

Significance
Many plasmids disseminating antibiotic resistance in bacteria encode toxin-antitoxin (TA) pairs, which are generally assumed to function as postsegregational killing (PSK) systems. A shortage of antibiotic alternatives and the link of TA pairs to PSK have stimulated the opinion that premature toxin activation could be used to kill these recalcitrant organisms in the clinic. Following previous observations, we confirm here that toxin Kid (killing determinant) and antitoxin Kis (killing suppressor) function as a rescue system that enforces the retention of plasmid R1 by host cells, and not as a PSK system. Because the rescue system is conserved in clinically worrisome plasmids, our findings advise against the activation of this TA pair as an antibiotic strategy to fight pathogens carrying these extrachromosomal DNAs.
Keywords: PemK, mRNA interferase, parD, plasmid stability, RNase
Abstract
Worldwide dissemination of antibiotic resistance in bacteria is facilitated by plasmids that encode postsegregational killing (PSK) systems. These produce a stable toxin (T) and a labile antitoxin (A) conditioning cell survival to plasmid maintenance, because only this ensures neutralization of toxicity. Shortage of antibiotic alternatives and the link of TA pairs to PSK have stimulated the opinion that premature toxin activation could be used to kill these recalcitrant organisms in the clinic. However, validation of TA pairs as therapeutic targets requires unambiguous understanding of their mode of action, consequences for cell viability, and function in plasmids. Conflicting with widespread notions concerning these issues, we had proposed that the TA pair kis-kid (killing suppressor-killing determinant) might function as a plasmid rescue system and not as a PSK system, but this remained to be validated. Here, we aimed to clarify unsettled mechanistic aspects of Kid activation, and of the effects of this for kis-kid–bearing plasmids and their host cells. We confirm that activation of Kid occurs in cells that are about to lose the toxin-encoding plasmid, and we show that this provokes highly selective restriction of protein outputs that inhibits cell division temporarily, avoiding plasmid loss, and stimulates DNA replication, promoting plasmid rescue. Kis and Kid are conserved in plasmids encoding multiple antibiotic resistance genes, including extended spectrum β-lactamases, for which therapeutic options are scarce, and our findings advise against the activation of this TA pair to fight pathogens carrying these extrachromosomal DNAs.
Plasmids serve as extrachromosomal DNA platforms for the reassortment, mobilization, and maintenance of antibiotic resistance genes in bacteria, enabling host cells to colonize environments flooded with antimicrobials and to take advantage of resources freed by the extinction of nonresistant competitors. Fueled by these selective forces and aided by their itinerant nature, plasmids disseminate resistance genes worldwide shortly after new antibiotics are developed, which is a major clinical concern (1–3). However, in antibiotic-free environments, such genes are dispensable. There, the cost that plasmid carriage imposes on cells constitutes a disadvantage in the face of competition from other cells and, because plasmids depend on their hosts to survive, also a threat to their own existence.
Many plasmids keep low copy numbers (CNs) to minimize the problem above, because it reduces burdens to host cells. However, this also decreases their chances to fix in descendant cells, a new survival challenge (4). To counteract this, plasmids have evolved stability functions. Partition systems pull replicated plasmid copies to opposite poles in host cells, facilitating their inheritance by daughter cells (5). Plasmids also bear postsegregational killing (PSK) systems, which encode a stable toxin and a labile antitoxin (TA) pair that eliminates plasmid-free cells produced by occasional replication or partition failures. Regular production of the labile antitoxin protects plasmid-containing cells from the toxin. However, antitoxin replenishment is not possible in cells losing the plasmid, and this triggers their elimination (5).
TA pairs are common in plasmids disseminating antibiotic resistance in bacterial pathogens worldwide (2, 6–10). The link of these systems to PSK and the exiguous list of alternatives in the pipeline have led some to propose that chemicals activating these TA pairs may constitute a powerful antibiotic approach against these organisms (5, 11–13). However, the appropriateness of these TA pairs as therapeutic targets requires unequivocal understanding of their function in plasmids. Although PSK systems encode TA pairs, not all TA pairs might function as PSK systems, as suggested by their abundance in bacterial chromosomes, where PSK seems unnecessary (14–16). Moreover, the observation that many plasmids bear several TA pairs (6–10) raises the intriguing question of why they would need more than one PSK system, particularly when they increase the metabolic burden that plasmids impose on host cells (17). Because PSK functions are not infallible, their gathering may provide a mechanism for reciprocal failure compensation, minimizing the number of cells that escape killing upon plasmid loss (5). Alternatively, some TA pairs may stabilize plasmids by mechanisms different from PSK, and their grouping might not necessarily reflect functional redundancy (18).
This may be the case in plasmid R1, which encodes TA pairs hok-sok (host killing-suppressor of killing) and kis(pemI)-kid(pemK) (19–23). Inconsistent with PSK, we had noticed that activation of toxin Kid occurred in cells that still contained R1, and that this happened when CNs were insufficient to ensure plasmid transmission to descendant cells. We also found that Kid cleaved mRNA at UUACU sites, which appeared well suited to trigger a response that prevented plasmid loss and increased R1 CNs without killing cells, as suggested by our results. In view of all this, we argued that Kid and Kis functioned as a rescue system for plasmid R1, and not as a PSK system (24). This proposal cannot be supported by results elsewhere, suggesting that Kid may cleave mRNA at simpler UAH sites (with H being A, C, or U) (25, 26), a view that has prevailed in the literature (14, 16, 27–29). Moreover, other observations indicate that our past experiments may have been inappropriate to conclude that Kid does not kill Escherichia coli cells (30, 31). Importantly, Kid, Kis, and other elements that we found essential for R1 rescue are conserved in plasmids conferring resistance to extended-spectrum β-lactamases, a worrying threat to human health (1, 6–10, 32). Therapeutic options to fight pathogens carrying these plasmids are limited, and activation of Kid may be perceived as a good antibiotic alternative. Because the potential involvement of this toxin in plasmid rescue advises against such approach, we aimed to ascertain here the mode of action; the effects on cells; and, ultimately, the function of Kid (and Kis) in R1.
