FFWT to LUS-S (black vs. green curves in Fig. 6). That all five segments are rather mobile in both FFWT and LUS-S leads us to conclude that they are easily adaptable rather than being highly stabilized in either conformation, and this helps keep the energy barrier to the conformation change low.
Figure 6.
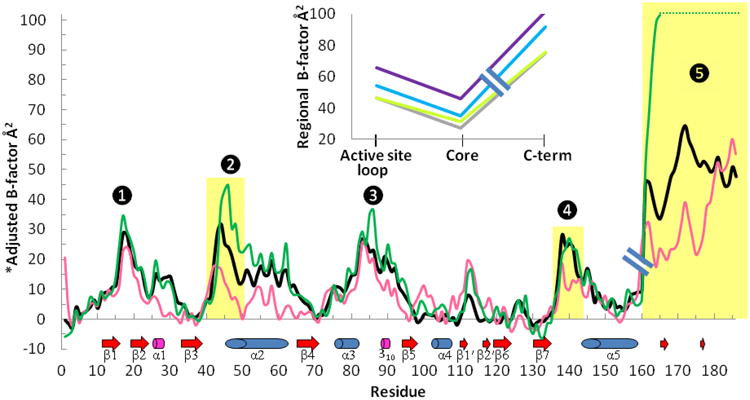
Mobility patterns in wild type StAhpC and CP→Ser structures. A hybrid plot of average main chain B-factors by residue is shown, with each color trace representing the B-factors for residues 1-160 of one chain and residues 161-186 of its dimer partner chain. Shown are the wild type FF AB′ dimer (black), the LUS-S AB′ dimer (PDB code 1yep; forest green), and a dimer from FF C46S with a solvent exposed active site (PDB code 1n8j KL′ dimer; pink). To normalize comparisons, the B-factors were adjusted by subtracting a trace-specific “core” B-value (defined as the 10th percentile B-factor of atoms in the chain contributing residues 1-160). Indicated below the plot are the positions of secondary structures (labeled). The peaks labeled 1 to 5 are the segments involved in the FF↔LU transition, with regions that move highlighted in gold. In C46S, the less-ordered N-terminus may be due to the lack of a chloride ion in this crystal form, and the low B-factors near residue 30 are due to a crystal packing interaction. Differences in the 100-130 region may be attributed to the unidentified ligand at the decamer-building interface (see Experimental Procedures, Fig. S3). Inset: Plotted are the average main chain B-factor for residues 40-50 (active site loop) and for residues 161′-186′ (C-term), along with the 10th percentile B-value of the active site loop containing chain (Core) for each of the four FFWT active sites: AB′ (grey), CD′ (lime green), DC′ (blue), and EE′sym (purple). The variation in absolute mobility among the four FFWT active sites relates to the crystal packing interactions, with for example, the EE′sym active site being the most disordered, because it has no crystal contacts (see also Fig. 1b).
Changes associated with the CP→Ser mutation
Positional differences in the FF active site
In addition to StAhpC, many structures of FF Prx enzymes have only been solved as CP→Ser mutants46,47,48,49,50,51,52,53 assuming that a serine in the reactive CP position faithfully mimics the active site. Given that an active site Cys→Ser mutation can alter the thermodynamics of a folded protein sufficiently to cause substantial rearrangement of the active site,54,55 we looked for differences caused by the C46S mutation by comparing the 4 FFWT active sites with the 20 independent FF active sites seen in crystals of the C46S mutant (FFC46S)10 (Fig. 7a). Although the FFWT and FFC46S structures are very similar (rmsd ∼0.35 Å for 186 Cα-atoms), the sets of FFWT and FFC46S structures cluster distinctly (Fig. 7a), revealing systematic active site differences. For clarity in this comparison, we refer to the C46S mutant structure as being “shifted” relative to the wild type.
Figure 7.
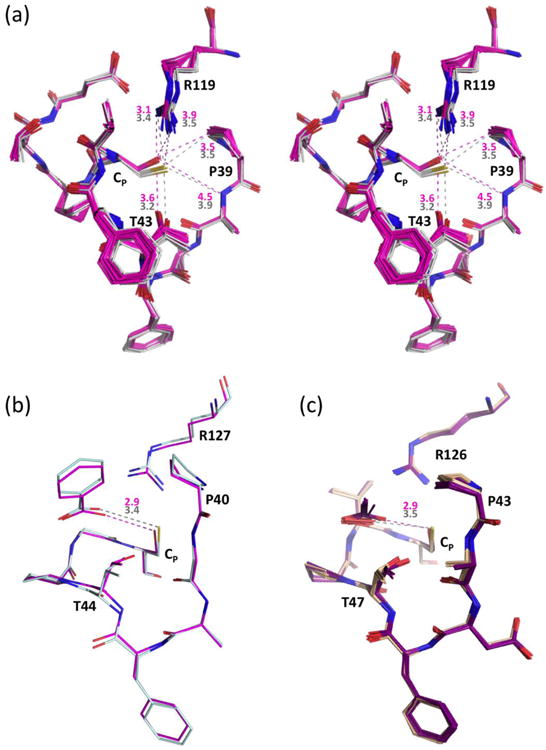
Active site changes due to CP→Ser mutations. (a) Stereo diagram of 20 chains of C46S (pink) overlaid onto the 4 chains of wild type StAhpC (solid white), with representative distances given in Å based on C46S Chain K (pink) and wild type Chain A (grey). Estimation of coordinate uncertainty by SFCHECK68 for both of these models are in the range of 0.2-0.3 Å. (b) Active site overlay of wild type HsPrxV (light blue, PDB code 1hd2) and its CP→Ser mutant (pink, PDB code 1urm) with color-coded distances to the bound benzoate indicated, which were chosen for comparison based on having resolution better than 2.0 Å, clear density for the ligands, and adoption of the same space group with similar unit cells. For 12 chains of wild type HsPrxV with benzoate, acetate, or DTT ligands (PDB codes 1hd2, 3mng, 1h4o, 1oc3, 2vl2, and 2vl3) the average Cys Sγ…ligand distance is 3.46 Å (c) Active site overlay of 10 chains from wild type ApTpx (light pink, peroxide-bound; PDB code 3a2v) and 10 chains from its CP→Ser mutant (purple, acetate bound; PDB code 3a2x) with the color-coded average distances indicated.
