

Abstract
Membranous nephropathy (MN), characterized by the presence of diffuse thickening of the glomerular basement membrane and subepithelial in situ immune complex disposition, is the most common cause of idiopathic nephrotic syndrome in adults, with an incidence of 5-10 per million per year. A number of studies have confirmed the relevance of several experimental insights to the pathogenesis of human MN, but the specific biomarkers of MN have not been fully elucidated. As a result, our knowledge of the alterations in histone methylation in MN is unclear. We used chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq) to analyze the variations in a methylated histone (H3K9me3) in peripheral blood mononuclear cells from 10 MN patients and 10 healthy subjects. There were 108 genes with significantly different expression in the MN patients compared with the normal controls. In MN patients, significantly increased activity was seen in 75 H3K9me3 genes, and decreased activity was seen in 33, compared with healthy subjects. Five positive genes, DiGeorge syndrome critical region gene 6 (DGCR6), sorting nexin 16 (SNX16), contactin 4 (CNTN4), baculoviral IAP repeat containing 3 (BIRC3), and baculoviral IAP repeat containing 2 (BIRC2), were selected and quantified. There were alterations of H3K9me3 in MN patients. These may be candidates to help explain pathogenesis in MN patients. Such novel findings show that H3K9me3 may be a potential biomarker or promising target for epigenetic-based MN therapies.
Keywords: ChIP-seq, Epigenetics, H3K9me3, Membranous nephropathy
Introduction
Membranous nephropathy (MN), characterized by the presence of diffuse thickening of the glomerular basement membrane and subepithelial in situ immune complex disposition, shows a set of distinct morphologic features in glomeruli, such as glomerular basement membrane spikes, that form along the periphery of all glomerular capillary loops, and subepithelial electron-dense deposits corresponding to granular IgG staining (1). MN occurs in all age groups and races and both sexes worldwide. Recently, the association of epigenetic modifications with MN has attracted little interest, but study of these relationships may uncover new mechanisms to explain the MN epiphenotype.
Epigenetics is the study of inherited modifications in gene expression and phenotype caused by mechanisms other than changes in the underlying DNA sequence. Unlike genetic polymorphisms, non-genetic partial and reversible DNA modifications can occur (2). Histone lysine methylation, which is one of the important epigenetic modifications, is believed to be part of a histone code and has been implicated in multiple biological processes including gene activation, silencing, X-chromosome inactivation, DNA repair, cell cycle control, and DNA methylation (3). Methylation occurs at five major lysine residues located within the amino-terminal histone tails (H3K4, H3K9, H3K27, H3K36, H4K20), and one lysine residue found within the globular histone domain (H3K79). Moreover, these lysines can be mono-, di- or trimethylated (4). Among the various histone lysine methylation patterns, this study focused on methylation at lysine 9 of histone H3 (H3K9), owing to its association with condensed and repressed chromatin (5). Trimethylated H3K9 (H3K9me3) is a posttranslational modification, and is commonly linked with both facultative and constitutive heterochromatin formation as well as transcriptional repression (6). Aberrant alterations in histone lysine methylation patterns that change chromatin structure could lead to dysregulated gene transcription and disease progression. Therefore, it is significant to investigate whether there is aberrant H3K9me3 in the nonphysiological MN environment. This may provide important clues to assist in the development of new treatments for MN as well as to give a deeper understanding of the pathogenesis of MN.
High-throughput techniques are very efficient methods to analyze large-scale gene expression and to simultaneously investigate the complex molecular basis of pathological processes at the genomic level (7). As one of the approaches, ChIP-seq enables the genome wide identification of binding sites of transcription factors and other DNA-binding proteins (8). With the emergence of high-throughput sequencing platforms like Illumina Genome Analyzer and SOLiD, and the availability of ChIP-grade antibodies, ChIP-seq has become one of the most widely used methods for determining functional elements in the genome (9). In general, when compared with ChIP-chip, ChIP-seq has some advantages, such as higher signal/noise ratios, lower cost, and smaller amounts of ChIP DNA for genome-wide analysis (10).
The understanding of cellular and molecular mechanisms involved in the pathogenesis of MN has come from studies carried out in the Heymann nephritis model of MN in the rat, which closely resembles the clinical and pathologic features of the human disease (11). A number of studies have confirmed the relevance of several experimental insights to the pathogenesis of human MN, but the specific biomarkers of MN have not been fully elucidated (7). Hence, in this study, we adopted ChIP-seq technology to profile and compare the variations in H3K9me3 at the genome-wide level in peripheral blood mononuclear cells (PBMCs) from MN patients and healthy controls to gain a better understanding of the pathogenic mechanisms in MN.