Results and Discussion
Kid Does Not Kill E. coli Cells.
R1 replication rates are proportional to the amount of protein RepA that the plasmid produces in host cells. Thus, overexpression of copA, an antisense RNA that impedes translation of repA, reduces R1 content in host cells as they divide and leads to the eventual production of plasmid-free descendants in the population (33). Two observations made when we used this strategy in E. coli cells carrying an R1 derivative bearing kis-kid argued against a PSK function for this TA pair (24). First, activation of Kid occurred in cells that still contained the plasmid; second, this inhibited growth of our cultures but did not kill cells, because they resumed proliferation when further expression of copA was discontinued.
A bacteriostatic and reversible effect had also been described for MazF, a chromosomal homolog of Kid (34). However, later results revealed that E. coli cells died upon prolonged exposure to MazF, and that this happened earlier in minimal medium than in the rich medium that we originally used in our experiments (30, 31). We thus decided to express copA in E. coli cells carrying mini-R1 plasmids bearing kis-kid (mR1KK), kis-kid18, encoding an inactive mutant of kid (mR1Ctrl), or hok-sok (mR1hs), now using minimal medium and doubling the length of our previous experiments. Production of copA stopped the growth of mR1KK and mR1hs cultures, indicating Kid and Hok activation in these samples (Fig. 1A). Hok kills cells by damaging their membrane (20, 21), and MazF had been involved in the induction of a similar phenotype in E. coli (35, 36). Thus, we analyzed the permeability of cells in our samples to propidium iodide (PI; an indicator of cell membrane damage and cell death). This showed that PI-permeable cell numbers increased considerably upon Hok activation but remained close to control values in cultures arrested by Kid (Fig. 1B). We also examined whether mR1KK-containing cells could resume growth when further expression of copA was discontinued. For this, aliquots from our mR1KK and mR1Ctrl samples in Fig. 1A were seeded at regular intervals on plates repressing further copA production, and the numbers of plasmid-carrying cells grown on these plates were compared with each other. Our results showed that the viability of cells arrested by Kid did not decrease during the experiment, and remained similar to that of control cells, confirming that prolonged exposure to Kid did not kill cells in minimal medium and supporting our proposal that the toxin is not part of a PSK system (Fig. 1C).
Fig. 1.
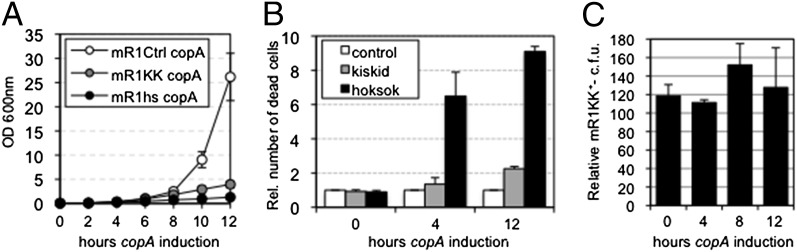
Kid does not kill E. coli. (A) Growth curves of DH10B cells carrying pBAD22copA plus either mR1KK, mR1hs, or mR1Ctrl and induced with arabinose to produce copA for the indicated times in minimal medium. (B) Relative (Rel.) number of dead (i.e., PI-permeable) cells in mR1KK (kiskid) and mR1hs (hoksok) samples from A, normalized against their control mR1Ctrl samples. (C) Proportion of cells carrying mR1KK that resume proliferation when sustained production of copA in A is ceased at the indicated times. Numbers are relative to those observed in control samples (i.e., cells carrying mR1Ctrl). n = 3; bars represent SEM.
Kid Inhibits Cell Division in E. coli and Does Not Halt Protein Synthesis Completely.
The experiments above delivered a puzzling result. Our cultures in Fig. 1 were started at an optical density (OD600) of 0.05, and 4 h later, the average OD600 in mR1Ctrl samples was 0.329, whereas that in mR1hs samples was 36% lower (i.e., 0.247). This, and the increase in dead cells observed in the latter case (Fig. 1B), indicated that maximal R1 dispersal had already occurred in cells, and that plasmid-free descendants were being produced and killed by Hok. However, although Kid activation occurs presegregationally (i.e., precedes activation of Hok), the average OD600 values in mR1KK and mR1Ctrl samples were almost identical (0.339 and 0.329) at the same time point, as if Kid had not become active yet.
To investigate this further, we expressed Kid in E. coli and followed individual cells under the microscope. In all 50 cases, examined cells producing the toxin stopped dividing but not growing in size (Fig. 2A). These observations explained why activation of Kid in our mR1KK samples was not paralleled by a decrease in OD600 relative to control samples at early time points. Moreover, although Kid cleaves mRNA and inhibits protein synthesis (24–26), the cell enlargement shown in Fig. 2A suggested that the toxin does not halt protein production completely in E. coli. To test this, we measured translation rates in the cells above before and at different time points after inducing Kid expression and compared them with those in control cells. This showed that Kid rapidly decreased protein production to 30% of that observed in control cells, and that this level of synthesis remained unchanged afterward (Fig. 2B). Analysis of radiolabeled extracts by 2D gel electrophoresis confirmed that inhibition was substantial but not complete, because cells arrested by Kid still produced several proteins (Fig. 2C).
Fig. 2.
