

Significance
The cellular response to hypoxia is regulated by hypoxia-inducible factor-1α and -2α (HIF-1α and -2α). We have discovered that filamin A (FLNA), a large cytoskeletal actin-binding protein, physically interacts with HIF-1α (but not with HIF-2α) and promotes tumor growth and angiogenesis. Hypoxia induces a calpain-dependent cleavage of FLNA to generate a fragment that enhances nuclear accumulation of HIF-1α and is corecruited to HIF-1α target promoters, resulting in enhanced gene expression. This mechanism helps to explain why FLNA is upregulated in certain tumors and offers opportunities in targeting the hypoxia signaling pathway therapeutically.
Keywords: cancer, subcellular localization, signal transduction
Abstract
The cellular response to hypoxia is regulated by hypoxia-inducible factor-1α and -2α (HIF-1α and -2α). We have discovered that filamin A (FLNA), a large cytoskeletal actin-binding protein, physically interacts with HIF-1α and promotes tumor growth and angiogenesis. Hypoxia induces a calpain-dependent cleavage of FLNA to generate a naturally occurring C-terminal fragment that accumulates in the cell nucleus. This fragment interacts with the N-terminal portion of HIF-1α spanning amino acid residues 1–390 but not with HIF-2α. In hypoxia this fragment facilitates the nuclear localization of HIF-1α, is recruited to HIF-1α target gene promoters, and enhances HIF-1α function, resulting in up-regulation of HIF-1α target gene expression in a hypoxia-dependent fashion. These results unravel an important mechanism that selectively regulates the nuclear accumulation and function of HIF-1α and potentiates angiogenesis and tumor progression.
Adaptation of organisms to low oxygen levels (hypoxia) is a fundamental biological process that is required in both physiological and pathophysiological situations. In response to hypoxia the hypoxia-inducible factors (HIFs) activate transcription of a large group of genes encoding proteins involved in angiogenesis, erythropoiesis, and glucose metabolism (1). The activity of HIF-1α or HIF-2α is mainly regulated at the level of protein stability (2, 3). The intracellular distribution of HIF-1α has also been shown to be regulated by hypoxia. At normoxia the protein can be detected in both the nuclear and cytoplasmic compartments, whereas at hypoxia HIF-1α shows an exclusively nuclear localization (4, 5).
Filamins are large actin-binding proteins that are widely expressed and stabilize 3D actin webs and link them to cellular membranes (6). Filamin A (FLNA; also named actin-binding protein-280) is the most abundant isoform and widely expressed variant in human tissues. The scaffolding of FLNA with cell surface receptors provides mechanical stability, maintains cell–cell and cell–matrix connections, and transmits stress signals to the actin skeleton during cellular locomotion. In addition, FLNA interacts with a wide range of proteins involved in signal transduction (6, 7). FLNA is a homodimer of 280 kDa containing an N-terminal actin-binding domain and 24 rod-domain repeats, interrupted by two short hinge (H1 and H2) segments. Calpain proteases have been shown to cleave the H1 and H2 sites in vitro, producing a C-terminal 90-kDa fragment (8). Several studies suggest that FLNA, generally regarded as a cytoplasmic architectural molecule, may act as a nuclear transcriptional modulator through this endogenous proteolytic fragment, which is accumulated in the cell nucleus (9, 10).
Here we report that tumors derived from melanoma cells lacking FLNA have impaired growth, decreased angiogenesis, and diminished expression of HIF-target gene expression compared with tumors expressing FLNA, leading to the discovery that FLNA interacts with HIF-1α but not HIF-2α. In fact, hypoxia induces cleavage of FLNA, generating a fragment that enhances accumulation of HIF-1α in the cell nucleus and is recruited to hypoxia-responsive element (HRE)-containing promoters. Thus, this study reveals a hitherto unknown mechanism of regulation of HIF-1α signaling in hypoxic cells that contributes to tumor growth and angiogenesis.
Results
Lack of FLNA Expression Reduces Tumor Size and Tumor Angiogenesis.
Analysis of FLNA expression in cancer cohorts shows that FLNA mRNA is deregulated in many forms of cancer (Tables S1 and S2) (www.oncomine.org) (11). Here we have investigated the relevance of FLNA for solid tumor growth. We injected melanoma M2 cells lacking FLNA expression or A7 cells stably expressing FLNA into SCID mice. As shown in Fig. 1A, tumor xenografts expressing FLNA (A7) showed a larger size than M2 cell-derived tumors. Tumors expressing FLNA grew faster than M2 tumors (Fig. 1B). Immunohistochemical analysis of the tumor vasculature showed that FLNA-expressing tumor xenografts had higher vascular density than tumors lacking FLNA (Fig. 1 C and D). Furthermore, the vessels in A7 cell-derived tumors were distorted and disorganized, with an increased vessel area and vessel diameter compared with M2 tumors. Tumor growth and angiogenesis correlated with FLNA protein levels in the tumors (Fig. 1E). These results show that FLNA expression promotes tumor growth and angiogenesis.
Fig. 1.

