

Abstract
Notch signaling is an evolutionarily ancient, highly conserved pathway important for deciding cell fate, cellular development, differentiation, proliferation, apoptosis, adhesion, and epithelial-to-mesenchymal transition. Notch signaling is also critical in mammalian cardiogenesis, as mutations in this signaling pathway are linked to human congenital heart disease. Furthermore, Notch signaling can repair myocardial injury by promoting myocardial regeneration, protecting ischemic myocardium, inducing angiogenesis, and negatively regulating cardiac fibroblast-myofibroblast transformation. This review provides an update on the known roles of Notch signaling in the mammalian heart. The goal is to assist in developing strategies to influence Notch signaling and optimize myocardial injury repair.
Keywords: Notch signaling, Congenital heart disease, Myocardial regeneration, Cardioprotection, Angiogenesis, Cardiac fibroblast-myofibroblast transformation
Introduction
The Notch signaling pathway is a phylogenetically conserved signaling pathway, which was first described by Morgan approximately 90 years ago (1). Notch signaling is modulated by glycosylation, differential intracellular trafficking, and ubiquitin-dependent degradation of individual components of the signaling pathway (2). In mammals, the Notch pathway consists of four Notch proteins (Notch 1-4) and five ligands, Jagged 1, Jagged 2, Delta-like 1, Delta-like 3, and Delta-like 4. Notch homologues of Notch are present in echinoderms, ascidians, nematodes, insects, and vertebrates. In all organisms, Notch signaling provides effective communication between adjacent cells to regulate cell fate decisions, cellular development, differentiation, proliferation, apoptosis, adhesion, and epithelial-to-mesenchymal transformation (EMT) (3). Furthermore, Notch signaling is involved in the development of the atrioventricular canal, the aortic valve, the ventricles, and the outflow tract during mammalian cardiogenesis (4).
Mutations that affect Notch signaling are associated with several types of congenital heart disease (5). Notch signaling can promote myocardial regeneration, protect the myocardium from ischemia, induce angiogenesis, and inhibit cardiac fibroblast to myofibroblast transformation (CMT). Each of these events supports cardiac repair following myocardial injury (6-9). In this review, we provide an update on the roles of Notch signaling in the mammalian heart to provide therapeutic insight into modulation of Notch signaling to optimize myocardial injury repair.
Notch signaling overview
Notch is a 300-kDa single-chain transmembrane protein with four homologous isoforms in mammals, identified as Notch 1-4. Notch is cleaved by proteases, and this posttranslational modification forms a functional heterodimer on the cell surface. The Notch extracellular domain consists of 36 epidermal growth factor-like repeats (EGFR) and three cysteine-rich LIN12/Notch repeats. EGFR is responsible for ligand binding to Notch, while the LIN12/Notch repeats prevent ligand-independent activation (10). The Notch intracellular domain (NICD) contains an RBP-Jκ (recombination signal binding protein for immunoglobulin Jκ region) associated molecular domain, six ankyrin/cdc10 repeats, two nuclear localization signals, a transcriptional activation domain, as well as a sequence rich in proline, glutamic acid, serine, and threonine on the C-terminus. The RBP-Jκ-associated molecular domain and ankyrin/cdc10 repeats interact with RBP-Jκ, and the transcriptional activation domain contains phosphorylation sites that may allow other intracellular signals to modulate Notch signaling. The domain with the sequence rich in proline, glutamic acid, serine, and threonine promotes protein instability (3).
Recently, three other functional regions of NICD have been identified. The first, the potential phosphorylated domain, located between the ankyrin repeats and the proline, glutamic acid, serine, and threonine-rich domain, can enhance binding of the NICD to RBP-Jκ. The second, a downregulation targeting sequence, is required for endocytic trafficking of Notch. The third, the WSSSSP motif, a C-terminal phosphorylation site is important for Notch turnover (Figure 1A) (1).
Figure 1. Schematic representation of Notch signaling. A, Notch receptor is composed of extracellular and intracellular domains. The extracellular domain consists of EGFR and LNR. The intracellular domain contains RAM23, DTS, ANK, NLS, PPD, TAD, PEST, and S4. The red arrows indicate cleavage sites: S1 (Furin), S2 (TACE), and S3/S4 (γ-secretase). B, Notch ligands have DSL and EGFR domains. C, Notch signaling transduction pathway ligand-receptor interaction leads to two successive cleavages at S2 by TACE and S3/4 by γ-secretase to release NICD. NICD translocates to the nucleus and forms a transcriptional activation complex after binding to MAML-1 and CSL. Prototypic target genes include Hes and the HRT family members. EGFR: epidermal growth-like repeats; LNR: LIN12/Notch repeats; RAM: RBP-Jκ associated molecule; DTS: downregulation targeting sequence; ANK: ankyrin/cdc10 repeats; NLS: nuclear localization signals; PPD: potential phosphorylated domain; TAD: transcriptional activation domain; PEST: sequence rich in proline, glutamic acid, serine, and threonine; S4: WSSSSP; TACE: TNF-α converting enzyme; DSL: Delta, Serrate, Lag2; NICD: Notch intracellular domain; MAML-1: Mastermind-like 1; CSL: C promoter-binding factor in humans, Suppressor of hairless in Drosophila, LAG in Caenorhabditis elegans, also called RBP-Jκ in mice; Hes: Hairy and enhancer of split; HRT: Hairy-related transcription; CoR: co-repressor; HDAC: histone deacetylase.