Material and Methods
Human subjects
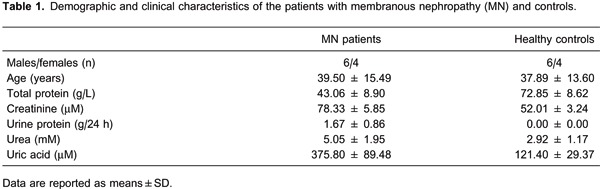
Twenty subjects were enrolled in the study, which included 10 MN patients and 10 healthy volunteers (Table 1). The diagnosis of MN was confirmed through pathological diagnosis and the presence of other clinical evidence. All patients in this study presenting to the Nephrology Department of the 181st Hospital had renal biopsies positive for MN and were diagnosed to be free of any other complications, active infections, diabetes mellitus as well as other diseases. Age-, race-, and gender-matched healthy controls were recruited by advertising. The control group was comprised of healthy volunteers diagnosed to be without any inflammatory diseases or conditions. Urine protein, uric acid and urea were higher in MN patients, but total protein was lower. The Ethics Committee of the 181st Hospital approved the study; peripheral blood samples were obtained with informed consent from all participating individuals.
Isolation of PBMCs
Blood samples were obtained from MN patients (n=10) and normal healthy donors (n=10). Blood (5 mL per subject) was diluted with equal volumes of phosphate-buffered saline (PBS). An equal volume of diluted blood was overlaid on Ficoll-Paque Plus in a 1:1 ratio and centrifuged at 2700 g for 25 min at 25°C. The PBMC layer was harvested and washed twice with PBS to remove plasma and Ficoll. We analyzed the total number of PBMCs using flow cytometry (Beckman Coulter, USA), the final concentration of PBMCs was adjusted to 1×107 cells/mL. These samples were then stored at -80°C until assayed.
Chromatin immunoprecipitation
The ChIP was performed according to described protocols (Epigentek, USA), with some modifications. The preparation of sheared chromatin (cell fixation, enzymatic shearing) was performed following the product manufacturer's instructions (Epigentek). Briefly, cells were fixed with 1% formaldehyde in flasks at room temperature for 10 min and subsequently washed with ice-cold PBS before a 0.1-M glycine solution was added to stop the fixation. The cells were scraped from the flask and homogenized with an ice-cold Dounce homogenizer (Active Motif, USA) to release the nuclei. The nuclei were then collected and resuspended in nucleus lysis buffer. The lysate was incubated with anti-K9 trimethylated histone H3 antibody (Epigentek) at 4°C overnight. The immune complex was treated with proteinase K for 2 h at 65°C and DNA was purified with a QIAquick PCR purification kit (Qiagen, USA).
Next-generation sequencing
All standard protocols for Illumina sequence preparation, sequencing, and quality control were provided by Illumina (USA). In short, DNA recovered from a conventional ChIP procedure was quantified using the QuantiFluor fluorometer (Promega, USA). The DNA integrity was verified using the Agilent Bioanalyzer 2100 (Agilent, USA). The DNA was then processed, including end repair, adaptor ligation, and size selection, using an Illumina sample prep kit following the manufacturer's instructions (Illumina). Final DNA libraries were validated and sequenced at 75 bp per sequence read, using an Illumina GAIIx sequencer at a depth of approximately 30 million sequences per sample.
ChIP-seq analysis
ChIP DNA-end repairing, adaptor ligation, and amplification were performed as described earlier (12). Fragments of about 100 bp (without linkers) were isolated from agarose gels and used for sequencing with a Solexa/Illumina 2G genetic analyzer (CD Genomics, USA). Sequencing tags were aligned to the bosTau4 Oct 2007 release of the reference genome using SOAP 2.21 (BGI, China) and Bowtie, an ultrafast memory-efficient short-read aligner (13). We considered those tags that aligned uniquely with less than two mismatches. For enriched-region (peak) identification (peak calling), we used the Model-based Analysis of ChIP-seq (MACS) algorithm (14) with the MACS version 1.4.0 software (BGI). A simple yet effective technique for the analysis of eukaryotes, MACS was designed to identify transcription factor binding sites and histone modification-enriched regions in ChIP-seq data sets, with or without control samples (15). Sequence reads that map to multiple sites in the human genome were removed. The MACS program was used to determine enriched H3K9me3 peaks using healthy volunteers as control. The parameters for MACS were: effective genome size=2.70e+09; tag size=49; model fold=10.30; P value cutoff=0.05. Peaks were displayed through the UCSC Genome Browser (http://genome.ucsc.edu/). The enriched peaks were annotated with the gene annotation using AmiGO 1.8. AmiGO is accessible online at the Gene Ontology (GO) website (http://www.geneontology.org/) for users to obtain the data provided by the GO Consortium (16). MEME 4.7.0 was used for motif discovery in the present study.