FLNA-expressing tumor xenografts grow faster and show increased angiogenesis and HIF-1α activity. (A) Gross view of tumor xenografts 31 d after injection of M2 or A7 cells into SCID mice. (B) FLNA-expressing tumor xenografts (A7) grow faster than tumor xenografts that do not express FLNA (M2). (C and D) FLNA-expressing tumor xenografts have increased angiogenesis (n = 6). (C) Tumor vasculature was studied with a whole-mount method using anti-CD31 and anti-NG2 antibodies to label the endothelial cells (red) and pericytes (green). (D) Tumor vessel area, density, and vessel diameter were evaluated (n = 6). (E) Expression of FLNA and β-actin in xenograft tumor samples was detected by immunoblotting. (F) FLNA-expressing tumor xenografts (A7) express higher levels of HIF-target genes (n = 3). Data are shown as mean ± SD. *P < 0.05 compared with corresponding samples of M2-derived tumor xenografts.
Tumor angiogenesis is regulated by the HIF pathway through up-regulation of several proangiogenic factors, including vascular endothelial growth factor-A (VEGF-A) (1). We therefore investigated HIF signaling in tumors derived from M2 or A7 cells. As shown in Fig. 1F, we detected higher levels of HIF target genes in FLNA-expressing tumors, indicating that HIF signaling is enhanced in tumors expressing FLNA.
FLNA Enhances HIF-Dependent Transcriptional Activity.
In functional assays FLNA significantly enhanced hypoxia-inducible reporter gene activity in A7 cells compared with M2 cells (Fig. 2A), indicating that FLNA increases the transactivation function of endogenous HIF proteins. We next investigated the impact of FLNA on the expression of endogenous HIF target genes (Fig. 2B). We found that HIF target genes showed higher mRNA levels in hypoxia in A7 cells than in M2 cells. Although to a lesser extent, the presence of FLNA also correlated with enhanced mRNA expression of some HIF target genes in normoxia. Compared with M2 cells, A7 cells secreted significantly more VEGF-A protein in both normoxia and hypoxia and showed a significant hypoxia-dependent induction response (Fig. 2C). Taken together these observations indicate that FLNA enhances the transactivation function of HIF proteins.
Fig. 2.

FLNA increases the HIF-α transactivation function. M2 and A7 cells were cultured under normoxic (N) or hypoxic (H) conditions. (A and B) HRE-driven luciferase reporter activity (A) and expression of HIF target genes (B) are higher in A7 cells compared with M2 cells. (C) A7 cells cultured in normoxia or hypoxia secrete more VEGF-A than M2 cells. (D and E) Silencing of FLNA reduces HIF target gene expression (D) and VEGF-A secretion (E) in A7 cells in both normoxia and hypoxia. A7 cells were transfected with a negative control siRNA (CTR) or a siRNA targeting FLNA (FLNA). Data are shown as mean ± SD of at least three independent experiments. *P < 0.05 compared with corresponding samples in M2 cells; #P < 0.05 compared with corresponding samples transfected with control siRNA.
Down-Regulation of FLNA Expression Inhibits the HIF Transactivation Function.
To further study the impact of FLNA on the HIF signaling pathway, we transfected into A7 cells specific siRNAs targeting FLNA. The expression of FLNA mRNA was reduced by siRNA transfection to approximately 60% (Fig. S1). Silencing FLNA resulted in significant reduction of VEGF-A and GLUT3 mRNA expression and VEGF-A secretion in both normoxia and hypoxia (Fig. 2 D and E). We also analyzed the expression of previously described HIF-2α–specific target genes, such as Cyclin D2, p27, E2F1, A2AR, and CITED2 (12–14). Among these genes only CITED2 showed hypoxia-dependent induction in M2 and A7 cells (Fig. S2A). However, silencing FLNA did not impair CITED2 expression in A7 and HEK 293T cells (Fig. S2 B and C). These observations argue against a role for FLNA in the regulation of HIF-2α specific target genes.
FLNA Physically Interacts with HIF-1α but Not HIF-2α.
On the basis of the observed regulation of HIF target gene expression by FLNA, we asked whether HIF-1α was able to interact with FLNA. To test this in an unbiased manner we performed yeast two-hybrid assays. We screened a yeast two-hybrid library prepared from human T-cell cDNA. Sequence analysis of the positive cDNA clones revealed four clones encoding FLNA starting at rod-domain repeat 20 or 22 (Fig. S3A). The interaction between HIF-1α and the C terminus of FLNA was confirmed in mammalian cells where NIH 3T3 cells were transfected with plasmids encoding the HA-tagged C terminus of FLNA spanning repeats 20–24 (HA-FLNA20-24) and FLAG-tagged HIF-1α (Fig. 3A). Interaction between endogenous proteins was also investigated using nuclear and cytosolic extracts from COS1 cells. Our results show that full-length FLNA interacts with HIF-1α in the cytoplasmic compartment, with no binding observed between nuclear proteins (Fig. 3B).
Fig. 3.

FLNA interacts with HIF-1α but not with HIF-2α. (A) FLAG-fused HIF-1α interacts with HA-tagged FLNA repeats 20–24 (HA-FLNA20-24) in NIH 3T3 cells. (B) Endogenous FLNA and HIF-1α interact in the cytoplasmic compartment. Nuclear (Nuc) and cytosolic (Cyt) extracts were prepared from COS1 cells cultured in normoxia (N) or hypoxia (H). (C) Identification of HIF-1α domains interacting with FLNA. HEK 293A cells were cotransfected with plasmids encoding HA-FLNa20-24 and FLAG-mHIF-1α (1-390), (392-622), (531-822), or pCMX as control. (D) HA-FLNA20-24 interacts with HIF-1α but not with HIF-2α. HEK 293A cells were cotransfected with plasmids encoding HA-FLNA20-24 and FLAG-mHIF-1α, FLAG-mHIF-2α, or pCMX as control. *Unspecific signal.