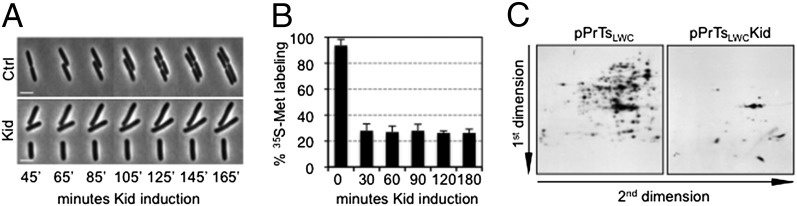
Kid inhibits cell division and does not halt protein synthesis completely in E. coli. (A) Time-lapse images of DH10B cells carrying thermosensitive expression vector pPrTsLWCKid (Lower) or its empty control [Ctrl (pPrTsLWC); Upper] upon incubation at the inducing temperature (42 °C) for the times shown. (Scale bar: 2 μm.) (B) Incorporation of [35S]methionine (35S-Met) in DH10B cells hosting pPrTsLWCKid, relative to that in cells carrying empty pPrTsLWC, after being cultured at 42 °C for the indicated times. (C) 2D gel analysis of protein extracts prepared from the pPrTsLWC and pPrTsLWCKid samples at the 30-min time point in B. n = 3; bars represent SEM.
Kid Does Not Inhibit Its Own Production or That of Kis and RepA.
Kid had been reported to cleave mRNA at UAA, UAC (26), and UAU sites (collectively, UAH sites) (25). However, we observed cleavage at UUACU sites but not at UAA and UAU sequences close to them, and our results also seemed incompatible with cleavage simply at UAC sites (ref. 24 and below). Because the notion that Kid cleaves mRNAs at UA(A/C/U) (14, 27), UA(A/C) (28), or UAC (16, 29) prevailed, we decided to investigate this issue in more detail. Resumption of cell growth in Fig. 1C suggested that cells arrested by Kid can produce Kis de novo, to reneutralize the toxin. To test this, we expressed Kid (or its inactive mutant Kid18) and Kis sequentially in E. coli. Induction of Kid (but not Kid18) inhibited cell growth, and subsequent expression of Kis relieved cells from the arrest (Fig. S1). Analysis of protein synthesis confirmed that cells arrested by Kid could produce Kis de novo, and that this reverted the inhibitory effect of the toxin (Fig. 3A). Moreover, we also observed that expression of Kid could be sustained in E. coli cells in the absence of Kis (Fig. 3B). The mRNA sequences of kis and kid bear eight and nine UAH (three and four UAC) sites, respectively, but no UUACU sequences; therefore, these results supported our views concerning the selectivity of Kid.
Fig. 3.

Kid restricts protein outputs in a UUACU-dependent manner. (A) Analysis of protein extracts from DH10B cells carrying expression vectors p177PraraKid and pTet-HS3FKis. Samples before induction (0 h), and at regular intervals after inducing Kid alone first, using arabinose (1–3 h), and then inducing Kis for another 3 h, using anhydrotetracycline (A-Tet) (3.25–6 h), were analyzed. Both 35S-labeled samples (Upper) and -unlabeled samples blotted with antibodies against Kid and Kis (Lower) are shown. WB, Western blot. (B) Analysis of 35S-labeled extracts from GCM2 cells, before (−) or at the indicated times after (with 0 h being immediately after) inducing them to produce Kid from pTet-Kid, using A-Tet. (C) Analysis of extracts from DH10B cells cotransformed with p177Prara-Kid18 (lanes 1–4) or p177Prara-Kid (lanes 5–8) and either pTet-H-EGFP-RepAr (lacking UUACU sites, lanes 1 and 2 and 5 and 6) or pTet-H-EGFP-RepAs (bearing UUACU sites; lanes 3 and 4 and 7 and 8). Odd lanes correspond to uninduced (−) samples. Even lanes correspond to samples induced (+) to produce Kid (or Kid18) for 1 h first and then His6-EGFP-RepAr (or His6-EGFP-RepAr) for another hour before labeling. Extracts from parallel unlabeled samples and blotted with an anti-EGFP antibody are also shown. (D) Analysis of 35S-labeled extracts from DH10B cells cotransformed with p177Prara-Kid plus either pTet-H-DnaBs (bearing UUACU sites, lanes 1 and 2) or pTet-H-DnaBr (lacking UUACU sites, lanes 3 and 4). Odd lanes correspond to uninduced (−) samples. Even lanes correspond to samples that were induced (+) to produce Kid alone for 1 h first, and then the corresponding DnaB variant, before labeling. Arrowheads (black for Kid and Kid18; white for Kis, RepA, and DnaB) mark the position of proteins in gels.
Next, we looked at RepA, a key player in the rescue model. R1 transcribes this gene from two promoters: PrcopB produces a copB-repA mRNA, whereas PrrepA produces a repA transcript. PrrepA is stronger than PrcopB, but the upstream product of the copB-repA mRNA (i.e., CopB) represses it, limiting RepA production and R1 replication rates (37). We had found that presegregational activation of Kid enabled cleavage of copB-repA mRNA at two intercistronic UUACU sites, and that this inhibited production of CopB and derepressed PrrepA (24). Because repA lacks UUACU sites, we had proposed that cells arrested by Kid should be able to produce RepA, but this remained to be validated. Here, we analyzed the expression of an EGFP-RepA fusion in cells arrested by Kid. The mRNA encoding egfp-repA bore 33 UAH sites but lacked UUACU sequences (Table S1), and our results confirmed that those cells produced as much EGFP-RepA as cells that had expressed Kid18 instead (Fig. 3C, lanes 2 and 6). Reassuringly, introduction of UUACU in the region linking egfp and repA turned the fusion gene very sensitive to Kid (Fig. 3C, lane 8).