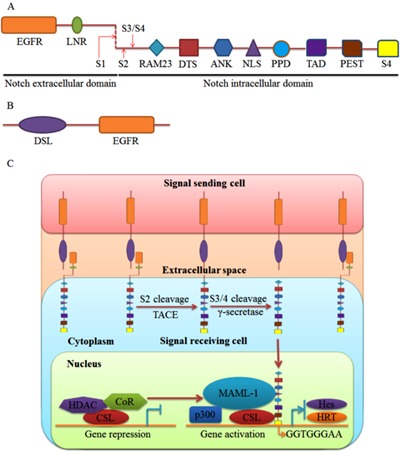
The Jagged and Delta family members were identified as Notch ligands in mammals. Both have a DSL (Delta, Serrate, and Lag 2) domain and an EGFR (Figure 1B). In addition, the Jagged family has a cysteine-rich domain (11). Upon ligand binding, Notch is activated and undergoes a conformational change that releases NICD from the cell membrane following two successive cleavages, in the extracellular transmembrane region by TNF-α converting enzyme, and in the cross-membrane area by γ-secretase complex. Following release, NICD translocates to the nucleus, where it binds to the transcription factor CSL (C promoter-binding factor L in humans, Suppressor of Hairless in Drosophila, LAG in Caenorhabditis elegans, also called RBP-Jκ in mice). When Notch signaling is not stimulated, the CSL protein binds to promoters on its target genes and recruits histone deacetylases and co-repressors to curb gene transcription. When Notch signaling is stimulated, NICD and CSL interact to displace the co-repressor and form a triple complex with Mastermind-Like 1. The triple complex stabilizes the binding of CSL/NICD to DNA, which recruits lipoprotein acetyltransferase p300 to upregulate target gene transcription. The prototypic target genes of Notch signaling contain the basic helix-loop-helix transcription factors Hairy and enhancer of split (Hes) and the Hairy-related transcription (HRT) factor family (Figure 1C) (12).
Regulatory role of Notch signaling during cardiac development
Notch signaling oversees pre-patterning of the cardiac mesoderm. In gastrula-stage embryos, with the assistance of estrogen receptor 9 and GATA 4 transcription factor, Notch regulates timing of heart field specification for early cardiogenesis, which is necessary for normal cardiac development (13). Moreover, in the endocardial lineage, Delta 4, Notch 1, and Notch 4 transcription begins at early gastrulation, while HRT 1 and HRT 2 are expressed in the endocardium or myocardium at various stages of cardiac development (14). Notch 1, Notch 2, and Delta-like 1 are required for the determination of the embryonic left-right axis and proper looping of the heart tube. Taken together, the roles of these factors reveal that Notch signaling is important during the entire process of cardiac development.
The atrioventricular canal is an essential structure for separating atrial and ventricular blood flow. Alterations in Notch signaling lead to abnormal development in the atrioventricular canal. Both bone morphogenetic protein-2 and T-box 2 transcription factor are uniquely expressed during atrioventricular canal formation. Deficiency of bone morphogenetic protein-2 in cardiac myocytes results in atrioventricular canal dysplasia. T-box 2 is a downstream target of bone morphogenetic protein-2 that helps the atrioventricular canal to express chamber-specific myocardial genes. Notch signaling coordinates atrioventricular canal development via Hairy/enhancer of split-related with YRPW motif (Hey) gene regulating the bone morphogenetic protein-2/T-box 2 pathway (15). Following atrioventricular canal formation, a subset of endocardial cells in the atrioventricular canal undergo EMT, which is a required step during early valve development. Matrix metalloproteinase-2 facilitates EMT-generated cell migration into the cardiac cushion. Inactivation of Notch signaling prevents EMT by decreasing matrix metalloproteinase-2 levels (16). VE-cadherin is an endothelial-specific adherens junction protein for maintenance of endothelial cell integrity. Snail is a well-known regulator of adherens junction protein. Notch signaling initiates EMT by Snail-mediated VE-cadherin repression (17). Moreover, Notch signaling also induces EMT by suppressing the vascular endothelial growth factor (VEGF) pathway via downregulating endothelial growth factor receptor (VEGFR) 2 (18). In addition, Notch signaling crosstalks with the PI3K/Akt pathway to phosphorylate endothelial nitric oxide synthase and initiate EMT in the atrioventricular canal (Figure 2) (19).
Figure 2. Summary of cardiac developmental aspects of Notch signaling. A, Notch signaling affects AVC development via BMP 2/Tbx 2 pathway. B, Notch signaling initiates EMT via MMP-2, snail/VE-cadherin, VEGFR2/VEGF, and PI3K/Akt/eNOS pathways. EMT is a required step for valve development. C, Notch signaling promotes ventricular trabecular formation dependent on NRG1-ErbB2/4, Ephrin B2/Eph B4, and BMP-10 signaling pathways. D, Notch signaling stimulates smooth muscle differentiation during OFT development. E, Notch signaling modulates coronary vessel morphogenesis in which the embryonic epicardium actively participates. F, Notch signaling regulates cardiac conduction system function via effects on the atrioventricular node. AVC: atrioventricular canal; BMP-2: bone morphogenetic protein-2; Tbx2: T-box 2; EMT: epithelial-to-mesenchymal transition; MMP-2: matrix metalloproteinase-2; VEGFR 2: vascular endothelial growth factor receptor 2; VEGF: vascular endothelial growth factor; BMP-10: bone morphogenetic protein-10; OFT: outflow tract.
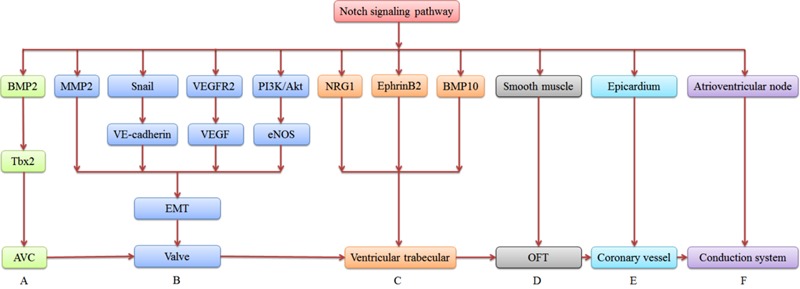
The Neuregulin 1-ErbB 2/4, EphrinB 2/EphrinB 4, and bone morphogenetic protein-10 signaling pathways are required for ventricular trabecular formation. Notch mutants show defective expression of Neuregulin 1, EphrinB 2, and bone morphogenetic protein-10. Notch signaling promotes myocardial trabecular proliferation, differentiation, and maturation by inducing bone morphogenetic protein-10 and EphrinB 2/EphrinB 4 expression (20). The outflow tract is an embryonic structure that connects the ventricles to the ascending aorta and pulmonary arteries, and gives rise to the aortic and pulmonary valves, the aortic arch, and the outflow tract septum. Jagged 1 propagates a wave of smooth muscle cell differentiation during the development of the aortic arch artery, contributing to the assembly of the arterial wall (21). Moreover, Notch 1 and 4 expression is restricted to endothelial cells, and Notch 2 and 3 are mainly observed in vascular smooth muscle cells. Their target genes, Hey 1, Hey 2, and Hey L, are all expressed in vascular smooth muscle cells and regulate aortic arch formation. Combined, this indicates that Notch signaling plays multiple, crucial roles in outflow tract development (Figure 2) (22).