To identify domain(s) in HIF-1α responsible for the observed interaction, we cotransfected HEK 293A cells with a set of truncated HIF-1α constructs (Fig. S3B) together with plasmids encoding FLNA20-24. The immunoprecipitation results revealed that FLNA interacts with the N-terminal portion of HIF-1α spanning amino acid residues 1–390 (Fig. 3C).
We next examined whether FLNA also interacts with HIF-2α. FLAG-tagged HIF-1α or HIF-2α was coexpressed with HA-FLNA20-24 in HEK 293A cells, and whole-cell extracts were used in the immunoprecipitation analysis. As shown in Fig. 3D, only HIF-1α was able to interact with FLNA20-24, whereas no binding was observed for HIF-2α, indicating that interaction of FLNA with members of the HIF family is specific for HIF-1α.
Hypoxia Enhances Calpain-Dependent Proteolytic Cleavage of FLNA.
Filamins are proteins highly susceptible to proteolysis. FLNA can be cleaved by calpain proteases at the H1 and H2 regions, generating 190-, 90-, and 10-kDa fragments (8). It has also been reported that FLNA can be cleaved by caspase and granzyme B during the process of apoptosis, generating distinct fragments from the ones resulting from calpain-dependent cleavage (15). In whole-cell extracts of A7 cells, in addition to a 280-kDa protein corresponding to full-length FLNA, we detected a ∼200-kDa fragment (Fig. 4A, Left) using an antibody against the N terminus of FLNA, and a ∼90-kDa fragment (Fig. 4A, Right) using an antibody against the C terminus of FLNA. These results suggest that FLNA is cleaved in the H1 and H2 domains by calpain proteases in A7 cells, generating a C-terminal fragment of FLNA (FLNACT). Interestingly, the cleavage was significantly enhanced by hypoxia treatment, resulting in increased generation of FLNACT (Fig. 4B, lane 1 and 2). Moreover, the cleavage could be inhibited in a dose-dependent manner by a calpain protease inhibitor, calpeptin (Fig. 4B, lanes 3 and 4). In contrast to calpeptin, a caspase-3/7 inhibitor, Ac-DEVD-CHO, had no effect on the cleavage (Fig. 4B, lane 5), indicating that the hypoxia-induced cleavage of FLNA is calpain-mediated and caspase-3/7-independent.
Fig. 4.

Hypoxia-inducible and calpain-dependent cleavage of FLNA increases HIF-1α nuclear localization. (A) The expression of FLNA in whole-cell extracts from M2 and A7 cells detected using an antibody against the N terminus of FLNA (Left) or C terminus of FLNA (Right). (B) Hypoxia enhances calpain-dependent and caspase-independent cleavage of FLNA. A7 cells were cultured in normoxia (N) or hypoxia (H) and treated with either vehicle, a calpain inhibitor calpeptin (+, 25 μM; ++, 50 μM), or a caspase-3/7 inhibitor, Ac-DEVD-CHO (10 μM), for 8 h. (C) GLUT1 mRNA expression is significantly reduced by inhibition of FLNA cleavage in A7 cells. Relative GLUT1 mRNA level in M2 and A7 cells treated as in B were analyzed. Data are shown as mean ± SD of at least three independent experiments. *P < 0.05. (D) FLNA increases the nuclear localization of endogenous HIF-1α. Nuclear (Nuc) and cytosolic (Cyt) extracts were prepared from M2 and A7 cells exposed to either normoxia (N) or hypoxia (H) for 8 h. (E) Inhibition of FLNA cleavage by calpeptin impairs nuclear accumulation of GFP-HIF-1α. M2 and A7 cells were cultured at normoxia or hypoxia (H) and treated with vehicle (Control), calpeptin (25 μM), or Ac-DEVD-CHO (10 μM) for 8 h. Quantifications are presented as described in SI Materials and Methods.
We also investigated the impact of hypoxia on calpain-mediated cleavage of FLNA in COS-1 cells. In agreement with the results obtained in A7 cells, cleavage of FLNA in COS-1 cells was up-regulated by hypoxia, and calpeptin treatment prevented the cleavage (Fig. S4A). Cell fractionation assays using COS-1 extracts also showed that FLNACT localized more in nuclear than in cytoplasmic compartment and the levels of nuclear FLNACT were increased by hypoxia (Fig. S4B).
To investigate whether hypoxia-induced cleavage of FLNA by calpain proteases has an impact on HIF-1α function, we treated M2 and A7 cells with either calpain or caspase-3/7 inhibitor. We found that the calpain inhibitor calpeptin significantly reduced expression of the HIF-1α target gene glucose transporter 1 (GLUT1) in A7 cells but had no effect in M2 cells. In contrast, inhibition of caspase3/7 by Ac-DEVD-CHO had no effect in A7 cells (Fig. 4C). These results suggest that calpain-dependent cleavage of FLNA enhanced the cellular response to hypoxia.
Cleavage of FLNA by Calpain Enhances Nuclear Accumulation of HIF-1α.