Despite conservation of the CP backbone position, Ser46-Oγ is shifted consistently by ∼1 Å compared to Cys46-Sγ, and is further from Ala40-NH, Thr43-Oγ, and Arg119-NH2, and closer to Arg119-NH1 (Fig. 7a). Also, the side chains of Arg119 and Pro39 shift slightly and Thr43 sits deeper in the active site pocket. Independent evidence for which of these shifts are due to the Cys→Ser mutation can be gleaned from the prototypical Prx substrate-bound structures of Aeropyrum pernix thiol peroxidase (ApTpx, a Prx6 family member despite the “Tpx” name;1 PDB code 3a2v)56 (Fig. 3a) and the diol inhibitor-bound Homo sapiens PrxV (HsPrxV; PDB code 3mng),24 which fortuitously have high resolution structures available for both the wild type and CP→Ser forms.1 In both cases, similar shifts are seen for the Ser, Arg, and Thr side chains, and these structures additionally show that bound ligands sit deeper in the active site of the CP→Ser mutants (Fig. 7b and c).
In rationalizing these common shifts, we assume, for a few reasons, that the CP residue in the wild type structures is present as the catalytically relevant thiolate: first, the StAhpC crystals are grown at pH 6.5, above the experimentally measured CP pKa of 5.9 +/- 0.1;23 second, the peroxide-bound ApTpx crystals (PDB code: 3a2v) were at pH 6.5 and catalytically active;24 third, all the HsPrxV structures, ranging from pH=5.6 to pH=8.0, adopt similar active site geometry;24 and fourth, the interactions between Sγ and its active site surroundings are compatible with the thiolate interpretation (Fig. 7). It is to be expected that a hydroxyl would not be able to perfectly mimic a thiolate, as the thiolate is both larger (van der Waals radius ∼0.7 Å greater), cannot donate a hydrogen bond, and with its more diffuse electron clouds is “softer,” making for rather different H-bonding properties.57,58 We attribute the Ser and Arg shifts to making the Arg119-NH1…Oγ H-bond shorter and better aligned in the mutant, and the Thr shift as filling in the space opened by the Sγ to Oγ change while improving the linearity of its H-bond with the backbone carbonyl.59 The ∼0.5 Å deeper penetration of ligands into the active sites of CP→Ser mutants (Fig. 7b and c) would be both due to the smaller van der Waals radius of oxygen and the formation of a Ser-OγH … ligand H-bond. Given such structural differences it should not be surprising that the C46S mutation may also have thermodynamic consequences.
B-factors and crystallizability indicate mutation-induced alterations to mobility
Beyond the changes in atom positions, we also observed striking differences between the B-factors of FFWT and a FFC46S active site that is not constrained by crystal contacts (Fig. 6). Relative to their respective cores, FFC46S has B-factors about half as high as the FFWT in all three regions of the structure that change positions in the FF↔LU transition (yellow highlights). This greater order of C46S provides evidence that in some way the CP-Ser mutation has a stabilizing effect on the FF active site, and we propose that this apparent stabilizing effect is at least in part due to the Ser-Arg hydrogen bond (Fig. 7a) being stronger than those formed by the sulfur atom.58 The ApTpx and HsPrxV CP→Ser structures do not share this difference in B-factor patterns, but this may be due to a stabilizing effect of the bound inhibitors and/or crystal packing interactions.
Additional evidence for a difference in the dynamic properties of C46S and wild type StAhpC is that we were not able to crystallize the reduced wild type enzyme in the C46S mutant P1 spacegroup,10 despite the near identity of their average structures. The explanation we propose is that even though the FF conformation is dominant for the reduced wild type enzyme, LU or partially LU conformations are present at a high enough level to prevent growth of the P1 crystals which have some packing interactions that are not compatible with the LU conformation. This is consistent with deuterium exchange analysis of reduced wild type StAhpC that revealed the active site loop as the least protected region.60 These differences in StAhpC induced by the CP→Ser mutation reinforce two things: first, one must be cautious inferring the properties of the wild type enzyme from studies of such variants, and second, that the wild type active site has not evolved to be as highly stable as possible, as it would be counter-productive to catalysis (see also below).