Notch signaling modulates coronary vessel morphogenesis, in which the embryonic epicardium actively participates. Notch signaling elements are differentially expressed throughout the proepicardial-epicardial-coronary transition phases and are required for vessel wall maturation during coronary vessel development (23). Deletion of RBP-Jκ in the epicardium leads to enlarged coronary venous and arterial beds. In cooperation with transforming growth factor-β (TGF-β), Notch signaling regulates coronary smooth muscle differentiation from epicardium-derived cells to assist in the formation of a functional coronary system (Figure 2) (24).
Of interest, Notch signaling regulates cardiac conduction system function, in particular the atrioventricular node. Inhibition of Notch signaling impedes the normal development of the atrioventricular node. This results in selective loss of slow-conducting cells. Activation of Notch signaling can induce ventricular pre-excitation, and this is observed in Wolff-Parkinson-White syndrome, a disease consisting of a fatal arrhythmia (25). Therefore, atrioventricular conduction disorders are closely related to dysfunction in the Notch signaling pathway (Figure 2).
Mutations in Notch signaling associated with congenital heart disease
Notch signaling impacts various aspects of heart development to such a degree that abnormal Notch signaling can induce a series of heart malformations. The most common malformations include bicuspid aortic valve disease and aortic valve calcification. Bicuspid aortic valve disease is caused by abnormal EMT due to Notch 1 mutations. There is a negative correlation between the longevity gene Sirt1 and Notch signaling, and Sirtuin1 is also an important factor for bicuspid aortic valve development (26). Runt-related transcription factor 2 is closely linked to the development of valvular calcification and regulates several osteogenic genes. Notch 1, Hey 1, and Hey 2 repress the function of runt-related transcription factor 2, and this effect describes aortic valve calcification observed when Notch is mutated (27). SRY-box, containing gene 9, prevents calcification occurring with Notch inhibition, suggesting that loss of Notch 1 signaling contributes to aortic valve calcification by an SRY-box containing gene 9-dependent mechanism (28). RBP-Jκ is essential for maintenance of valve homeostasis, and RBP-Jκ deletion in the aortic valve endothelium can trigger valve degenerative disease by upregulation of VEGFR2 and TGF-β1 (Table 1) (29).
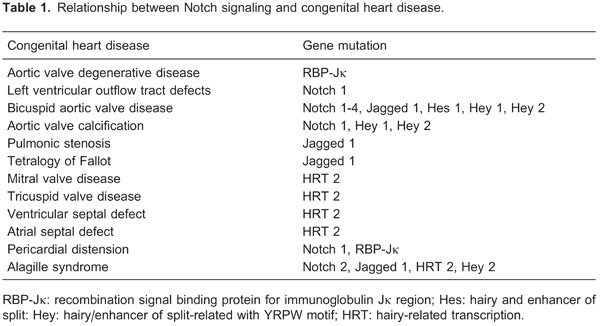
Alagille syndrome is an autosomal dominant genetic disorder. The most frequent Alagille syndrome cardiovascular defect is peripheral pulmonary stenosis, and 13% of Alagille syndrome expresses as tetralogy of Fallot. Mutations in Notch 2, Jagged 1, HRT 2, and Hey 2 loci are associated with Alagille syndrome (Table 1) (30).
In addition, Notch 1 and RBP-Jκ mutations lead to severe pericardial distension. HRT 2 mutations induce tricuspid valve stenosis and regurgitation, mitral valve regurgitation, ventricular septal defect, and secundum atrial septal defect. Notch 1 missense alleles are associated with left ventricular outflow tract defects (31). Jagged 1 mutations result in tetralogy of Fallot or pulmonic stenosis (Table 1).
Facilitating role of Notch signaling in myocardial regeneration
Notch signaling influences tissue formation and morphogenesis by fine-tuning the balance between the progenitor cell pool and the number of differentiating progeny (32). Notch is activated upon co-culture of endothelial progenitor cells with neonatal rat cardiomyocytes, amplifying endothelial progenitor cell differentiation into cardiomyocytes. When γ-secretase is inhibited, the cardiac marker proteins α-sarcomeric actin and troponin T are significantly suppressed in endothelial progenitor cells (33). Simultaneously, cardiomyocyte proliferative capacity is enhanced when immature cardiomyocytes are co-cultured with mesenchymal stromal cells. The mechanism of enhanced proliferative capacity involves Notch 1 activation by Jagged 1 expressed in adjacent mesenchymal stromal cells. The proliferative capability is significantly attenuated when Jagged 1 is silenced (34). Moreover, Notch 1 also regulates cardiac progenitor cells fate. N1ICD and RBP-Jκ form a complex that initiates and upregulates NK2 homeobox 5 in cardiac progenitor cells to promote the formation of cycling myocytes (Figure 3) (6).