Results
ChIP-seq
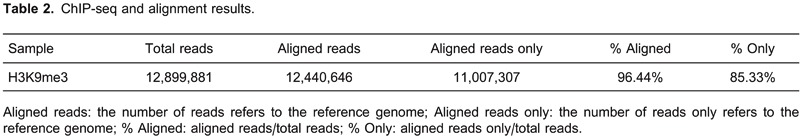
Using specific antibodies, we performed a ChIP-seq on two samples: histone H3 lysine 9 trimethylation of PBMCs from healthy controls and from MN patients. We used Bowtie (USA), an ultrafast memory-efficient short read aligner (17), to align sequencing reads to the reference genome, and the alignment results are presented in Table 2. All ChIP-seq data aligned well with the reference genome. This process resulted in a total of 12,899,881 reads and 12,440,646 (96.44%) aligned reads, covering 539,358,043 bases.
Genome-wide distributions of histone H3 lysine 9 trimethylation of PBMCs from MN patients
MACS 1.4.0 was used for enriched region (peak) identification, with the arguments set as follows: bandwidth=273, model fold=10.30, P value cutoff=0.05. The range for calculating the regional lambda was 5000 bp. The ChIP-seq for the MN sample generated 217 enriched regions (peaks), total length was 192,047 bp. The peak distribution of the ChIP-seq reads of H3K9me3 in MN patients exhibited a typical priority distribution pattern (Figures 1 and 2), with peak distances ranging from 150 to 200 bp.
Figure 1. ChIP-seq peak distribution (distance) of H3K9me3 from membranous nephropathy patients.
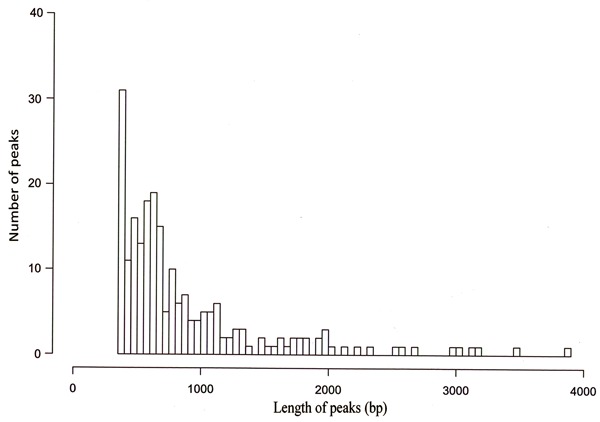
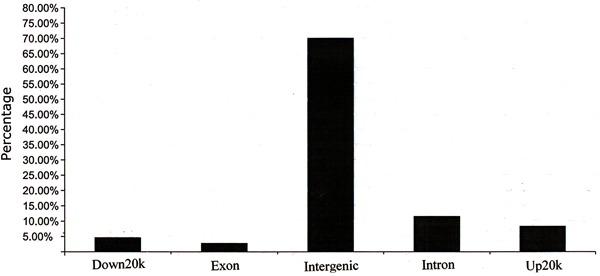
Figure 2. Genome-wide distribution of peaks relative to annotated genes.
GO analysis of peaks relative to annotated genes
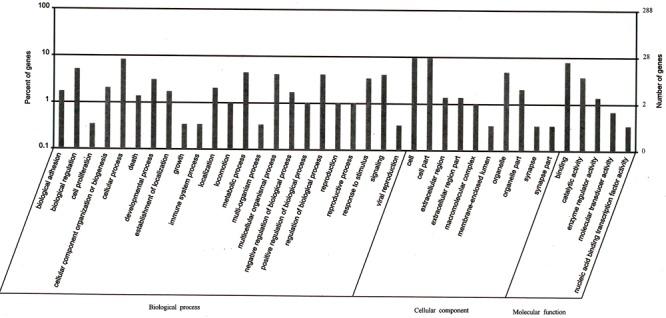
To further understand the functions of the annotated genes related to peaks, they were functionally classified using GO terminology. In terms of the GO database, the differentially expressed proteins encoded by these genes were divided into three groups: cellular components, molecular function, and biological processes. The 86 genes included in the GO analysis are shown in Figure 3. From the cellular component categories, we could determine the subcellular location of 28 annotated gene products that were cellular components; 14 located on organelles. Details of the cellular component categories are available in Supplementary Table S1. Molecular function ontology revealed a binding function in 25 and catalytic activity in 12. Genes with enzyme-regulatory activity comprised 13.3% of the total. Details of the molecular function ontology are available in Supplementary Table S2. A total of 23 genes were involved in cellular processes and 15 involved in biological regulation. Moreover, there were 13 genes involved in metabolic processes, 10 involved in regulation of biological processes, and 5 involved in component organization. Details of the biological process ontology are available in Supplementary Table S3.