Structural characterization of the StAhpC CR→Ala mutant
Recently a study of HsPrxIV crystals that could accommodate both the FF and LU conformations showed that a resolving Cys→Ala mutant (HsPrxIV-C245A) adopted the FF conformation when reduced, but was LU after a 5 min 1 mM peroxide treatment said to generate the CP-SOH form.27 Because of the apparent resistance of the CP-SOH to further oxidation, the authors concluded that once the newly-formed CP-SOH transitions to LU, “it does not reform the FF conformation until the peroxidatic cysteine is reduced.”27 However, we note that a Western blot of similar treatments in that study showed an un-quantified portion of the sample was hyperoxidized to CP-SO2/3, and the identification of the CP-SOH state was only based on interpreting an electron density map that for some chains showed alternate conformations for the CP side chain. To address this important point further, we sought to use our StAhpC crystal form to crystallographically define thermodynamic impacts on the FF↔LU equilibrium of both a CR→Ala mutation and CP-SOH formation.
β-mercaptoethanol (BME) as a crystallization aid
We generated the equivalent StAhpC CR→Ala mutant (C165A), which as expected was active in the presence of a suitable reductant. As purified, the C165A variant did not readily crystallize, but crystal growth was strongly promoted by treatment with 100 mM hydrogen peroxide, and the resulting structure showed the protein in the LU conformation in what, based on the electron density, we originally modeled as the CP-SOH form (data not shown). However, mass spectrometric analyses of the crystals (Fig. S2) showed them to be a roughly 60:40 mix of a BME adduct (presumably a mixed disulfide with CP) and Cp-SO3 despite the lack of clear density for these modifications. Using this insight we created a C165A BME-adduct crystal by exposure to only μM levels of peroxide in the presence of added BME. We suggest based on this experience that, especially for CR-mutants or 1-Cys Prxs, trapping an otherwise dynamic Prx enzyme (e.g. 26) in an LU, mixed-disulfide state (for instance, with BME) may enhance conformational homogeneity and serve as a generally useful crystallization aid.
The structure of reduced C165A
The BME-adduct crystal was soaked with DTT to yield a 1.9 Å resolution structure of reduced StAhpC C165A (Table 1; Fig. S2a). In this C165ADTT structure, the BA′ active site (in which the C-terminal region cannot fold) is like WTDTT adopting a 50:50 mix of the LUSH and LUC-term conformations. However, for the other four active sites, the active site loops are visibly in the FF conformation with an unmodified CP side chain (Fig. S2b), but the C-termini have very weak electron density and were modeled at ∼60% FF (Table 2). As little, if any, CP oxidation is present, the ∼40% unfolding of the C-termini must reflect a destabilization caused by the C165A mutation itself. A mutation of a Cys→Ala in a buried, non-polar environment would be expected to be destabilizing.61
Time course of C165A oxidation implies a dynamic CP-SOH form
To seek conditions for generating pure C165A CP-SOH for crystallographic study, we used mass spectrometry to characterize its peroxide-driven CP oxidation. The time course reveals that S-BME and SOH forms appear early, and that as the BME is used up, the SOH peak smoothly decreases while the SO2- peak increases (Fig. 8). This conversion implies that the StAhpC CP-SOH form is not locked into the LU conformation, but dynamically adopts the FF conformation from which it reacts with a second peroxide to form CP-SO2-. Given this behavior, we were not able to create a pure C165A CP-SOH species for structural analysis.
Figure 8.
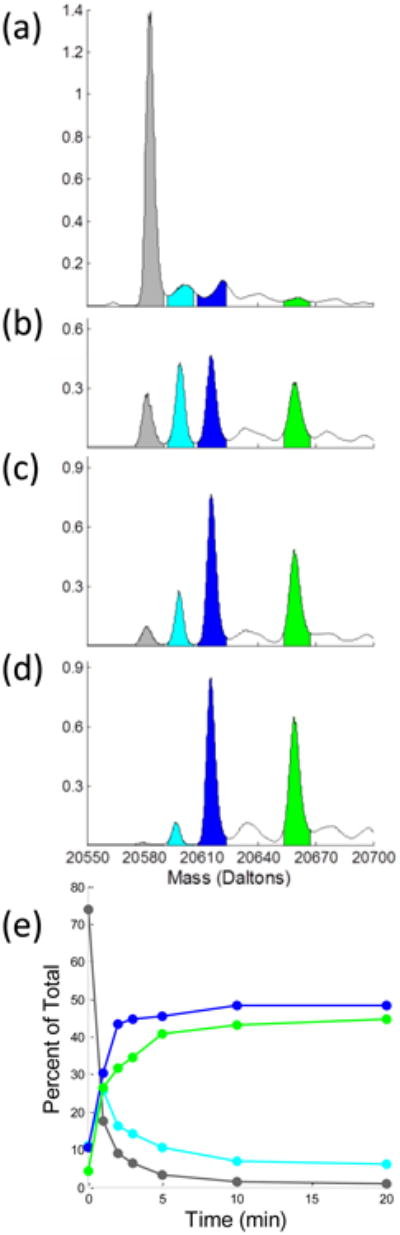
Modifications of C165A StAhpC by 1 mM hydrogen peroxide. (a-d) Mass spectra of purified C165A StAhpC starting material and after treatment with 1 mM peroxide for 1, 3, and 20 min, respectively (see Experimental Procedures). The theoretical weight of C165A is 20584.32 Da (C928H1427N241O283S3), and for modifications: 20600.32 Da (+1 Oxygen) 20616.32 Da (+2 Oxygens), 20660.44 Da (BME adduct, + C2H4OS). Experimental peaks have values of 20584 Da (grey), 20599 Da (cyan), 20615 Da (blue), and 20659 Da (green). Inferred identities of the observed species assume that additions are associated with the CP residue. The y-axes are normalized so that the summed areas of the four colored peaks is a constant. (e) Integrated normalized populations of four prominent species as a function of time. We suspect that the time=0 populations attributed to SOH (cyan) and SO2- (blue) are overestimates, because the peak centers are not at the expected mass and they may be satellite peaks caused by some non-covalent ligand binding to the fully reduced protein. Fortunately, these areas are small and their inclusion does not substantially impact the conclusions.