Figure 3. Regulatory role of Notch signaling for myocardial regeneration. A, Notch signaling amplifies EPC differentiation into cardiomyocytes through Jagged 1, NICD and Wnts. B, MSCs enhance cardiomyocyte proliferative capacity through Jagged 1 and NICD. C, Notch signaling promotes the differentiation of CPCs into cardiomyocytes through Jagged 1, NICD, RBP-Jκ, and Nkx2.5. D, Notch signaling expands the proportion of CSCs differentiating into cardiomyocytes through Jagged 1, NICD, and Nkx2.5. E, Notch 1 signaling stimulates proliferation of immature cardiomyocytes through Jagged 1, NICD, and cyclin D1. F, Notch signaling activates cell cycle reentry of quiescent cardiomyocytes through NICD, RBP-Jκ, and cyclin D1. EPCs: endothelial progenitor cells; NICD: Notch intracellular domain; Wnts: Wnt signaling; MSCs: mesenchymal stromal cells; CPCs: cardiac progenitor cells; RBP-Jκ: recombination signal binding protein for immunoglobulin Jκ region; Nkx2.5: NK2 homeobox 5; CSCs: cardiac stem cells.
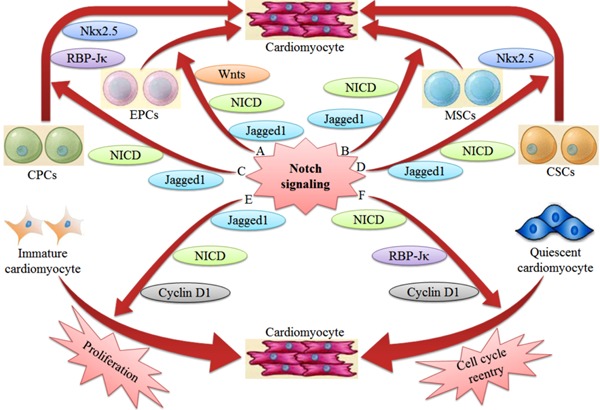
Notch 1 signaling is a critical determinant of cardiac stem cell proliferation and differentiation. N1ICD overexpression in neonatal cardiac progenitor cells significantly expands the proportion of transit-amplifying myocytes. Blocking Notch signaling with a γ-secretase inhibitor results in a 62% reduction in amplifying myocytes and a 54% decrease in myocyte numbers, resulting in life-threatening dilated cardiomyopathy (35). The high levels of Notch 1 and Jagged 1 in immature cardiomyocytes not only protect immature cardiomyocytes from apoptosis, but also markedly stimulate the proliferation of immature cardiomyocytes (36). Cyclin D1 is induced in quiescent cardiomyocytes by the ectopic expression of Notch 2, which translocates to the nucleus and interacts with cyclin-dependent kinases 4 and 6 to phosphorylate retinoblastoma protein and activates cell cycle reentry and progression (37). The upregulation of Notch 1 in the hypertrophic heart controls the adaptive response of the heart to stress, not only limiting the extent of the hypertrophic response but also contributing to cell survival in cardiomyocytes (Figure 3) (38).
Notch signaling is cardioprotective in the ischemic myocardium
Activation of Notch signaling following tissue injury has been documented in various tissues. Notch 1 controls the response to injury in the adult heart by limiting myocyte hypertrophy, enhancing myocyte survival, promoting precursor proliferation, controlling cardiogenic differentiation, and reducing interstitial fibrosis (38). A dynamic Notch 1 signaling response is required to activate the adult epicardium and produce a multipotent cell population that stimulates fibrosis for cardiac injury repair (39). Based on the Akt-mTOR-Stat 3-Notch 1 cascade, Notch 1 signaling elicits cardioprotection against chronic myocardial ischemia (40). The level of hypoxia-inducible factor-1α is upregulated in ischemic or hypoxic myocardium, reducing the damage induced by myocardial ischemia reperfusion injury through inhibition of opening of the mitochondrial permeability transition pore. This effect depends on decreasing reactive oxygen species levels. Notch signaling activates the hypoxia pathway through the Hes 1/signal transducers and activators of transcription 3 (Stat 3) pathway, inducing the transcriptional activation of the hypoxia-inducible factor-1α gene (41). The gradual accumulation of hypoxia-inducible factor-1α in ischemic tissue not only initiates Hes 1 transcription (42), but also interacts with and stabilizes NICD (43). The canonical Notch signaling pathway protects hepatocytes from ischemia reperfusion injury by repressing reactive oxygen species production through Janus kinase 2/Stat 3 signaling (44). Therefore, effective regulation of Notch signaling would be expected to reduce myocardial ischemia reperfusion injury. The levels of Notch 1 and Hes 1 decline in cardiomyocytes during postnatal cardiac development and are activated and restored in the injured adult myocardium. With the stimulation by hepatocyte growth factor, Hes 1 levels stimulate proliferation, providing cardioprotection under the action of the Hepatocyte growth factor/Notch 1/c-Met/Akt network (45). Almost all of the Notch receptors and ligands are present at varying levels in the injured myocardium during post-infarction remodeling, indicating a regulatory role for Notch signaling in the functional recovery of the ischemic myocardium (46). Notch 1 activated bone marrow-derived cells also mediate cardiac repair, which not only stimulates bone marrow-derived cell recruitment into myocardial infarcts, but also promotes neovascularization into the infarct border area. Transplanted mesenchymal stem cells that overexpress NICD decrease infarct size and improve cardiac function, demonstrating that activated Notch 1 signaling accelerates the cardiac functional recovery following myocardial infarction (47).