Finally, two UUACU sites cleaved by Kid in the dnaB gene of E. coli (24) were mutated silently (i.e., to UUACC), and we compared the expression of DnaB from the mutant and the WT genes in cells arrested by the toxin. The elimination of UUACU sites (without changing their central UAC sequences) stabilized production of DnaB in cells expressing Kid (Fig. 3D). Thus, our observations do not support the view that Kid targets UAH sites. Interestingly, evidence elsewhere suggests that the toxin may display “relaxed” selectivity under less physiological conditions, such as occurs with restriction endonucleases. The latter cleave DNA at sequences that are similar, but not identical, to their canonical targets when enzyme-to-substrate ratios are increased 100-fold (38). Similarly, although cleavage of AUACA sites by Kid has been reported in vitro, this required toxin/mRNA ratios 100-fold higher than those required for UUACU cleavage (39).
It is worth mentioning that the coding sequences of proteins identified in our 2D gels contain, altogether, 373 UAH (109 UAC) sites but only two UUACU sequences (Table S1). Therefore, the presence of UUACU in genes does not necessarily imply that it will not be expressed in cells arrested by Kid. Folding into ds mRNA structures, which are not cleaved by Kid (25), may protect these sites from cleavage. Additionally, production of WT DnaB from a strong promoter was not inhibited completely in the presence of Kid (Fig. 3D), suggesting that high transcription rates help sensitive mRNAs to escape cleavage, at least partially. Mechanisms like these may operate to facilitate replenishment of short-lived proteins encoded in sensitive mRNAs in cells arrested by Kid, if they are essential.
Kid Uncouples DNA Replication and Cell Division in E. coli.
We also looked at R1 molecules in cells arrested by Kid, for which we cloned an array of 240 TetR binding sites in a mini-R1 derivative (mR1TetO240). This plasmid was introduced in ILO1, an E. coli strain with 240 LacI binding sites integrated close to the chromosomal origin of replication (oriC), together with pWX6, a vector expressing TetR-YFP and LacI-CFP, to facilitate tracking of mR1tetO240 and oriC by fluorescence microscopy (40). Induction of Kid expression from another plasmid inhibited cell division and led to a progressive increase of mR1tetO240- and oriC-foci numbers in arrested cells. As a consequence, individual cells containing four (or more) R1 foci and four oriC foci accumulated over time in these samples, a phenotype that was not observed in the absence of Kid expression (Fig. 4A).
Fig. 4.
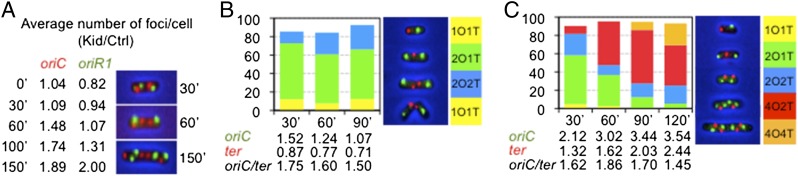
Kid uncouples DNA replication and cell division in E. coli. (A) Average numbers of oriC- (red) and oriR1- (green) foci in ILO1 cells carrying plasmids pWX6, mR1tetO240, and pPrTsLWCKid (Kid) and cultured at 42 °C for the indicated times. Values are normalized against those in parallel control samples carrying pWX6, mR1tetO240, and empty pPrTsLWC (Ctrl). Representative images of cells at different time points are shown. (B) Relative abundance of oriC-ter (OT) phenotypes observed in ILO6 cells carrying plasmids pWX6 and pPrTsLWC (Ctrl), after culturing them at 42 °C for the indicated times. Images of representative phenotypes in this sample are shown, and the average number of oriC (green) and ter (red) foci per cell, as well as oriC/ter ratios per cell and time point, are indicated. (C) Same as in B but in cells carrying pWX6 and pPrTsLWCKid. Average numbers of foci result from dividing the total number of each type of foci by the total number of cells (minimum of 600) analyzed per sample and time point.
All proteins needed to replicate R1, except RepA, are encoded by the host. To find out whether they were available for plasmid rescue, we followed chromosomal replication in single cells arrested by Kid. For this, we used ILO6, an E. coli strain bearing 240 LacI- and 240 TetR-binding sites close to oriC and the chromosome’s termination locus (ter), respectively, and, as before, also transformed with pWX6. In the absence of Kid expression, these cells progressed normally through the cell cycle, moving from a situation where only one oriC and one ter were detected (1O1T; Fig. 4B) to one in which a new round of replication had started, producing two oriC and one ter signals (2O1T; Fig. 4B). A third population of cells containing two oriC and two ter signals (2O2T; Fig. 4B) was also observed. These had completed chromosome replication, and in most cases, cell membrane invagination at midcell positions was evident. Completion of cytokinesis produced two identical cells, each containing one oriC and one ter (1O1T). No other major phenotype was identified, and the percentage of cells of each type remained fairly constant in asynchronously growing cell populations, which doubled every 20–25 min, approximately the gap between time points.
However, Kid induced additional phenotypes in these cells. The first one corresponded to elongated cells containing two oriC and two ter loci but no sign of cell membrane invagination (2O2T; Fig. 4C). Slightly longer cells containing four oriC and two ter (4O2T; Fig. 4C), and even longer cells with four oriC and four ter (4O4T; Fig. 4C), were also observed. The oriC/ter ratios also remained fairly constant in these cells, indicating that they were able to complete chromosome replication. However, progression from phenotype 2O2T to 1O1T was barely observed, and phenotypes 4O2T and 4O4T accumulated with time in these samples. The latter are equivalent to the elongated phenotype shown in Fig. 2A, highlighting the statistical significance of that result. Our observations in Fig. 4C confirmed that cells arrested by Kid still produce all proteins required to replicate R1, and revealed that the toxin uncouples cell division and DNA replication in E. coli. Thus, activation of Kid triggers two overlapping responses contributing to R1 survival: Inhibition of cell division avoids the appearance of plasmid-free cells, and stimulation of R1 replication restores safe CNs in arrested cells.