The facile hyperoxidation of the StAhpC C165A variant is an expected result, and makes all the more surprising the report that HsPrxIV-C245A27 resists hyperoxidation sufficiently to be crystallized as a stable CP-SOH form. Although the two proteins may behave this differently, another possibility is that despite the electron density map evidence, the peroxide-treated HsPrxIV-C245A structure was not CP-SOH, but a mixture of CP-SOH, CP-SO2-, CP-SO3- and/or mixed disulfide forms. Our experience noted above with the structure of C165A treated with 100 mM peroxide (where we had satisfactorily modeled CP-SOH into the electron density maps of protein that was mixture CP-SO3- and a BME adduct) shows that electron density alone in an exposed region of a structure may be insufficient to differentiate between CP-SOH and higher oxidation states. While we agree that CP-SOH formation may indeed promote unfolding of the active site, we suggest that mass spectrometric characterization of the peroxide-treated HsPrxIV-C245A is needed before firm conclusions are drawn.
A conceptual model for understanding the impact of Prx modifications on activity
The changes in conformation and equilibrium populations that we have observed highlight to what extent even slight modifications in or near the Prx active site can alter the enzyme's properties. Although the variety of effects appears complex, a simple thermodynamic model for how the various modifications stabilize or destabilize the FF active site can account for the range of structural changes seen and help in understanding their implications for function (Fig. 9). The six curves illustrate how with changes in interaction energies of just a few kcal/mol, the FF↔LU equilibrium can shift from 1000:1 to 1:100. The crystal structures analyzed here show a hierarchy of FF↔LU equilibria (Table 2) that allow us to assign various StAhpC forms to the different curves (Fig. 9). Beginning with the most ordered FF active site, the StAhpC C46S mutant seems in the range of the orange to red curves (99.9 – 99% FF). The four FFWT active sites that are visibly FF (but less so than C46S) might be represented by the orange to yellow curves (99-90% FF), while the equivalent active sites in the C165ADTT structure — with the C-terminal region destabilized relative to wild type — would be between the yellow and green curves (90-50% FF). The BA′ active site of the WTDTT structure with its ∼50:50 mix of LUSH/LUC-term conformations is described by the green curve. Lastly, the BME adduct that strongly destabilizes the FF active site loop would be represented by the violet curve or above (≥99% LU). Two key insights from these analyses are that (1) for wild type StAhpC the FF:LU free energy difference is not large, so even minor changes can substantially impact the enzyme's properties, and (2) given this sensitive positioning, the FF-stabilizing CP→Ser mutation and the FF-destabilizing CR→Ala mutation may each sufficiently perturb the FF↔LU equilibrium that the measured properties of those mutants will not reliably report on the properties of the wild-type enzyme.
Figure 9.

A thermodynamic framework for understanding the impact of Prx modifications on its function via alteration of the active site loop FF↔LU equilibrium. Plotted is an illustrative series of six possible free energy changes associated with the LU to FF transition with each 1.36 kcal/mol increment in their relative stabilities corresponding to a 10-fold change in the FF/LU equilibrium ratio at 298 K. The transition state barrier is not to scale, but for simplicity, we have drawn it as relatively low, consistent with the rapid dynamics seen for some Prxs.26 Based on the relative strength of density observed for the FF and LU conformations in various crystal forms and active sites, we have roughly assigned the structures discussed here to the different curves. Along this series, StAhpC forms discussed in the text are shown at the point where we approximate their FF↔LU equilibria. As illustrated by the arrows, modifications that destabilize the FF active site will lower the initial rate of reaction with peroxides while protecting against hyperoxidation, whereas modifications that stabilize the FF will slow disulfide formation and increase the enzyme's susceptibility to hyperoxidation. These effects could, in theory, also be induced by inhibitors that stabilized either the LU or FF conformation.
Though each Prx enzyme will have distinct properties, these concrete examples of the linkage between sequence, structure, and thermodynamics can be applied to understand the impact of how sequence variations and physiological Prx posttranslational modifications such as Thr/Ser phosphorylation and lysine acetylation, many of which occur in or affect the FF C-terminal region,10,11,13,16,17, 18,19 may regulate both the enzyme's activity, and/or its sensitivity to hyperoxidation by altering the FF↔LU equilibrium. Indeed, mutations disrupting in the C-terminal region of certain Prxs allowed them to retain activity and become much less sensitive to inactivating hyperoxidation.62,63 Similarly, in vivo proteolytic C-terminal truncation20 or the acetylation of HsPrx1 at Lys19718, have both been reported to lower the enzyme's susceptibility to hyperoxidation, as would be expected if they destabilize the C-terminal packing.