Dual role of Notch signaling in the induction of angiogenesis
Several components of Notch signaling, including Notch 1, Notch 4, Jagged 1, and Delta-like 4, are expressed in vascular endothelial cells (48). During early embryogenesis, Notch signaling modulates the migration of angioblasts from the lateral mesoderm towards the dorsal aorta, inducing their endothelial cell specification. During later stages, Notch signaling controls endothelial cell specification into arterial or venous identities. Moreover, Notch 1 is required for arterial endothelium differentiation and vessel wall maturation during coronary vessel development (23). Activated Notch 1 mediates vascular smooth muscle proliferation and neointimal formation through Hey 2 expression following vascular injury (49). The increased secretion of VEGF from vascular endothelial cells induces angiogenesis to meet the increasing needs of ischemic tissue perfusion during ischemia or hypoxia. Delta-like 4/Notch is essential for the proper angiogenic response by regulating sprouting angiogenesis and coordinating the interaction between inflammation and angiogenesis (50). Delta-like 4 and Jagged 1 serve opposing functions in the regulation of neovascularization. With the stimulation of VEGF, high levels of Delta-like 4 are expressed in endothelial cells to inhibit excessive angiogenesis by downregulating VEGFRs (51). Alternatively, Jagged 1 antagonizes Delta-like 4-mediated restraint during neovascularization to promote new vessel growth (52). Notch signaling, therefore, serves dual roles in the induction of angiogenesis. N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (DAPT), a γ-secretase inhibitor, promotes angiogenesis by enhancing the responsiveness of endothelial cells to VEGF (53). Influenced by diabetes, the sprouting capability of aortic endothelial cells is reduced. While DAPT permits enhanced proliferation, migration, and sprouting of aortic endothelial cells, combining VEGF and DAPT treatment for ischemic tissue damage can increase blood vessel density and improve tissue perfusion. This indicates that modulating Notch signaling enhances neovascularization and perfusion recovery for ischemic disease (54). In contrast to these results, DAPT has also been shown to inhibit VEGF-induced endothelial cell proliferation, migration, and survival, resulting in reduced neovascularization. N1ICD overexpression reverses DAPT inhibition by stimulating crosstalk with the PI3K/Akt signaling pathway (55). Therefore, an in-depth understanding of the regulatory mechanisms of Notch signaling in neovascularization has great significance for the functional repair of the injured myocardium.
Inhibitory effect of Notch signaling on CMT
Cardiac fibroblasts are a major cell type in the heart, accounting for 60-70% of the total number of heart cells. Under physiological conditions, cardiac fibroblasts produce an appropriate amount of extracellular matrix proteins, such as collagen, to maintain structural integrity and normal functioning of the heart. The myocardium responds to injury by depositing excessive collagen to form a myocardial fibrous scar. This fibrotic response not only impedes ventricular contraction and relaxation, but also causes non-synchronous contraction due to myocardial electrical uncoupling. All of these responses are important in heart failure and can cause malignant arrhythmias, resulting in sudden cardiac death. Therefore, limiting the myocardial fibrotic response may prove to be an important therapeutic approach for heart failure. Recent studies have shown that Notch participates in the occurrence and progression of tissue fibrosis, as Notch gene induction can stimulate fibrosis by activating alpha-smooth muscle actin (α-SMA) transcription and promoting myofibroblast transformation (56). Inhibition of Notch signaling by DAPT can significantly reduce profibrotic factor expression, including interleukin (IL)-4, IL-6, and connective tissue growth factor. It can also reduce the number of myofibroblasts, decrease hydroxyproline content, and prevent or even regress fibrosis (57). Blocking Notch signaling has been shown to inhibit fibrosis in the skin, liver, and kidney, indicating that these effects cross organ systems (58,59).
Excessive collagen deposition is regulated by myofibroblasts, which are a specialized fibroblast type with a high capacity for collagen synthesis and sustained expression of α-SMA. CMT is a critical event in the initiation of myocardial fibrosis that can further impair cardiac function following myocardial injury. Notch signaling regulates myofibroblast transformation. NICD overexpression can induce the differentiation of lung fibroblasts into myofibroblasts (60). Activated Notch signaling also facilitates myofibroblast differentiation from alveolar epithelial cells via the TGF-β/Smad3 pathway (59). However, Notch 3 overexpression causes repression of TGF-β1, facilitating fibroblast differentiation into myofibroblasts. Notch signaling is also involved in CMT. Notch receptors 1, 2, 3, and 4 are all expressed in cardiac fibroblasts, and the expression of Notch 1, 3, and 4 is decreased in TGF-β1-induced CMT. DAPT promotes CMT in a time-dependent manner, leading to an increase in α-SMA expression and collagen synthesis (8). Thus, Notch signaling may reduce myocardial fibrosis following myocardial injury by negatively regulating the degree of CMT. However, the specific mechanisms of CMT warrant further investigation.
Conclusions and perspectives
Notch signaling is highly relevant for proper myocardial function and response to injury. Through a complex regulatory network (Figure 2), Notch signaling is involved in almost every aspect of cardiac development. However, the mechanisms whereby Notch signaling functions in cardiac cardiogenesis have not been fully elucidated, and warrant further study.
Congenital heart disease is the most common of human birth defects, and abnormal Notch signaling is a major contributor (Table 1). Therefore, developing prenatal diagnostic techniques targeted at detecting Notch signaling deficits will advance our ability to detect impaired fetal development. This ability to identify and reduce the occurrence of congenital heart disease at a very early stage will increase the number of viable deliveries.
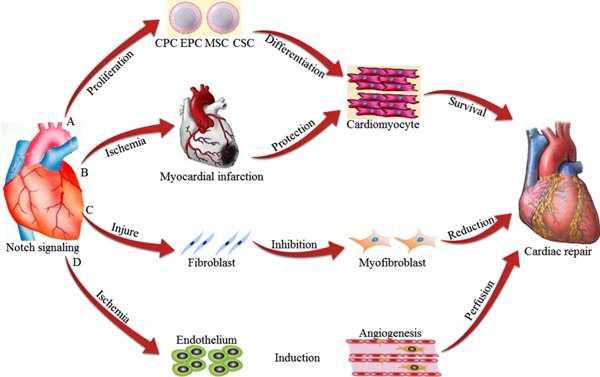
Cardiovascular disease is a significant threat to human health, and myocardial repair capacity is currently extremely limited. Enhancing our ability to repair the injured myocardium and improve cardiac function following infarction remains a major clinical challenge. Notch signaling regulates stem cell differentiation into cardiomyocytes (Figure 3), providing a new opportunity for the regeneration and repair of the myocardium. In addition, Notch signaling interacts with other signaling pathways to ameliorate myocardial dysfunction, inducing angiogenesis to enhance myocardial perfusion, and inhibiting CMT to reduce additional damage resulting from cardiac repair (Figure 4). Overall, the combined functions of Notch signaling mediate functional repair of the myocardium during injury, although the specific molecular mechanisms remain to be clarified. Developing new drugs based on Notch signaling would be expected to create novel options for the treatment of cardiovascular disease.