Kid Inhibits Divisome Assembly and Septum Formation in E. coli.
Cells arrested by Kid did not initiate membrane invagination (Figs. 2C and 4C), indicating that they could not start cell division. Thus, we examined cells expressing Kid, Kis and Kid or none of these proteins by EM. This confirmed that cells arrested by the toxin reached sizes close to 4–5 μm but did not initiate septation, a process that was clearly visible in cells from the other samples (Fig. 5A).
Fig. 5.

Kid inhibits septum formation and divisome assembly in E. coli. (A) scanning EM (SEM) and transmission EM (TEM) images of 4–5 μm-long DH10B cells carrying either pPrTsHC and pPrTsLWC (Ctrl), pPrTsHC and pPrTsLWCKid (Kid), or pPrTsLWCKid and pPrTsHCKis (KidKis), and cultured at 42 °C for 2 h before analysis. The presence or absence of septum in cells is indicated with green and red arrows, respectively. (B) Primer extension of ftsZ-egfp and zapA-egfp mRNAs isolated from DH4FZGFP and DH4ZAGFP cells carrying plasmid pPrTsMCKid (Kid) or its parental control vector pPrTsMC (Ctrl), cultured at 42 °C for 30 min before analysis. Red arrows mark mRNA cleavage products, with the sequence cleaved shown on the axis. (C) Time-lapse bright-field (Top) and fluorescent (Middle) images of DH4FZGFP cells carrying pPrTsMCKid (Kid) or its parental control vector pPrTsMC (Ctrl). The images correspond to the same cells, imaged before (0′) and after (90′) placing them at 42 °C. (Bottom) Magnified views of cells stained with membrane dye FM4-64 (red) and demonstrating the distribution of FtsZ-EGFP (green) in Ctrl and Kid samples at the 90′ time point are also shown. (D) Same as in C but using DH4ZAGFP cells to examine ZapA-EGFP. (Scale bar: 2 μm.)
Cell division in E. coli starts with the assembly of dynamic ring-like FtsZ polymers that serve as a scaffold for the construction of a “divisome” at midcell positions when nucleoid separation begins (41). Many mRNAs encoding divisomal proteins contain UUACU sites, and we analyzed whether Kid cleaved two of them. We fused egfp to the 3′ end of ftsZ or zapA genes in E. coli and induced the expression of Kid in the resulting strains (DH4FZGFP and DH4ZAGFP, respectively). Cleavage of ftsZ-egfp and zapA-egfp transcripts at UUACU sites in cells arrested by Kid, but not in control cells, was confirmed by primer extension (Fig. 5B). We also monitored FtsZ-EGFP and ZapA-EGFP proteins in these samples using fluorescent microscopy. Time-lapse experiments showed that FtsZ formed sharp rings at midcell positions in control cells, before division started. In contrast, cells expressing Kid formed diffused FtsZ structures at midcell positions that, when magnified, appeared to be spirals rather than single, sharply defined rings (Fig. 5C) and oscillated rapidly back and forth between midcell positions and the cell poles (Fig. S2). ZapA also assembled in sharp ring-like structures at midcell positions, with a pattern resembling that of FtsZ, in control samples. However, in cells expressing Kid, ZapA formed static two-dot–like structures associated with the cell membrane at midcell positions or at the cell poles (Fig. 5D). ZapA is important for the spatiotemporal tuning of FtsZ polymers and for the establishment of physical links between the divisome and the replicating chromosome that help to coordinate cell division with nucleoid segregation (42). It is noteworthy that ZapA induces the association of FtsZ protofilaments into large bundles (43), and cells deficient in this protein cannot coalesce FtsZ spirals into midcell rings and show cytokinesis defects (44). Thus, although pleiotropic effects are likely, our results suggest one way in which Kid inhibits the assembly of a functional divisome in E. coli.
Rescue System Is Conserved in Clinically Worrisome Plasmids.
Our results in this work clarify the function of kis-kid, supporting its role in R1 rescue and explaining how this TA pair contributes to plasmid survival (Fig. 6). When R1 replication rates provide enough plasmid copies to allow their segregation to descendant cells, Kid remains neutralized by Kis and cells proliferate normally (Fig. 6, CN OK). However, if insufficient plasmid replication compromises this, Kid becomes active presegregationally (Fig. 6, CN too low). Cleavage of mRNAs at UUACU sites restricts protein outputs selectively, inhibiting cell division and avoiding plasmid loss but keeping cells alive and capable of replicating DNA. Because activation of Kid occurs in cells that still contain R1, the toxin also cleaves copB-repA mRNA at two UUACU sites upstream of repA. This reduces CopB levels and derepresses transcription of a monocistronic repA mRNA that lacks those sites, enabling higher production of the initiator protein and increasing R1 replication rates to accomplish rescue.
Fig. 6.

Kis-kid and hok-sok function sequentially and coordinately to facilitate survival of plasmid R1. When R1 CNs ensure plasmid transmission to both descendant cells, Kid remains neutralized and parental cells proliferate normally (1). However, Kid becomes active when R1 copy numbers are insufficient to warrant plasmid transmission to both daughter cells. This arrests parental cell division and stimulates RepA synthesis to rescue the plasmid. Once the latter is accomplished, the system resets, allowing host cells to proliferate again (2). If rescue fails, plasmid-free cells arise, and they are killed by Hok (3). White circles represent R1; light blue, red, yellow, and black spheres represent Kis, Kid, RepA, and Hok, respectively; and green rings and dark blue rods represent the divisome and the nucleoid, respectively.