Targeting the prokaryotic Prx C-terminal region for drug design
Although it is most common to target the active sites of proteins for drug therapy, inhibitors that trap enzymes in conformations in which catalysis cannot occur offer an alternative strategy. A recent example is a small molecule inhibitor that holds a tumor-associated mutant of isocitrate dehydrogenase 1 in an open and un-reactive conformation.64 As Prxs contain a universally conserved active site, yet exhibit highly divergent C-terminal regions, especially between human and prokaryotic homologs,1 this latter strategy could be very useful for developing inhibitors selective for pathogen Prxs.10,44 In particular, our observations here lead us to suggest three novel strategies for developing antibiotics that could diminish prokaryotic redox defenses through blocking Prx activity. First, molecules that disrupt the C-terminal packing (indicated by the upward arrow in Fig. 9) would decrease the amount of FF enzyme present and thus inhibit the enzyme's peroxidase activity. Second, and somewhat counter-intuitive, even more effective could be molecules capable of highly stabilizing the FF C-terminus of prokaryotic Prxs (indicated by the downward arrow in Fig. 9). These would strongly shift the equilibrium toward FF to promote (potentially to 100%) the hyperoxidation of CP so that inactive CP-SO2/3- states would accumulate. Third, in a special case that combines these effects, an inhibitor that both destabilized the FF active site and also blocked resolution of the CP-SOH state (similarly to the behavior of the StAhpC-C165A mutant) would both decrease peroxidase activity and promote formation of the hyperoxidized forms. Because few prokaryotes possess a sulfiredoxin-type enzyme capable of reversing hyperoxidation of the peroxidatic cysteine,11 the latter two approaches could be extremely effective, as the hyperoxidation to CP-SO2/3- would cause permanent inactivation.65
Supplementary Material
Acknowledgments
The mass spectrometric analyses were enabled by the Oregon State University Environmental Health Science Center supported by NIEHS grant #P30 ES000210, and crystallographic data collection was done at the Advanced Light Source (ALS), supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Footnotes
This study was supported in part by National Institutes of Health grant RO1 GM050389 to L.B.P. and P.A.K.
Abbreviations: Prx - peroxiredoxin, StAhpC - Salmonella typhimurium alkyl hydroperoxide reductase component C, FF - fully folded, LU - locally unfolded, CP - peroxidatic cysteine, CR - resolving cysteine, LUS-S - locally unfolded disulfide form, LUSH - locally unfolded thiol form, LUC-term - C-terminally unfolded form, WTDTT - DTT-treated wild type structure, C165ADTT - DTT-treated C165A structure, FFWT - fully folded wild type, FFC46S - fully folded C46S mutant, HsPrxIV - Homo sapiens PrxIV, HsPrxI - Homo sapiens PrxI, HsPrxV - Homo sapiens PrxV, ApTpx - Aeropyrum pernix thiol peroxidase, PDB - Protein Data Bank, DTT - 1 -4-dithiothreitol, BME - β-mercaptoethanol, MES - 2-(N-morpholino)ethanesulfonic acid, PBS - phosphate buffered saline, ALS - Advanced Light Source, NCS - non-crystallographic symmetry, TLS - translation libration screw, AML - artificial mother liquor.
Supporting Information: Mass spectrometric data of wild type StAhpC crystals showing the shift from dimer to monomer when soaked with DTT (Figure S1). Mass spectrometric and crystallographic characterization of the C165A mutant crystals (Figure S2). Uninterpreted ligand density at the decamer-building interfaces (Figure S3). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hall A, Nelson K, Poole LB, Karplus PA. Structure-based Insights into the Catalytic Power and Conformational Dexterity of Peroxiredoxins. Antioxid & Redox Signaling. 2011;15:795–815. doi: 10.1089/ars.2010.3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol. 2008;4:278–286. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- 3.Cox AG, Winterbourn CC, Hampton MB. Mitochondrial peroxiredoxin involvement in antioxidant defense and redox signaling. Biochem J. 2010;425:313–325. doi: 10.1042/BJ20091541. [DOI] [PubMed] [Google Scholar]
- 4.Chen L, Xie Q, Nathan C. Alkyl Hydroperoxide Reductase Subunit C (AhpC) Protects Bacterial and Human Cells against Reactive Nitrogen Intermediates. Mol Cell. 1998;1:795–805. doi: 10.1016/s1097-2765(00)80079-9. [DOI] [PubMed] [Google Scholar]