Figure 4. Cardiac repair mediated through Notch signaling. A, Notch signaling promotes stem cell differentiation into cardiomyocytes. B, Notch signaling has protective effects on the ischemic myocardium. C, Notch signaling negatively regulates CMT. D, Notch signaling induces angiogenesis. CMT: cardiac fibroblast to myofibroblast transformation; CPC: cardiac progenitor cell; EPC: endothelial progenitor cell; MSC: mesenchymal stromal cell; CSC: cardiac stem cell.
Acknowledgments
We thank Medjaden Bioscience Limited for assistance in the preparation of this manuscript. Research supported in part by grants from the National Natural Science Foundation of China (#81260024), the Natural Science Foundation of Jiangxi Province (#20122BAB205026, #20132BAB205033) and the Graduate Innovation Fund of Jiangxi Province (#YC2011-B013).
Footnotes
First published online December 12, 2013.
References
- 1.Borggrefe T, Oswald F. The Notch signaling pathway: Transcriptional regulation at Notch target genes. Cell Mol Life Sci. 2009;66:1631–1646. doi: 10.1007/s00018-009-8668-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tien AC, Rajan A, Bellen HJ. A Notch updated. J Cell Biol. 2009;184:621–629. doi: 10.1083/jcb.200811141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwanbeck R, Martini S, Bernoth K, Just U. The Notch signaling pathway: Molecular basis of cell context dependency. Eur J Cell Biol. 2011;90:572–581. doi: 10.1016/j.ejcb.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 4.Penton AL, Leonard LD, Spinner NB. Notch signaling in human development and disease. Semin Cell Dev Biol. 2012;23:450–457. doi: 10.1016/j.semcdb.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pompa JL de la, Epstein JA. Coordinating tissue interactions: Notch signaling in cardiac development and disease. Dev Cell. 2012;22:244–254. doi: 10.1016/j.devcel.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boni A, Urbanek K, Nascimbene A, Hosoda T, Zheng H, Delucchi F, et al. Notch1 regulates the fate of cardiac progenitor cells. Proc Natl Acad Sci U S A. 2008;105:15529–15534. doi: 10.1073/pnas.0808357105. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 7.Gude N, Sussman M. Notch signaling and cardiac repair. J Mol Cell Cardiol. 2012;52:1226–1232. doi: 10.1016/j.yjmcc.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fan YH, Dong H, Pan Q, Cao YJ, Li H, Wang HC. Notch signaling may negatively regulate neonatal rat cardiac fibroblast-myofibroblast transformation. Physiol Res. 2011;60:739–748. doi: 10.33549/physiolres.932149. [DOI] [PubMed] [Google Scholar]
- 9.Kume T. Ligand-dependent Notch signaling in vascular formation. Adv Exp Med Biol. 2012;727:210–222. doi: 10.1007/978-1-4614-0899-4_16. [DOI] [PubMed] [Google Scholar]
- 10.Wang MM. Notch signaling and Notch signaling modifiers. Int J Biochem Cell Biol. 2011;43:1550–1562. doi: 10.1016/j.biocel.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.D'Souza B, Miyamoto A, Weinmaster G. The many facets of Notch ligands. Oncogene. 2008;27:5148–5167. doi: 10.1038/onc.2008.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Molle V, Campagna S, Bessin Y, Ebran N, Saint N, Molle G. First evidence of the pore-forming properties of a keratin from skin mucus of rainbow trout (Oncorhynchus mykiss, formerly Salmo gairdneri) Biochem J. 2008;411:33–40. doi: 10.1042/BJ20070801. [DOI] [PubMed] [Google Scholar]
- 13.Miazga CM, McLaughlin KA. Coordinating the timing of cardiac precursor development during gastrulation: A new role for Notch signaling. Dev Biol. 2009;333:285–296. doi: 10.1016/j.ydbio.2009.06.040. [DOI] [PubMed] [Google Scholar]
- 14.Pompa JL de la. Notch signaling in cardiac development and disease. Pediatr Cardiol. 2009;30:643–650. doi: 10.1007/s00246-008-9368-z. [DOI] [PubMed] [Google Scholar]
- 15.Rutenberg JB, Fischer A, Jia H, Gessler M, Zhong TP, Mercola M. Developmental patterning of the cardiac atrioventricular canal by Notch and Hairy-related transcription factors. Development. 2006;133:4381–4390. doi: 10.1242/dev.02607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischer A, Steidl C, Wagner TU, Lang E, Jakob PM, Friedl P, et al. Combined loss of Hey1 and HeyL causes congenital heart defects because of impaired epithelial to mesenchymal transition. Circ Res. 2007;100:856–863. doi: 10.1161/01.RES.0000260913.95642.3b. [DOI] [PubMed] [Google Scholar]
- 17.Niessen K, Fu Y, Chang L, Hoodless PA, McFadden D, Karsan A. Slug is a direct Notch target required for initiation of cardiac cushion cellularization. J Cell Biol. 2008;182:315–325. doi: 10.1083/jcb.200710067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Noseda M, McLean G, Niessen K, Chang L, Pollet I, Montpetit R, et al. Notch activation results in phenotypic and functional changes consistent with endothelial-to-mesenchymal transformation. Circ Res. 2004;94:910–917. doi: 10.1161/01.RES.0000124300.76171.C9. [DOI] [PubMed] [Google Scholar]