Activation of Kid is triggered by degradation of Kis, and because the latter is required to repress Prkis-kid, new synthesis of a UUACU-less kis-kid mRNA occurs in arrested cells. Because Kis neutralizes Kid at a 1:2 ratio, the transcript above supplies the antitoxin in excess, blocking newly produced Kid immediately and preactivated Kid progressively, and resetting the system when safe copy numbers are restored. However, the system is not infallible, and some cells divide despite Kid activation, producing plasmid-free descendants (Fig. 6, rescue failure). These are killed by Hok, which is induced postsegregationally. Thus, rather than being functionally redundant, kis-kid and hok-sok operate in a coordinated, noninterfering manner that facilitates survival of R1 in bacterial populations. The mRNA encoding Hok lacks UUACU sites, which allows production of this toxin in cells escaping the arrest by Kid; sok contains one UUACU site, but this is protected from Kid in a ds mRNA structure that is essential to neutralize Hok (45). Confirming the latter, we verified that Kid does not induce premature activation of the hok-sok TA pair (Fig. S3), which would kill cells in which the rescue system is still operating.
Interestingly, R1 bears another presegregational stability function, partition system parA (also known as par or parMRC), which, together with hok-sok, reduces R1 loss to 10−6 per cell per generation, a rate already considered equivalent to complete stability (19). Our results explain why despite the above, R1 also encodes kis-kid. Partition can only function in cells with two plasmid copies, and these numbers may not be reached before cell division if R1 replicates inefficiently. Thus, kis-kid functions as a fail-safe mechanism that becomes active when parA cannot contribute to plasmid stability, deploying a response that avoids plasmid loss and stimulates R1 replication until parA can function again.
Our results have important clinical implications. The contribution of plasmids to antibiotic resistance in bacterial pathogens has led to the proposal that agents inhibiting the replication of these extrachromosomal DNAs, or activating their PSK systems, could be exploited to kill these organisms directly and selectively (11–13). Our work raises concerns against this approach in the case of R1 and its kis-kid TA pair. Inhibition of R1 replication would activate the rescue system and, instead of killing cells, induce growth arrest from which bacteria can recover upon treatment cessation. Moreover, disruption of Kid–Kis complexes would activate the genetic circuitry involved in R1 rescue, and production of new Kid and Kis could titrate out the therapeutic agent used, thus facilitating resistance. Importantly, conserved kis-kid and all other elements required for R1 rescue (i.e., UUACU-positive copB-repA and UUACU-free repA mRNAs) are present in plasmids encoding extended-spectrum β-lactamases, which is a major clinical concern worldwide for which therapeutic alternatives are scarce (1, 3, 6–10), suggesting that the system also operates there. Worth noting, plasmids conferring vancomycin resistance to enterococci also encode fairly conserved Kid and Kis homologs, and their pharmacological activation has also been proposed as a strategy for tailored therapy against these worrisome pathogens (11). Our results advise that the mode of action and function of these TA pairs should be unambiguously defined before such an approach is put into practice.
Materials and Methods
A complete description of materials and methods is provided in SI Materials and Methods, including a list of all oligonucleotides (Table S2), plasmids (Table S3), and strains (Table S4) used in this work. Cells were grown in M9 medium plus all amino acids minus methionine. Glucose (0.2%) was used in all preinocula, and maintained in cultures in Figs. 2 B and C and 3B, but it was changed to 0.5% glycerol in all other cases. When appropriate, ampicillin (100 μg/mL), kanamycin (50 μg/mL), and chloramphenicol (10 μg/mL) were also used in cultures. Antibiotics were maintained in cultures when protein production was induced from expression vectors. Strain and plasmid(s) combinations used are as indicated in figure legends and in Tables S3 and S4. PI-permeable cells were quantified by FACS, using a LIVE/DEAD kit (Invitrogen). Ectopic expression of Kid was induced using A-Tet (0.2 μg/mL; Fig. 3B), arabinose (0.02%; Fig. 3 A, C, and D and Fig. S1), or temperature upshift (42 °C; Figs. 2, 4, and 5 and Fig. S2), depending on experiments. Expression of Kis, EGFP-RepA, and DnaB was always induced with A-Tet (0.2 μg/mL) and either 3 h (Kis; Fig. 3A and Fig. S1) or 1 h (EGFP-RepA and DnaB; Fig. 3 C and D) after inducing Kid expression. For in vivo labeling, 5 μCi/mL [35S]methionine was incorporated for 2 min into cells at 42 °C (Fig. 2 B and C) or 37 °C (Fig. 3). Polyclonal Kid and Kis antibodies (Fig. 2A) or a commercial monoclonal EGFP antibody (Fig. 3C) was used for Western blotting. Optical microscopy was performed in life cell imaging chambers using slides covered with 1% agarose in PBS (snapshots in Fig. 4) or in M9 medium plus all amino acids minus methionine plus 0.2% glucose (time-lapse images in Figs. 2A and 5 C and D). EM was performed on cells fixed with 0.4% glutaraldehyde. Primer extension was carried out as done by Pimentel et al. (24), using 50 μg of total RNA per sample.
Supplementary Material
Acknowledgments
We thank T. Mills and S. Penrhyn-Lowe, J. Skepper and I. Bolton, and S. Peak-Chew and F. Begum for assistance with the confocal microscope, Scanning EM/transmission EM, and MS, respectively. This work was supported by the Medical Research Council (Programme Grant MC_U105365008 to G.d.l.C.-M.), a Förderung der wissenschaftlichen Forschun (FWF) Erwin Schrödinger Fellowship (J2648) to C.A.A., a Spanish Ramón y Cajal Program Grant (RYC2009 04341) to J.A.B., and the Wellcome Trust (Grant WT099204AIA to D.J.S.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1308241111/-/DCSupplemental.