- 5.Fang FC. Perspectives series: host/pathogen interactions. Mechanisms of nitric oxide-related antimicrobial activity. J Clin Invest. 1997;99:2818–2825. doi: 10.1172/JCI119473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seaver LC, Imlay JA. Alkyl Hydroperoxide Reductase Is the Primary Scavenger of Endogenous Hydrogen Peroxide in Escherichia coli. J Bacteriol. 2001;183:7173–7181. doi: 10.1128/JB.183.24.7173-7181.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cosgrove K, et al. Catalase (KatA) and Alkyl Hydroperoxide Reductase (AhpC) Have Compensatory Roles in Peroxide Stress Resistance and Are Required for Survival, Persistence, and Nasal Colonization in Staphylococcus aureus. J Bacteriol. 2007;189:1025–1035. doi: 10.1128/JB.01524-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baker LMS, Raudonikiene A, Hoffman PS, Poole LB. Essential Thioredoxin-Dependent Peroxiredoxin System from Helicobacter pylori: Genetic and Kinetic Characterization. J Bacteriol. 2001;183:1961–1973. doi: 10.1128/JB.183.6.1961-1973.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mehta NS, Benoit SL, Mysore J, Maier RJ. In vitro and in vivo characterization of alkyl hydroperoxide reductase mutant strains of Helicobacter hepaticus. Biochim Biophys Acta. 2007;1770:257–265. doi: 10.1016/j.bbagen.2006.09.022. [DOI] [PubMed] [Google Scholar]
- 10.Wood ZA, Poole LB, Karplus PA. Peroxiredoxin Evolution and the Regulation of Hydrogen Peroxide Signaling. Science. 2003;300:650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 11.Rhee SG, Woo HA, Kil IS, Bae SH. Peroxiredoxin Functions as a Peroxidase and a Regulator and Sensor of Local Peroxides. J Biol Chem. 2012;287:4403–4410. doi: 10.1074/jbc.R111.283432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giorgio M, Trinei M, Migliaccio E, Pelicci PG. Hydrogen peroxide: a metabolic byproduct or a common mediator of ageing signals? Nat Rev Mol Cell Biol. 2007;8:722–728. doi: 10.1038/nrm2240. [DOI] [PubMed] [Google Scholar]
- 13.Rhee SG, Yang KS, Kang SW, Woo HA, Chang TS. Controlled Elimination of Intracellular H2O2: Regulation of Peroxiredoxin, Catalase, and Glutathione Peroxidase via Post-translational Modification. Antiox & Redox Signaling. 2005;7:619–626. doi: 10.1089/ars.2005.7.619. [DOI] [PubMed] [Google Scholar]
- 14.Haque A, Andersen JN, Salmeen A, Barford D, Tonks NK. Conformation-Sensing Antibodies Stabilize the Oxidized Form of PTP1B and Inhibit Its Phosphatase Activity. Cell. 2011;147:185–198. doi: 10.1016/j.cell.2011.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kil IS, et al. Feedback Control of Adrenal Steroidogenesis via H2O2-Dependent, Reversible Inactivation of Peroxiredoxin III in Mitochondria. Mol Cell. 2012;46:584–594. doi: 10.1016/j.molcel.2012.05.030. [DOI] [PubMed] [Google Scholar]
- 16.Zykova TA, et al. T-LAK Cell-originated Protein Kinase (TOPK) Phosphorylation of Prx1 at Ser-32 Prevents UVB-induced Apoptosis in RPMI7951 Melanoma Cells through the Regulation of Prx1 Peroxidase Activity. J Biol Chem. 2010;285:29138–29146. doi: 10.1074/jbc.M110.135905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang TS, Jeong W, Choi SY, Yu S, Kang SW, Rhee SG. Regulation of Peroxiredoxin I Activity by Cdc2-mediated Phosphorylation. J Biol Chem. 2002;277:25370–25376. doi: 10.1074/jbc.M110432200. [DOI] [PubMed] [Google Scholar]
- 18.Parmigiani RB, Xu WS, Venta-Perez G, Erdjument-Bromage H, Yaneva M, Tempst P, Marks PA. HDAC6 is a specific deacetylase of peroxiredoxins and is involved in redox regulation. PNAS. 2008;105:9633–9638. doi: 10.1073/pnas.0803749105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park JW, Piszczek G, Rhee SG, Chock PB. Glutathionylation of Peroxiredoxin I Induces Decamer to Dimers Dissociation with Concomitant Loss of Chaperone Activity. Biochemistry. 2011;50:3204–3210. doi: 10.1021/bi101373h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schröder E, Willis AC, Ponting CP. Porcine natural-killer-enhancing factor-B: oligomerisation and identification as a calpain substrate in vitro. Biochim Biophys Acta. 1998;1383:279–291. doi: 10.1016/s0167-4838(97)00217-3. [DOI] [PubMed] [Google Scholar]
- 21.Nelson KJ, Knutson ST, Soito L, Klomsiri C, Poole LB, Fetrow JS. Analysis of the peroxiredoxin family: Using active-site structure and sequence information for global classification and residue analysis. Proteins: Struct Funct Bioinform. 2010;79:947–964. doi: 10.1002/prot.22936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wood ZA, Poole LB, Hantgan RR, Karplus PA. Dimers to doughnuts: redox-sensitive oligomerization of 2-cysteine peroxiredoxins. Biochemistry. 2002;41:5493–5504. doi: 10.1021/bi012173m. [DOI] [PubMed] [Google Scholar]
- 23.Nelson KJ, Parsonage D, Hall A, Karplus PA, Poole LB. Cysteine pKa Values for the Bacterial Peroxiredoxin AhpC. Biochemistry. 2008;47:12860–12868. doi: 10.1021/bi801718d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hall A, Parsonage D, Poole LB, Karplus PA. Structural Evidence that Peroxiredoxin Catalytic Power Is Based on Transition-State Stabilization. J Mol Biol. 2010;402:194–209. doi: 10.1016/j.jmb.2010.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perkins A, Gretes MC, Nelson KJ, Poole LB, Karplus PA. Mapping the Active Site Helix-to-Strand Conversion of CxxxxC Peroxiredoxin Q Enzymes. Biochemistry. 2012;51:7638–7650. doi: 10.1021/bi301017s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ådén J, et al. Extraordinary μs–ms backbone dynamics in Arabidopsis thaliana peroxiredoxin Q. Biochim Biophys Acta (BBA) - Proteins and Proteomics. 2011;1814:1880–1890. doi: 10.1016/j.bbapap.