The HIF-1α signaling pathway is tightly regulated by hypoxia through several complex mechanisms involving protein degradation and control of subcellular localization. We found that HIF-1α degradation was not affected by the presence of FLNA (Fig. S5). We next investigated whether FLNA affects the intracellular distribution of HIF-1α. In hypoxia, we observed higher HIF-1α protein levels in the cytoplasm of M2 cells compared with A7 cells, whereas A7 cells showed slightly higher levels of HIF-1α protein in the nucleus (Fig. 4D). The quality of subcellular fractionation was controlled by investigating levels of YY1, a nuclear marker protein, and paxillin, a cytosolic marker protein. These results suggest that FLNA facilitates the nuclear accumulation of HIF-1α in hypoxia.
We also analyzed the subcellular localization of GFP-HIF-1α. Approximately 75–79% of M2 cells showed a predominantly nuclear distribution (N>C) of GFP-HIF-1α in both normoxia and hypoxia (Fig. 4E). In contrast to M2 cells but in agreement with earlier observations using other cell lines (4, 16), we observed an increase in the number of A7 cells with an exclusively nuclear (N) distribution of GFP-HIF-1α after hypoxia treatment (from 15% to 71%). These observations indicate that FLNA facilitated the nuclear accumulation of GFP-HIF-1α in hypoxia.
We next investigated whether FLNA cleavage affected the nuclear accumulation of HIF-1α in hypoxia. As shown in Fig. 4E, inhibition of calpain-dependent cleavage by calpeptin decreased the nuclear accumulation of GFP-HIF-1α in A7 cells (from 71% to 45%) but had no significant effect in M2 cells. Inhibition of caspase 3/7 by Ac-DEVD-CHO had no effect on the subcellular distribution of GFP-HIF-1α in either M2 or A7 cells.
Treatment of COS-1 cells with calpeptin under hypoxic conditions decreased the percentage of cells exhibiting exclusively nuclear (N) distribution of GFP-HIF-1α (Fig. S4C). Concomitantly, calpeptin decreased HRE-driven reporter gene activity in a dose-dependent manner in COS1 cells (Fig. S4D). Taken together, these results suggest that calpain-dependent cleavage of FLNA enhances the nuclear accumulation of HIF-1α, resulting in an increased transactivation function of HIF-1α at hypoxia.
FLNA Has No Impact on HIF-1α Function in Cells Lacking Cleavage of FLNA by Calpain Proteases.
To assess the relevance of calpain-mediated cleavage of FLNA, we investigated the presence of FLNACT in distinct cell lines, including the human osteosarcoma cell line U2OS and HEK 293T and HeLa cells (Fig. 5A and Fig. S6). In both U2OS and 293T cells, FLNACT was detected and up-regulated in response to hypoxia. In contrast, FLNA cleavage was not detected in HeLa cells in either normoxia or hypoxia. These results indicate that cleavage of FLNA is cell type-dependent and can be enhanced by hypoxia.
Fig. 5.

Cleavage of FLNA regulates HIF-1α function. U2OS, HEK 293T, and HeLa cells were transfected with a negative control siRNA (CTR) or an siRNA targeting FLNA (FLNA). The cells were cultured under normoxic (N) or hypoxic (H) conditions for 8 h. (A) Calpain-dependent cleavage of FLNA is impaired in HeLa cells. (B) Silencing FLNA affects HIF-1α transactivation activity in U2OS and HEK 293T cells but not in HeLa cells. Relative mRNA levels of VEGF-A and BNIP3 were analyzed using quantitative RT-PCR. Data are shown as mean ± SD of at least three independent experiments. *P < 0.05.
Interestingly, whereas in U2OS and HEK 293T cells silencing FLNA significantly decreased hypoxia-induced HIF target gene expression, in HeLa cells inhibition of FLNA expression had no effect on hypoxia-mediated up-regulation of VEGF-A and BNIP3 expression (Fig. 5B). These results show that the effect of FLNA on the HIF-1α transactivation function correlates with calpain-dependent cleavage of FLNA.
The C-Terminal Fragment of FLNA Plays an Important Role in Regulating Nuclear Accumulation and the Transactivation Function of HIF-1α.
To further investigate how the hypoxia-regulated cleavage of FLNA affects HIF-1α signaling, we generated FLAG-tagged constructs of wild-type FLNA (FLNA), a cleavage-resistant FLNA mutant in which amino acids 1741–1777 of FLNA containing the H1 domain have been deleted (ΔH1), and a truncated C-terminal FLNA fragment containing repeats 16–24 (FLNA16-24) (Fig. 6A). We further generated cell lines stably expressing FLAG-tagged FLNA, ΔH1, or FLNA16-24, and cells transfected with vector only were used as control. As shown in Fig. 6B, the stable cell lines expressed similar levels of wild-type FLNA, ΔH1, or FLNA16-24, respectively.
Fig. 6.

The C-terminal fragment of FLNA enhances nuclear accumulation and the transactivation function of HIF-1α. (A) Schematic representation of wild-type FLNA (FLNA), the cleavage-resistant FLNA mutant where H1 domain is deleted (ΔH1), and the truncated C-terminal FLNA containing repeats 16–24 (FLNA16-24). (B) Expression of wild-type FLNA and mutants in cell lines stably expressing FLNA, ΔH1, and FLNA16-24. Control cells were transfected with empty vector. (C) Nuclear localization of GFP-HIF-1α in hypoxia is increased in FLNA- and FLNA16-24-expressing cells. Quantifications are presented as described in SI Materials and Methods. (D and E) Transactivation activity of HIF-1α is up-regulated in FLNA- and FLNA16-24-expressing cells. HRE-driven luciferase reporter assay (D) and GLUT1 expression analyzed by quantitative RT-PCR analysis (E) were performed in the stable cell lines cultured in either normoxia (N) or hypoxia (H). Data are shown as mean ± SD of at least three independent experiments. *P < 0.05 compared with corresponding samples of control cells.