References
- 1.Coque TM, et al. Dissemination of clonally related Escherichia coli strains expressing extended-spectrum beta-lactamase CTX-M-15. Emerg Infect Dis. 2008;14(2):195–200. doi: 10.3201/eid1402.070350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carattoli A. Resistance plasmid families in Enterobacteriaceae. Antimicrob Agents Chemother. 2009;53(6):2227–2238. doi: 10.1128/AAC.01707-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Novais A, et al. Characterization of globally spread Escherichia coli ST131 isolates (1991 to 2010) Antimicrob Agents Chemother. 2012;56(7):3973–3976. doi: 10.1128/AAC.00475-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paulsson J. Multileveled selection on plasmid replication. Genetics. 2002;161(4):1373–1384. doi: 10.1093/genetics/161.4.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sengupta M, Austin S. Prevalence and significance of plasmid maintenance functions in the virulence plasmids of pathogenic bacteria. Infect Immun. 2011;79(7):2502–2509. doi: 10.1128/IAI.00127-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boyd DA, et al. Complete nucleotide sequence of a 92-kilobase plasmid harboring the CTX-M-15 extended-spectrum beta-lactamase involved in an outbreak in long-term-care facilities in Toronto, Canada. Antimicrob Agents Chemother. 2004;48(10):3758–3764. doi: 10.1128/AAC.48.10.3758-3764.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Périchon B, et al. Sequence of conjugative plasmid pIP1206 mediating resistance to aminoglycosides by 16S rRNA methylation and to hydrophilic fluoroquinolones by efflux. Antimicrob Agents Chemother. 2008;52(7):2581–2592. doi: 10.1128/AAC.01540-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woodford N, et al. Complete nucleotide sequences of plasmids pEK204, pEK499, and pEK516, encoding CTX-M enzymes in three major Escherichia coli lineages from the United Kingdom, all belonging to the international O25:H4-ST131 clone. Antimicrob Agents Chemother. 2009;53(10):4472–4482. doi: 10.1128/AAC.00688-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mnif B, et al. Molecular characterization of addiction systems of plasmids encoding extended-spectrum beta-lactamases in Escherichia coli. J Antimicrob Chemother. 2010;65(8):1599–1603. doi: 10.1093/jac/dkq181. [DOI] [PubMed] [Google Scholar]
- 10.Bonnin RA, Poirel L, Carattoli A, Nordmann P. Characterization of an IncFII plasmid encoding NDM-1 from Escherichia coli ST131. PLoS ONE. 2012;7(4):e34752. doi: 10.1371/journal.pone.0034752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moritz EM, Hergenrother PJ. Toxin-antitoxin systems are ubiquitous and plasmid-encoded in vancomycin-resistant enterococci. Proc Natl Acad Sci USA. 2007;104(1):311–316. doi: 10.1073/pnas.0601168104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Williams JJ, Hergenrother PJ. Exposing plasmids as the Achilles’ heel of drug-resistant bacteria. Curr Opin Chem Biol. 2008;12(4):389–399. doi: 10.1016/j.cbpa.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Williams JJ, Hergenrother PJ. Artificial activation of toxin-antitoxin systems as an antibacterial strategy. Trends Microbiol. 2012;20(6):291–298. doi: 10.1016/j.tim.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fu Z, Tamber S, Memmi G, Donegan NP, Cheung AL. Overexpression of MazFsa in Staphylococcus aureus induces bacteriostasis by selectively targeting mRNAs for cleavage. J Bacteriol. 2009;191(7):2051–2059. doi: 10.1128/JB.00907-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hayes F, Van Melderen L. Toxins-antitoxins: Diversity, evolution and function. Crit Rev Biochem Mol Biol. 2011;46(5):386–408. doi: 10.3109/10409238.2011.600437. [DOI] [PubMed] [Google Scholar]
- 16.Yamaguchi Y, Park JH, Inouye M. Toxin-antitoxin systems in bacteria and archaea. Annu Rev Genet. 2011;45:61–79. doi: 10.1146/annurev-genet-110410-132412. [DOI] [PubMed] [Google Scholar]
- 17.Cooper TF, Heinemann JA. Postsegregational killing does not increase plasmid stability but acts to mediate the exclusion of competing plasmids. Proc Natl Acad Sci USA. 2000;97(23):12643–12648. doi: 10.1073/pnas.220077897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de la Cueva-Méndez G, Pimentel B. Gene and cell survival: Lessons from prokaryotic plasmid R1. EMBO Rep. 2007;8(5):458–464. doi: 10.1038/sj.embor.7400957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gerdes K, Larsen JE, Molin S. Stable inheritance of plasmid R1 requires two different loci. J Bacteriol. 1985;161(1):292–298. doi: 10.1128/jb.161.1.292-298.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerdes K, Rasmussen PB, Molin S. Unique type of plasmid maintenance function: Postsegregational killing of plasmid-free cells. Proc Natl Acad Sci USA. 1986;83(10):3116–3120. doi: 10.1073/pnas.83.10.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gerdes K, et al. Mechanism of postsegregational killing by the hok gene product of the parB system of plasmid R1 and its homology with the relF gene product of the E. coli relB operon. EMBO J. 1986;5(8):2023–2029. doi: 10.1002/j.1460-2075.1986.tb04459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bravo A, Ortega S, de Torrontegui G, Díaz R. Killing of Escherichia coli cells modulated by components of the stability system ParD of plasmid R1. Mol Gen Genet. 1988;215(1):146–151. doi: 10.1007/BF00331316. [DOI] [PubMed] [Google Scholar]