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 27.Cao Z, Tavender TJ, Roszak AW, Cogdell RJ, Bulleid NJ. Crystal structure of reduced and of oxidized peroxiredoxin IV enzyme reveals a stable oxidized decamer and a non-disulfide-bonded intermediate in the catalytic cycle. J Biol Chem. 2011;286:42257–42266. doi: 10.1074/jbc.M111.298810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parsonage D, Karplus PA, Poole LB. Substrate specificity and redox potential of AhpC, a bacterial peroxiredoxin. PNAS. 2008;105:8209–8214. doi: 10.1073/pnas.0708308105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parsonage D, Youngblood DS, Sarma GN, Wood ZA, Karplus PA, Poole LB. Analysis of the Link between Enzymatic Activity and Oligomeric State in AhpC, a Bacterial Peroxiredoxin. Biochemistry. 2005;44:10583–10592. doi: 10.1021/bi050448i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baba T, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Studier FW. Protein production by auto-induction in high-density shaking cultures. Protein Expression Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 32.Poole LB. Flavin-Dependent Alkyl Hydroperoxide Reductase from Salmonella typhimurium. 2. Cystine Disulfides Involved in Catalysis of Peroxide Reduction. Biochemistry. 1996;35:65–75. doi: 10.1021/bi951888k. [DOI] [PubMed] [Google Scholar]
- 33.Sarma GN, Nickel C, Rahlfs R, Fischer M, Becker K, Karplus PA. Crystal Structure of a Novel Plasmodium falciparum 1-Cys Peroxiredoxin. J Mol Biol. 2005;346:1021–1034. doi: 10.1016/j.jmb.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 34.Mizohata E, Sakai H, Fusatomi E, Terada T, Murayama K, Shirouzu M, Yokoyama S. Crystal structure of an archaeal peroxiredoxin from the aerobic hyperthermophilic crenarchaeon Aeropyrum pernix K1. J Mol Biol. 2005;354:317–329. doi: 10.1016/j.jmb.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 35.Battye TGG, Kontogiannis L, Johnson O, Powell HR, Leslie AGW. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr Sect D: Biol Crystallogr. 2011;67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karplus PA, Diederichs K. Linking Crystallographic Model and Data Quality. Science. 2012;336:1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Driggers CM, Cooley RB, Sankaran B, Hirschberger LL, Stipanuk MH, Karplus PA. Cysteine Dioxygenase Structures from pH 4 to 9: Consistent Cys-Persulfenate Formation at Intermediate pH and a Cys-Bound Enzyme at Higher pH. J Mol Biol. 2013;425:3121–3136. doi: 10.1016/j.jmb.2013.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Evans PR, Murshudov GN. How good are my data and what is the resolution? Acta Crystallogr Sect D: Biol Crystallogr. 2013;69:1204–1214. doi: 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smart OS, et al. Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr Sect D: Biol Crystallogr. 2012;68:368–380. doi: 10.1107/S0907444911056058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 41.Afonine PV, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr Sect D: Biol Crystallogr. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rhoads TW, et al. Measuring copper and zinc superoxide dismutase from spinal cord tissue using electrospray mass spectrometry. Anal Biochem. 2011;415:52–58. doi: 10.1016/j.ab.2011.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rhoads TW, Williams JR, Lopez NI, Morré JT, Bradford CS, Beckman JS. Using Theoretical Protein Isotopic Distributions to Parse Small-Mass-Difference Post-Translational Modifications via Mass Spectrometry. J Am Soc Mass Spectrom. 2013;24:115–124. doi: 10.1007/s13361-012-0500-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nelson KJ, Parsonage D, Karplus PA, Poole LB. Evaluating peroxiredoxin sensitivity toward inactivation by peroxide substrates. Meth Enzymol. 2013;527:21–40. doi: 10.1016/B978-0-12-405882-8.00002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kitano K, Niimura Y, Nishiyama Y, Miki K. Stimulation of Peroxidase Activity by Decamerization Related to Ionic Strength: AhpC Protein from Amphibacillus xylanus. J Biochem. 1999;126:313–319. doi: 10.1093/oxfordjournals.jbchem.a022451. [DOI] [PubMed] [Google Scholar]
- 46.Matsumura T, et al. Dimer-Oligomer Interconversion of Wild-type and Mutant Rat 2-Cys Peroxiredoxin Disulfide Formation at Dimer-Dimer Interfaces is not Essential for Decamerization. J Biol Chem. 2008;283:284–293. doi: 10.1074/jbc.M705753200. [DOI] [PubMed] [Google Scholar]
- 47.Smeets A, Loumaye E, Clippe A, Rees JF, Knoops B, Declercq JP. The crystal structure of the C45S mutant of annelid Arenicola marina peroxiredoxin 6 supports its assignment to the mechanistically typical 2-Cys subfamily without any formation of toroid-shaped decamers. Protein Sci. 2008;17:700–710. doi: 10.1110/ps.073399308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hall A, Sankaran B, Poole LB, Karplus PA. Structural Changes Common to Catalysis in the Tpx Peroxiredoxin Subfamily. J Mol Biol. 2009;393:867–881. doi: 10.1016/j.jmb.2009.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stehr M, Hecht HJ, Jäger T, Flohé L, Singh M. Structure of the inactive variant C60S of Mycobacterium tuberculosis thiol peroxidase. Acta Crystallogr Sect D: Biol Crystallogr. 2006;62:563–567. doi: 10.1107/S0907444906008249. [DOI] [PubMed] [Google Scholar]