We next examined the subcellular distribution of GFP-HIF-1α in these cell lines (Fig. 6C). We found that in the majority of control cells and in cells stably expressing ΔH1, GFP-HIF-1α showed a predominantly nuclear distribution (N>C) in both normoxia and hypoxia (70–86%). However, in cells stably expressing FLNA, hypoxia significantly increased the exclusively nuclear distribution (N) of GFP-HIF-1α (from 31% to 74%). Similar results were also obtained in FLNA16-24-expressing cells (from 25% to 68%). These observations clearly indicate that FLNA16-24 is able to recapitulate the effect of full-length FLNA on the intracellular distribution of HIF-1α by increasing the nuclear accumulation of the protein in hypoxia. Furthermore, preventing the generation of the C-terminal fragment of FLNA by deletion of the H1 domain completely abrogated FLNA-mediated induction of nuclear accumulation of HIF-1α.
The transactivation function of endogenous HIF-1α was assessed using hypoxia-inducible reporter gene assays in the stable cell lines (Fig. 6D). Reporter gene activity at hypoxia was approximately fivefold higher in FLNA and FLNA16-24-expressing cells than in control cells. However, there was no significant difference between cells expressing ΔH1 and control cells. Moreover, FLNA and FLNA16-24, but not ΔH1-expressing cells, had more GLUT1 mRNA expression in hypoxia than control cells (Fig. 6E). Taken together, these results suggest that the cleavage of FLNA at the H1 domain by calpain proteases and the generation of the C-terminal fragment of FLNA are essential for the effect of full-length FLNA on the HIF-1α transactivation function in hypoxia.
FLNA Is Recruited to the Promoters of HIF Target Genes in Hypoxia.
Because our results show that the C-terminal fragment of FLNA increases nuclear accumulation of HIF-1α and its transactivation function, we further investigated whether FLNA is recruited to the promoters of HIF target genes in ChIP experiments. Our results demonstrate that in M2 cells, HIF-1α was present within the HRE-containing region of the VEGF-A promoter at hypoxia, whereas no FLNA binding was detected (Fig. 7A). In contrast to M2 cells, in A7 cells both HIF-1α and FLNA were detected in a hypoxia-dependent manner within the same region of the VEGF-A promoter. In addition, recruitment of HIF-1α to the VEGF-A promoter at normoxia was significantly enhanced in A7 cells, compared with M2 cells. Thus, enhanced recruitment of HIF-1α at normoxia in A7 cells correlates positively with the increase in target gene expression observed in Fig. 2, suggesting that FLNA may also have a role on the regulation of basal HIF-1α activity occurring at normoxia.
Fig. 7.
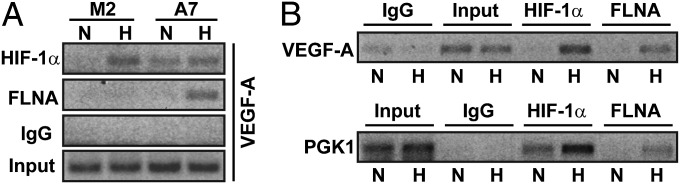
FLNA is recruited to promoters of HIF-1α target genes at hypoxia. M2, A7 cells (A), and COS-1 cells (B) were cultured at normoxia (N) or hypoxia (H) for 8 h. Soluble chromatin was immunoprecipitated using antibodies against HIF-1α or the C terminus of FLNA, or control IgG, and analyzed by PCR using primers spanning HRE-containing regions of VEGF-A or PGK1 promoters. (A) Both HIF-1α and FLNA are recruited to the HRE-containing regions of the VEGF-A promoter in FLNA-expressing A7 cells under hypoxic treatment. (B) HIF-1α and FLNA are detected on VEGF-A and PGK1 promoters in COS-1 cells cultured under hypoxic conditions.
In COS-1 cells that express endogenous FLNA, both HIF-1α and FLNA were present on HRE-containing regions of VEGF-A and PGK1 promoters under hypoxic conditions (Fig. 7B). Taken together, these results suggest that FLNA is recruited to the HIF-1α transcriptional complex on HRE-containing promoters, correlating with an increase in HIF-1α–dependent transcriptional activity.
Discussion
In the present study we have identified FLNA as a previously unidentified regulator of the HIF signaling pathway. We show that FLNA contributes to tumor growth and angiogenesis. This correlates with FLNA-dependent regulation of HIF target genes. A hypoxia-inducible nuclear cleavage product of FLNA binds to the N terminus of HIF-1α but not HIF-2α, increases nuclear localization of HIF-1α, and it is recruited to HRE-containing promoters and enhances the HIF-1α transactivation function. In conclusion, these results indicate that FLNA is important for efficient induction of HIF target genes that regulate tumor growth.