- 23.Tsuchimoto S, Ohtsubo H, Ohtsubo E. Two genes, pemK and pemI, responsible for stable maintenance of resistance plasmid R100. J Bacteriol. 1988;170(4):1461–1466. doi: 10.1128/jb.170.4.1461-1466.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pimentel B, Madine MA, de la Cueva-Méndez G. Kid cleaves specific mRNAs at UUACU sites to rescue the copy number of plasmid R1. EMBO J. 2005;24(19):3459–3469. doi: 10.1038/sj.emboj.7600815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang J, Zhang Y, Zhu L, Suzuki M, Inouye M. Interference of mRNA function by sequence-specific endoribonuclease PemK. J Biol Chem. 2004;279(20):20678–20684. doi: 10.1074/jbc.M314284200. [DOI] [PubMed] [Google Scholar]
- 26.Muñoz-Gómez AJ, Lemonnier M, Santos-Sierra S, Berzal-Herranz A, Díaz-Orejas R. RNase/anti-RNase activities of the bacterial parD toxin-antitoxin system. J Bacteriol. 2005;187(9):3151–3157. doi: 10.1128/JB.187.9.3151-3157.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamaguchi Y, Park JH, Inouye M. MqsR, a crucial regulator for quorum sensing and biofilm formation, is a GCU-specific mRNA interferase in Escherichia coli. J Biol Chem. 2009;284(42):28746–28753. doi: 10.1074/jbc.M109.032904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu MX, Zhang X, Li EL, Feng YJ. Recent advancements in toxin and antitoxin systems involved in bacterial programmed cell death. Int J Microbiol. 2010;2010:781430. doi: 10.1155/2010/781430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee MW, Rogers EE, Stenger DC. Xylella fastidiosa plasmid-encoded PemK toxin is an endoribonuclease. Phytopathology. 2012;102(1):32–40. doi: 10.1094/PHYTO-05-11-0150. [DOI] [PubMed] [Google Scholar]
- 30.Amitai S, Yassin Y, Engelberg-Kulka H. MazF-mediated cell death in Escherichia coli: A point of no return. J Bacteriol. 2004;186(24):8295–8300. doi: 10.1128/JB.186.24.8295-8300.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kolodkin-Gal I, Engelberg-Kulka H. Induction of Escherichia coli chromosomal mazEF by stressful conditions causes an irreversible loss of viability. J Bacteriol. 2006;188(9):3420–3423. doi: 10.1128/JB.188.9.3420-3423.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arias CA, Murray BE. Antibiotic-resistant bugs in the 21st century—A clinical super-challenge. N Engl J Med. 2009;360(5):439–443. doi: 10.1056/NEJMp0804651. [DOI] [PubMed] [Google Scholar]
- 33.Jensen RB, Grohmann E, Schwab H, Díaz-Orejas R, Gerdes K. Comparison of ccd of F, parDE of RP4, and parD of R1 using a novel conditional replication control system of plasmid R1. Mol Microbiol. 1995;17(2):211–220. doi: 10.1111/j.1365-2958.1995.mmi_17020211.x. [DOI] [PubMed] [Google Scholar]
- 34.Pedersen K, Christensen SK, Gerdes K. Rapid induction and reversal of a bacteriostatic condition by controlled expression of toxins and antitoxins. Mol Microbiol. 2002;45(2):501–510. doi: 10.1046/j.1365-2958.2002.03027.x. [DOI] [PubMed] [Google Scholar]
- 35.Kolodkin-Gal I, Verdiger R, Shlosberg-Fedida A, Engelberg-Kulka H. A differential effect of E. coli toxin-antitoxin systems on cell death in liquid media and biofilm formation. PLoS ONE. 2009;4(8):e6785. doi: 10.1371/journal.pone.0006785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Erental A, Sharon I, Engelberg-Kulka H. Two programmed cell death systems in Escherichia coli: An apoptotic-like death is inhibited by the mazEF-mediated death pathway. PLoS Biol. 2012;10(3):e1001281. doi: 10.1371/journal.pbio.1001281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Light J, Riise E, Molin S. Transcription and its regulation in the basic replicon region of plasmid R1. Mol Gen Genet. 1985;198(3):503–508. doi: 10.1007/BF00332947. [DOI] [PubMed] [Google Scholar]
- 38.Wei H, Therrien C, Blanchard A, Guan S, Zhu Z. The Fidelity Index provides a systematic quantitation of star activity of DNA restriction endonucleases. Nucleic Acids Res. 2008;36(9):e50. doi: 10.1093/nar/gkn182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Diago-Navarro E, et al. A mutagenic analysis of the RNase mechanism of the bacterial Kid toxin by mass spectrometry. FEBS J. 2009;276(17):4973–4986. doi: 10.1111/j.1742-4658.2009.07199.x. [DOI] [PubMed] [Google Scholar]
- 40.Wang X, Possoz C, Sherratt DJ. Dancing around the divisome: Asymmetric chromosome segregation in Escherichia coli. Genes Dev. 2005;19(19):2367–2377. doi: 10.1101/gad.345305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de Boer PA. Advances in understanding E. coli cell fission. Curr Opin Microbiol. 2010;13(6):730–737. doi: 10.1016/j.mib.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Espéli O, et al. A MatP-divisome interaction coordinates chromosome segregation with cell division in E. coli. EMBO J. 2012;31(14):3198–3211. doi: 10.1038/emboj.2012.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mohammadi T, et al. The GTPase activity of Escherichia coli FtsZ determines the magnitude of the FtsZ polymer bundling by ZapA in vitro. Biochemistry. 2009;48(46):11056–11066. doi: 10.1021/bi901461p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dajkovic A, Pichoff S, Lutkenhaus J, Wirtz D. Cross-linking FtsZ polymers into coherent Z rings. Mol Microbiol. 2010;78(3):651–668. doi: 10.1111/j.1365-2958.2010.07352.x. [DOI] [PubMed] [Google Scholar]
- 45.Franch T, Gultyaev AP, Gerdes K. Programmed cell death by hok/sok of plasmid R1: Processing at the hok mRNA 3′-end triggers structural rearrangements that allow translation and antisense RNA binding. J Mol Biol. 1997;273(1):38–51. doi: 10.1006/jmbi.1997.1294. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.