- 50.Choi J, et al. Crystal structure of the C107S/C112S mutant of yeast nuclear 2-Cys peroxiredoxin. Proteins: Struct Funct Bioinform. 2005;61:1146–1149. doi: 10.1002/prot.20704. [DOI] [PubMed] [Google Scholar]
- 51.D'Ambrosio K, Limauro D, Pedone E, Galdi I, Pedone C, Bartolucci S, De Simone G. Insights into the catalytic mechanism of the Bcp family: Functional and structural analysis of Bcp1 from Sulfolobus solfataricus. Proteins: Struct Funct Bioinform. 2009;76:995–1006. doi: 10.1002/prot.22408. [DOI] [PubMed] [Google Scholar]
- 52.Limauro D, D'Ambrosio K, Langella E, De Simone G, Galdi L, Pedone C, Pedone E, Bartolucci S. Exploring the catalytic mechanism of the first dimeric Bcp: Functional, structural and docking analyses of Bcp4 from Sulfolobus solfataricus. Biochimie. 2010;92:1435–1444. doi: 10.1016/j.biochi.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 53.Liao SJ, Yang CY, Chin KH, Wang AHJ, Chou SH. Insights into the Alkyl Peroxide Reduction Pathway of Xanthomonas campestris Bacterioferritin Comigratory Protein from the Trapped Intermediate–Ligand Complex Structures. J Mol Biol. 2009;390:951–966. doi: 10.1016/j.jmb.2009.05.030. [DOI] [PubMed] [Google Scholar]
- 54.Melchers J, Diechtierow M, Feher K, Sinning I, Tews I, Krauth-Siegel RL, Muhle-Goll C. Structural Basis for a Distinct Catalytic Mechanism in Trypanosoma brucei Tryparedoxin Peroxidase. J Biol Chem. 2008;283:30401–30411. doi: 10.1074/jbc.M803563200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Muhle-Goll C, Füller F, Ulrich AS, Krauth-Siegel RL. The conserved Cys76 plays a crucial role for the conformation of reduced glutathione peroxidase-type tryparedoxin peroxidase. FEBS Lett. 2010;584:1027–1032. doi: 10.1016/j.febslet.2010.01.054. [DOI] [PubMed] [Google Scholar]
- 56.Nakamura T, Kado Y, Yamaquchi T, Matsumara H, Ishikawa K, Inoue T. Crystal Structure of Peroxiredoxin from Aeropyrum pernix K1 Complexed with its Substrate, Hydrogen Peroxide. J Biochem. 2010;147:109–115. doi: 10.1093/jb/mvp154. [DOI] [PubMed] [Google Scholar]
- 57.Zhou P, Tian F, Lv F, Shang Z. Geometric characteristics of hydrogen bonds involving sulfur atoms in proteins. Proteins: Struct Funct Bioinform. 2009;76:151–163. doi: 10.1002/prot.22327. [DOI] [PubMed] [Google Scholar]
- 58.Platts JA, Howard ST, Bracke BRF. Directionality of Hydrogen Bonds to Sulfur and Oxygen. J Am Chem Soc. 1996;118:2726–2733. [Google Scholar]
- 59.Baker EN, Hubbard RE. Hydrogen bonding in globular proteins. Prog Bioph Mol Biol. 1984;44:97–179. doi: 10.1016/0079-6107(84)90007-5. [DOI] [PubMed] [Google Scholar]
- 60.Nirudodhi S, Parsonage D, Karplus PA, Poole LB, Maier CS. Conformational studies of the robust 2-Cys peroxiredoxin Salmonella typhimurium AhpC by solution phase hydrogen/deuterium (H/D) exchange monitored by electrospray ionization mass spectrometry. Intern J Mass Spectr. 2011;302:93–100. doi: 10.1016/j.ijms.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.González-Mondragón E, Zubillaga RA, Saavedra E, Chanez-Cardenas ME, Perez-Montfort R, Hernandez-Arana A. Conserved Cysteine 126 in Triosephosphate Isomerase Is Required Not for Enzymatic Activity but for Proper Folding and Stability. Biochemistry. 2004;43:3255–3263. doi: 10.1021/bi036077s. [DOI] [PubMed] [Google Scholar]
- 62.Sayed AA, Williams DL. Biochemical Characterization of 2-Cys Peroxiredoxins from Schistosoma mansoni. J Biol Chem. 2004;279:26159–26166. doi: 10.1074/jbc.M401748200. [DOI] [PubMed] [Google Scholar]
- 63.Koo KH, et al. Regulation of Thioredoxin Peroxidase Activity by C-terminal Truncation. Arch Biochem Biophys. 2002;397:312–318. doi: 10.1006/abbi.2001.2700. [DOI] [PubMed] [Google Scholar]
- 64.Wang F, et al. Targeted Inhibition of Mutant IDH2 in Leukemia Cells Induces Cellular Differentiation. Science. 2013;340:622–626. doi: 10.1126/science.1234769. [DOI] [PubMed] [Google Scholar]
- 65.Rhee SG, Jeong W, Chang TS, Woo HA. Sulfiredoxin, the cysteine sulfinic acid reductase specific to 2-Cys peroxiredoxin: its discovery, mechanism of action, and biological significance. Kidney International. 2007;72:S3–S8. doi: 10.1038/sj.ki.5002380. [DOI] [PubMed] [Google Scholar]
- 66.Davis IW, Murray LW, Richardson JS, Richardson DC. MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Research. 2004;32:W615–W619. doi: 10.1093/nar/gkh398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Harding MM, Nowicki MW, Walkinshaw MD. Metals in protein structures: a review of their principal features. Crystallography Reviews. 2010;16:247–302. [Google Scholar]
- 68.Vaguine AA, Richelle J, Wodak SJ. SFCHECK: a unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystallogr Sect D: Biol Crystallogr. 1999;55:191–205. doi: 10.1107/S0907444998006684. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.