Similar to HIF-1α in this study, more than 100 binding partners interact with FLNA (10). Although the majority of FLNA-interacting partners are membrane-associated or cytoplasmic proteins, several studies have provided evidence that FLNA interacts with nuclear proteins and participates in their signaling pathways. These proteins include transcription factors such as the androgen receptor, Smads, and FOXC1 (9, 17–19) and proteins involved in DNA repair [e.g., BRCA2 (20)]. In our study we show that FLNA interacts with HIF-1α and regulates the localization and transactivation of this transcription factor.
Previous studies have shown that the C-terminal fragment of FLNA is required for the nuclear translocation of the androgen receptor (17), and the nuclear localization of the FLNACT fragment restores the responsiveness of prostate cancer cells to androgens (21). In addition, wingless-related MMTV integration site 5A (Wnt5A)-driven cell migration has been shown to be dependent on FLNA cleavage (22). Our results show that the exclusively nuclear localization of HIF-1α and regulation of HIF-1α target gene expression is dependent on the generation of the FLNACT fragment. Furthermore, expression of FLNACT in cells lacking FLNA reconstituted the effect of FLNA on the HIF-1α signaling pathway. These observations unravel an unexpected role of the FLNACT as a previously unidentified regulator of HIF-1α function. Moreover, this mode of regulation is specific for HIF-1α and does not affect HIF-2α function.
We identified the N-terminal portion of HIF-1α as the interface interacting with FLNA. This region shows the highest level of homology between HIF-1α and HIF-2α (85% and 70% amino acid sequence identity in bHLH and PAS domains, respectively). However, these domains are known to show a high degree of plasticity, possibly explaining the molecular basis for partner protein specificity. Besides FLNA other proteins, including SEPT9_v1 and Sp1, have been shown to specifically interact with the N-terminal region of HIF-1α (23, 24), suggesting that specific regulation of HIF-α proteins with distinct partners is a common theme in the hypoxia-signaling pathway.
Treatment of cells with Wnt5A or IGFBP5 has been shown to induce FLNA cleavage by regulating calpain activity or FLNA phosphorylation, respectively (22, 25). Our study indicates hypoxia as another signal inducing cleavage of FLNA. Hypoxia is known to increase cytosolic calcium (26) and has been shown to up-regulate calpain protease activity in several cell types, such as pulmonary arterial endothelial cells (27). The ability of calpain to cleave FLNA is also regulated by phosphorylation (10–25). Several kinases, including PKA, Pak1, and RSK, as well as phosphatases such as calcineurin, have been proposed to regulate FLNA phosphorylation (10). In addition to up-regulation of calpain activity, hypoxia may have an impact on the phosphorylation status of FLNA, thus underlying cell type-specific differences in generating the cleavage product in hypoxia. Further studies are necessary to address this question.
Calpain activity has been shown to degrade HIF-2α in response to intermittent hypoxia, a mechanism that has no impact on HIF-1α protein levels (28). Here we show that HIF-1α–dependent signaling is up-regulated as a consequence of increased calpain activity, indicating that calpain can impact on HIF-α proteins by completely distinct mechanisms leading to opposite outcomes.
We show that FLNA expression leads to up-regulation of VEGF-A expression, secretion, and consequent induction of angiogenesis. Our results give a rationale to previous observations in lung cancer and peripheral cholangiocarcinoma whereby FLNA expression correlates with VEGF-A expression (29, 30).
The role of FLNA in tumor invasiveness and metastases suggested by in vitro cell motility studies is currently under scrutiny (10, 22, 31–33). However, other roles for FLNA in cancer have been proposed. In prostate cancer cells it has been shown that nuclear FLNACT promotes sensitization of cells to antiandrogen therapy (21), a mechanism that may explain why down-regulation of FLNA observed in certain cancer cohorts could confer an advantage to prostate cancer development (Table S2). In a K-Ras–induced lung adenocarcinoma mouse model we have shown that FLNA expression contributes to tumor growth (34). In addition, the reported role of FLNA in BRCA2-mediated DNA repair suggests that FLNA may modulate cancer genomic instability (35). Here we show that FLNA contributes to tumor growth by increasing HIF-1α activity. It is possible that FLNA has an impact on tumor growth in cancers in which calpain-dependent cleavage of FLNA occurs. Analysis of FLNA expression in cancer cohorts shows that FLNA mRNA is up-regulated in several types of cancer (Table S1). Protein expression levels and the cleavage state of FLNA remain to be investigated to assess the role of FLNA in these types of cancer. Other roles of FLNA, such as the above-mentioned mechanism in prostate cancer (21), could explain why FLNA is down-regulated in some types of cancer (Table S2).
Our unexpected discovery of FLNA as a regulator of HIF-1α function supports the concept that the actin-cross-linking protein FLNA also exists as a nuclear protein (36). However, our results show that the nuclear localization of FLNA does not always suppress tumor growth, as has been recently proposed (37). On the basis of our observations, we propose a model in which FLNA mediates a previously unrecognized mechanism of regulation of HIF-1α function and thus controls adaptive responses to hypoxia (Fig. S7). HIF-1α interacts with full-length FLNA in the cytoplasm and under hypoxic conditions, calpain protease activity is up-regulated, resulting in increased cleavage of the C-terminal fragment of FLNA that translocates to the nucleus together with HIF-1α. This, in turn, facilitates the nuclear accumulation of HIF-1α and corecruitment to HRE-containing target genes. The functional consequence of this interaction is an enhanced transactivation function of HIF-1α that facilitates tumor growth by regulating tumor cell metabolism and inducing tumor angiogenesis. This HIF-1α–specific mechanism offers opportunities in targeting the hypoxia-signaling pathway therapeutically.