- 19.Chang AC, Fu Y, Garside VC, Niessen K, Chang L, Fuller M, et al. Notch initiates the endothelial-to-mesenchymal transition in the atrioventricular canal through autocrine activation of soluble guanylyl cyclase. Dev Cell. 2011;21:288–300. doi: 10.1016/j.devcel.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 20.Grego-Bessa J, Luna-Zurita L, Monte G del, Bolos V, Melgar P, Arandilla A, et al. Notch signaling is essential for ventricular chamber development. Dev Cell. 2007;12:415–429. doi: 10.1016/j.devcel.2006.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manderfield LJ, High FA, Engleka KA, Liu F, Li L, Rentschler S, et al. Notch activation of Jagged1 contributes to the assembly of the arterial wall. Circulation. 2012;125:314–323. doi: 10.1161/CIRCULATIONAHA.111.047159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.High FA, Zhang M, Proweller A, Tu L, Parmacek MS, Pear WS, et al. An essential role for Notch in neural crest during cardiovascular development and smooth muscle differentiation. J Clin Invest. 2007;117:353–363. doi: 10.1172/JCI30070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Monte G del, Casanova JC, Guadix JA, MacGrogan D, Burch JB, Perez-Pomares JM, et al. Differential Notch signaling in the epicardium is required for cardiac inflow development and coronary vessel morphogenesis. Circ Res. 2011;108:824–836. doi: 10.1161/CIRCRESAHA.110.229062. [DOI] [PubMed] [Google Scholar]
- 24.Grieskamp T, Rudat C, Ludtke TH, Norden J, Kispert A. Notch signaling regulates smooth muscle differentiation of epicardium-derived cells. Circ Res. 2011;108:813–823. doi: 10.1161/CIRCRESAHA.110.228809. [DOI] [PubMed] [Google Scholar]
- 25.Rentschler S, Harris BS, Kuznekoff L, Jain R, Manderfield L, Lu MM, et al. Notch signaling regulates murine atrioventricular conduction and the formation of accessory pathways. J Clin Invest. 2011;121:525–533. doi: 10.1172/JCI44470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sciacca S, Pilato M, Mazzoccoli G, Pazienza V, Vinciguerra M. Anti-correlation between longevity gene SirT1 and Notch signaling in ascending aorta biopsies from patients with bicuspid aortic valve disease. Heart Vessels. 2013;28:268–275. doi: 10.1007/s00380-012-0238-5. [DOI] [PubMed] [Google Scholar]
- 27.Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, et al. Mutations in Notch1 cause aortic valve disease. Nature. 2005;437:270–274. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
- 28.Acharya A, Hans CP, Koenig SN, Nichols HA, Galindo CL, Garner HR, et al. Inhibitory role of Notch1 in calcific aortic valve disease. PloS One. 2011;6:e27743. doi: 10.1371/journal.pone.0027743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Z, Feng L, Wang CM, Zheng QJ, Zhao BJ, Yi W, et al. Deletion of rbp-j in adult mice leads to the onset of aortic valve degenerative diseases. Mol Biol Rep. 2012;39:3837–3845. doi: 10.1007/s11033-011-1162-y. [DOI] [PubMed] [Google Scholar]
- 30.Kamath BM, Bauer RC, Loomes KM, Chao G, Gerfen J, Hutchinson A, et al. Notch2 mutations in Alagille syndrome. J Med Genet. 2012;49:138–144. doi: 10.1136/jmedgenet-2011-100544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Riley MF, McBride KL, Cole SE. Notch1 missense alleles associated with left ventricular outflow tract defects exhibit impaired receptor processing and defective emt. Biochim Biophys Acta. 2011;1812:121–129. doi: 10.1016/j.bbadis.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fre S, Huyghe M, Mourikis P, Robine S, Louvard D, Artavanis-Tsakonas S. Notch signals control the fate of immature progenitor cells in the intestine. Nature. 2005;435:964–968. doi: 10.1038/nature03589. [DOI] [PubMed] [Google Scholar]
- 33.Koyanagi M, Bushoven P, Iwasaki M, Urbich C, Zeiher AM, Dimmeler S. Notch signaling contributes to the expression of cardiac markers in human circulating progenitor cells. Circ Res. 2007;101:1139–1145. doi: 10.1161/CIRCRESAHA.107.151381. [DOI] [PubMed] [Google Scholar]
- 34.Sassoli C, Pini A, Mazzanti B, Quercioli F, Nistri S, Saccardi R, et al. Mesenchymal stromal cells affect cardiomyocyte growth through juxtacrine Notch-1/Jagged-1 signaling and paracrine mechanisms: Clues for cardiac regeneration. J Mol Cell Cardiol. 2011;51:399–408. doi: 10.1016/j.yjmcc.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 35.Urbanek K, Cabral-da-Silva MC, Ide-Iwata N, Maestroni S, Delucchi F, Zheng H, et al. Inhibition of Notch1-dependent cardiomyogenesis leads to a dilated myopathy in the neonatal heart. Circ Res. 2010;107:429–441. doi: 10.1161/CIRCRESAHA.110.218487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Collesi C, Zentilin L, Sinagra G, Giacca M. Notch1 signaling stimulates proliferation of immature cardiomyocytes. J Cell Biol. 2008;183:117–128. doi: 10.1083/jcb.200806091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Campa VM, Gutierrez-Lanza R, Cerignoli F, Diaz-Trelles R, Nelson B, Tsuji T, et al. Notch activates cell cycle reentry and progression in quiescent cardiomyocytes. J Cell Biol. 2008;183:129–141. doi: 10.1083/jcb.200806104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Croquelois A, Domenighetti AA, Nemir M, Lepore M, Rosenblatt-Velin N, Radtke F, et al. Control of the adaptive response of the heart to stress via the Notch1 receptor pathway. J Exp Med. 2008;205:3173–3185. doi: 10.1084/jem.20081427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Russell JL, Goetsch SC, Gaiano NR, Hill JA, Olson EN, Schneider JW. A dynamic Notch injury response activates epicardium and contributes to fibrosis repair. Circ Res. 2011;108:51–59. doi: 10.1161/CIRCRESAHA.110.233262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ge W, Ren J. mTOR-STAT3-Notch signalling contributes to ALDH2-induced protection against cardiac contractile dysfunction and autophagy under alcoholism. J Cell Mol Med. 2012;16:616–626. doi: 10.1111/j.1582-4934.2011.01347.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee JH, Suk J, Park J, Kim SB, Kwak SS, Kim JW, et al. Notch signal activates hypoxia pathway through hes1-dependent src/signal transducers and activators of transcription 3 pathway. Mol Cancer Res. 2009;7:1663–1671. doi: 10.1158/1541-7786.MCR-09-0191. [DOI] [PubMed] [Google Scholar]