Materials and Methods
Tumor growth and angiogenesis were studied using tumor xenograft model in SCID mice. Gene expression was analyzed by quantitative RT-PCR. Detailed methods for tumor xenograft model, cell culture, plasmid construction and transfection, RNA interference, protein and RNA extraction, immunoprecipitation, immunoblotting, ChIP, VEGF-A secretion, and visualization of fluorescent proteins are given in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Y. Ohta for the gift of M2 cell line, K. Xie for pGL3-V2274, and K. Watanabe for pHA-FLNA20-24. This work was partly supported by grants from the Swedish Heart-Lung Foundation (to L.M.A. and L.P.), Swedish Research Council (to L.P., J.B., and T.P.), Swedish Cancer Society (to L.M.A., L.P., and T.P.), Sahlgrenska Hospital Funds (to L.M.A.), Göran Gustafsson’s Foundation (J.B.), Torsten and Ragnar Söderberg’s Foundation (to J.B. and L.P.), and The Board of Research at Karolinska Institutet (to X.Z.), and funded by a core grant by the Singapore Ministry of Education to the Cancer Science Institute of Singapore (to L.P.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1320815111/-/DCSupplemental.
References
- 1.Semenza GL. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci. 2012;33(4):207–214. doi: 10.1016/j.tips.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cockman ME, et al. Hypoxia inducible factor-alpha binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J Biol Chem. 2000;275(33):25733–25741. doi: 10.1074/jbc.M002740200. [DOI] [PubMed] [Google Scholar]
- 3.Tanimoto K, Makino Y, Pereira T, Poellinger L. Mechanism of regulation of the hypoxia-inducible factor-1 alpha by the von Hippel-Lindau tumor suppressor protein. EMBO J. 2000;19(16):4298–4309. doi: 10.1093/emboj/19.16.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kallio PJ, et al. Signal transduction in hypoxic cells: Inducible nuclear translocation and recruitment of the CBP/p300 coactivator by the hypoxia-inducible factor-1alpha. EMBO J. 1998;17(22):6573–6586. doi: 10.1093/emboj/17.22.6573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Depping R, et al. Nuclear translocation of hypoxia-inducible factors (HIFs): Involvement of the classical importin alpha/beta pathway. Biochim Biophys Acta. 2008;1783(3):394–404. doi: 10.1016/j.bbamcr.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 6.Stossel TP, et al. Filamins as integrators of cell mechanics and signalling. Nat Rev Mol Cell Biol. 2001;2(2):138–145. doi: 10.1038/35052082. [DOI] [PubMed] [Google Scholar]
- 7.Zhou AX, Hartwig JH, Akyürek LM. Filamins in cell signaling, transcription and organ development. Trends Cell Biol. 2010;20(2):113–123. doi: 10.1016/j.tcb.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 8.Gorlin JB, et al. Human endothelial actin-binding protein (ABP-280, nonmuscle filamin): A molecular leaf spring. J Cell Biol. 1990;111(3):1089–1105. doi: 10.1083/jcb.111.3.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loy CJ, Sim KS, Yong EL. Filamin-A fragment localizes to the nucleus to regulate androgen receptor and coactivator functions. Proc Natl Acad Sci USA. 2003;100(8):4562–4567. doi: 10.1073/pnas.0736237100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakamura F, Stossel TP, Hartwig JH. The filamins: Organizers of cell structure and function. Cell Adhes Migr. 2011;5(2):160–169. doi: 10.4161/cam.5.2.14401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rhodes DR, et al. Oncomine 3.0: Genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007;9(2):166–180. doi: 10.1593/neo.07112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahmad A, et al. Adenosine A2A receptor is a unique angiogenic target of HIF-2alpha in pulmonary endothelial cells. Proc Natl Acad Sci USA. 2009;106(26):10684–10689. doi: 10.1073/pnas.0901326106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aprelikova O, Wood M, Tackett S, Chandramouli GV, Barrett JC. Role of ETS transcription factors in the hypoxia-inducible factor-2 target gene selection. Cancer Res. 2006;66(11):5641–5647. doi: 10.1158/0008-5472.CAN-05-3345. [DOI] [PubMed] [Google Scholar]
- 14.Gordan JD, Bertout JA, Hu CJ, Diehl JA, Simon MC. HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell. 2007;11(4):335–347. doi: 10.1016/j.ccr.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Browne KA, Johnstone RW, Jans DA, Trapani JA. Filamin (280-kDa actin-binding protein) is a caspase substrate and is also cleaved directly by the cytotoxic T lymphocyte protease granzyme B during apoptosis. J Biol Chem. 2000;275(50):39262–39266. doi: 10.1074/jbc.C000622200. [DOI] [PubMed] [Google Scholar]
- 16.Ruas JL, Poellinger L, Pereira T. Functional analysis of hypoxia-inducible factor-1 alpha-mediated transactivation. Identification of amino acid residues critical for transcriptional activation and/or interaction with CREB-binding protein. J Biol Chem. 2002;277(41):38723–38730. doi: 10.1074/jbc.M205051200. [DOI] [PubMed] [Google Scholar]