- 42.Chen J, Imanaka N, Griffin JD. Hypoxia potentiates Notch signaling in breast cancer leading to decreased e-cadherin expression and increased cell migration and invasion. Br J Cancer. 2010;102:351–360. doi: 10.1038/sj.bjc.6605486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qiang L, Wu T, Zhang HW, Lu N, Hu R, Wang YJ, et al. Hif-1alpha is critical for hypoxia-mediated maintenance of glioblastoma stem cells by activating Notch signaling pathway. Cell Death Differ. 2012;19:284–294. doi: 10.1038/cdd.2011.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu HC, Qin HY, He F, Wang L, Fu W, Liu D, et al. Canonical Notch pathway protects hepatocytes from ischemia/reperfusion injury in mice by repressing reactive oxygen species production through jak2/stat3 signaling. Hepatology. 2011;54:979–988. doi: 10.1002/hep.24469. [DOI] [PubMed] [Google Scholar]
- 45.Gude NA, Emmanuel G, Wu W, Cottage CT, Fischer K, Quijada P, et al. Activation of Notch-mediated protective signaling in the myocardium. Circ Res. 2008;102:1025–1035. doi: 10.1161/CIRCRESAHA.107.164749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oie E, Sandberg WJ, Ahmed MS, Yndestad A, Laerum OD, Attramadal H, et al. Activation of Notch signaling in cardiomyocytes during post-infarction remodeling. Scand Cardiovasc J. 2010;44:359–366. doi: 10.3109/14017431.2010.511256. [DOI] [PubMed] [Google Scholar]
- 47.Li Y, Hiroi Y, Ngoy S, Okamoto R, Noma K, Wang CY, et al. Notch1 in bone marrow-derived cells mediates cardiac repair after myocardial infarction. Circulation. 2011;123:866–876. doi: 10.1161/CIRCULATIONAHA.110.947531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bridges E, Oon CE, Harris A. Notch regulation of tumor angiogenesis. Future Oncol. 2011;7:569–588. doi: 10.2217/fon.11.20. [DOI] [PubMed] [Google Scholar]
- 49.Li Y, Takeshita K, Liu PY, Satoh M, Oyama N, Mukai Y, et al. Smooth muscle Notch1 mediates neointimal formation after vascular injury. Circulation. 2009;119:2686–2692. doi: 10.1161/CIRCULATIONAHA.108.790485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Al Haj Zen A, Oikawa A, Bazan-Peregrino M, Meloni M, Emanueli C, Madeddu P. Inhibition of delta-like-4-mediated signaling impairs reparative angiogenesis after ischemia. Circ Res. 2010;107:283–293. doi: 10.1161/CIRCRESAHA.110.221663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harrington LS, Sainson RC, Williams CK, Taylor JM, Shi W, Li JL, et al. Regulation of multiple angiogenic pathways by DII4 and Notch in human umbilical vein endothelial cells. Microvasc Res. 2008;75:144–154. doi: 10.1016/j.mvr.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 52.Benedito R, Roca C, Sorensen I, Adams S, Gossler A, Fruttiger M, et al. The Notch ligands DII4 and Jagged1 have opposing effects on angiogenesis. Cell. 2009;137:1124–1135. doi: 10.1016/j.cell.2009.03.025. [DOI] [PubMed] [Google Scholar]
- 53.Cao L, Arany PR, Wang YS, Mooney DJ. Promoting angiogenesis via manipulation of vegf responsiveness with Notch signaling. Biomaterials. 2009;30:4085–4093. doi: 10.1016/j.biomaterials.2009.04.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cao L, Arany PR, Kim J, Rivera-Feliciano J, Wang YS, He Z, et al. Modulating Notch signaling to enhance neovascularization and reperfusion in diabetic mice. Biomaterials. 2010;31:9048–9056. doi: 10.1016/j.biomaterials.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Takeshita K, Satoh M, Ii M, Silver M, Limbourg FP, Mukai Y, et al. Critical role of endothelial Notch1 signaling in postnatal angiogenesis. Circ Res. 2007;100:70–78. doi: 10.1161/01.RES.0000254788.47304.6e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aoyagi-Ikeda K, Maeno T, Matsui H, Ueno M, Hara K, Aoki Y, et al. Notch induces myofibroblast differentiation of alveolar epithelial cells via transforming growth factor-{beta}-smad3 pathway. Am J Respir Cell Mol Biol. 2011;45:136–144. doi: 10.1165/rcmb.2010-0140oc. [DOI] [PubMed] [Google Scholar]
- 57.Dees C, Zerr P, Tomcik M, Beyer C, Horn A, Akhmetshina A, et al. Inhibition of Notch signaling prevents experimental fibrosis and induces regression of established fibrosis. Arthritis Rheum. 2011;63:1396–1404. doi: 10.1002/art.30254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen Y, Zheng S, Qi D, Zheng S, Guo J, Zhang S, et al. Inhibition of Notch signaling by a gamma-secretase inhibitor attenuates hepatic fibrosis in rats. PloS One. 2012;7:e46512. doi: 10.1371/journal.pone.0046512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bielesz B, Sirin Y, Si H, Niranjan T, Gruenwald A, Ahn S, et al. Epithelial Notch signaling regulates interstitial fibrosis development in the kidneys of mice and humans. J Clin Invest. 2010;120:4040–4054. doi: 10.1172/JCI43025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu T, Hu B, Choi YY, Chung M, Ullenbruch M, Yu H, et al. Notch1 signaling in FIZZ1 induction of myofibroblast differentiation. Am J Pathol. 2009;174:1745–1755. doi: 10.2353/ajpath.2009.080618. [DOI] [PMC free article] [PubMed] [Google Scholar]