- 17.Ozanne DM, et al. Androgen receptor nuclear translocation is facilitated by the f-actin cross-linking protein filamin. Mol Endocrinol. 2000;14(10):1618–1626. doi: 10.1210/mend.14.10.0541. [DOI] [PubMed] [Google Scholar]
- 18.Sasaki A, Masuda Y, Ohta Y, Ikeda K, Watanabe K. Filamin associates with Smads and regulates transforming growth factor-beta signaling. J Biol Chem. 2001;276(21):17871–17877. doi: 10.1074/jbc.M008422200. [DOI] [PubMed] [Google Scholar]
- 19.Berry FB, O’Neill MA, Coca-Prados M, Walter MA. FOXC1 transcriptional regulatory activity is impaired by PBX1 in a filamin A-mediated manner. Mol Cell Biol. 2005;25(4):1415–1424. doi: 10.1128/MCB.25.4.1415-1424.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yuan Y, Shen Z. Interaction with BRCA2 suggests a role for filamin-1 (hsFLNa) in DNA damage response. J Biol Chem. 2001;276(51):48318–48324. doi: 10.1074/jbc.M102557200. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, et al. A 90 kDa fragment of filamin A promotes Casodex-induced growth inhibition in Casodex-resistant androgen receptor positive C4-2 prostate cancer cells. Oncogene. 2007;26(41):6061–6070. doi: 10.1038/sj.onc.1210435. [DOI] [PubMed] [Google Scholar]
- 22.O’Connell MP, et al. Wnt5A activates the calpain-mediated cleavage of filamin A. J Invest Dermatol. 2009;129(7):1782–1789. doi: 10.1038/jid.2008.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amir S, Wang R, Simons JW, Mabjeesh NJ. SEPT9_v1 up-regulates hypoxia-inducible factor 1 by preventing its RACK1-mediated degradation. J Biol Chem. 2009;284(17):11142–11151. doi: 10.1074/jbc.M808348200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.To KK, Sedelnikova OA, Samons M, Bonner WM, Huang LE. The phosphorylation status of PAS-B distinguishes HIF-1alpha from HIF-2alpha in NBS1 repression. EMBO J. 2006;25(20):4784–4794. doi: 10.1038/sj.emboj.7601369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abrass CK, Hansen KM. Insulin-like growth factor-binding protein-5-induced laminin gamma1 transcription requires filamin A. J Biol Chem. 2010;285(17):12925–12934. doi: 10.1074/jbc.M109.061754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Toescu EC. Hypoxia sensing and pathways of cytosolic Ca2+ increases. Cell Calcium. 2004;36(3-4):187–199. doi: 10.1016/j.ceca.2004.02.019. [DOI] [PubMed] [Google Scholar]
- 27.Zhang J, Patel JM, Block ER. Hypoxia-specific upregulation of calpain activity and gene expression in pulmonary artery endothelial cells. Am J Physiol. 1998;275(3 Pt 1):L461–L468. doi: 10.1152/ajplung.1998.275.3.L461. [DOI] [PubMed] [Google Scholar]
- 28.Nanduri J, et al. Intermittent hypoxia degrades HIF-2alpha via calpains resulting in oxidative stress: implications for recurrent apnea-induced morbidities. Proc Natl Acad Sci USA. 2009;106(4):1199–1204. doi: 10.1073/pnas.0811018106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Uramoto H, Akyürek LM, Hanagiri T. A positive relationship between filamin and VEGF in patients with lung cancer. Anticancer Res. 2010;30(10):3939–3944. [PubMed] [Google Scholar]
- 30.Guedj N, et al. Comparative protein expression profiles of hilar and peripheral hepatic cholangiocarcinomas. J Hepatol. 2009;51(1):93–101. doi: 10.1016/j.jhep.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 31.Kim H, McCulloch CA. Filamin A mediates interactions between cytoskeletal proteins that control cell adhesion. FEBS Lett. 2011;585(1):18–22. doi: 10.1016/j.febslet.2010.11.033. [DOI] [PubMed] [Google Scholar]
- 32.Zhou AX, et al. Filamin a mediates HGF/c-MET signaling in tumor cell migration. Int J Cancer. 2011;128(4):839–846. doi: 10.1002/ijc.25417. [DOI] [PubMed] [Google Scholar]
- 33.Xu Y, et al. Filamin A regulates focal adhesion disassembly and suppresses breast cancer cell migration and invasion. J Exp Med. 2010;207(11):2421–2437. doi: 10.1084/jem.20100433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nallapalli RK, et al. Targeting filamin A reduces K-RAS-induced lung adenocarcinomas and endothelial response to tumor growth in mice. Mol Cancer. 2012;11:50. doi: 10.1186/1476-4598-11-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yue J, et al. The cytoskeleton protein filamin-A is required for an efficient recombinational DNA double strand break repair. Cancer Res. 2009;69(20):7978–7985. doi: 10.1158/0008-5472.CAN-09-2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uribe R, Jay D. A review of actin binding proteins: new perspectives. Mol Biol Rep. 2009;36(1):121–125. doi: 10.1007/s11033-007-9159-2. [DOI] [PubMed] [Google Scholar]
- 37.Savoy RM, Ghosh PM. The dual role of filamin A in cancer: can’t live with (too much of) it, can’t live without it. Endocr Relat Cancer. 2013;20(6):R341–R356. doi: 10.1530/ERC-13-0364. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.