Figure 3. Distribution of peaks relative genes by Gene Ontology (GO) analysis (AmiGO 1.8). The lateral axis represents the GO terminology. The left vertical axis represents the proportion of the related genes. The right vertical axis represents the number of the related genes.
Motif analysis of peaks relative to annotated genes
The detection of functional DNA motifs has been greatly facilitated by the availability of high-throughput functional genomic data that provide direct or indirect evidence for gene regulation (18). MEME is an efficient de novo motif discovery tool for large-scale genomic sequence data, the motif is believable when E-values <0.05. Using MEME 4.7.0, five motifs were searched for the ChIP-seq dataset (Table 3, Figure 4). A de novo search for H3K9 binding motifs indicated that histone modification at different locations changed the sequence-based binding preference of histone H3.
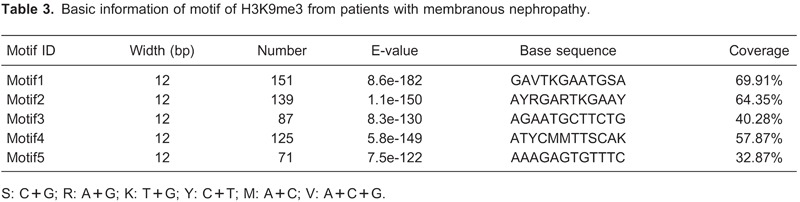
Figure 4. ChIP-seq motif logo. The lateral axis indicates locus of motif. The total height of the vertical axis reflects the conservation of the motif. The height of each base represents probability of the base.
Comparison of H3K9me3 status between MN patients and healthy subjects
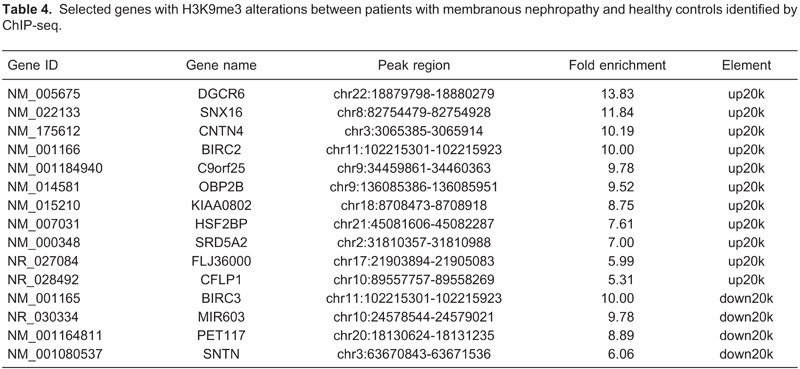
By applying the above analysis procedure to the sequence results, we found that 108 genes displayed significant differences in H3K9me3 in MN patients compared with healthy subjects. Among these genes, 75 displayed increased H3K9me3 and 33 genes showed decreased H3K9me3 expression. The H3K9me3 alterations of 15 selected genes that showed the largest differences are reported in Table 4. Detailed lists of genes with H3K9me3 alterations are provided in Supplementary Table S4. Among these genes, DGCR6, SNX16, CNTN4, BIRC2 and BIRC3 were selected. These five genes showed the largest differences, and they may be associated with MN.
Discussion
Modifications of histone tails were thought to specify a code that regulates the expression of genes. Trimethylated H3K9 (H3K9me3) is a highly conserved histone post-translational modification and is commonly linked to both facultative and constitutive heterochromatin formation and transcriptional repression. H3K9 methylation was initially studied as a marker of epigenetic silencing and constitutive heterochromatin, which was found close to the centromeres. To cover these regions, we used ChIP-seq in this study, which enables the genome-wide identification of binding sites of transcription factors and other DNA-binding proteins. H3K9me3 was first described several years ago (19), but still little is known about its subtle interrelationship with other epigenetic modifications and its potential functional significance in human disease. Consequently, we selected H3K9me3 as the target of investigation using a ChIP-seq strategy, and tested the hypothesis that H3K9me3 was associated with the pathogenesis of membranous nephropathy.
In the present study, we focused on the trimethylation status of H3K9 in MN patients and healthy subjects. The identified candidate genes with significant methylation differences are available in Supplementary Table S4. Overall, these include genes associated with immunity, cell signal transduction, protein transcription and synthesis of channels and transporters, and extracellular matrix, etc.
Among the identified candidate genes, we found that DGCR6 displayed increased H3K9me3 in MN patients compared with healthy controls. The DiGeorge critical region 6 (DGCR6) gene exists as two highly homologous copies (DGCR6 and DGCR6L) on chromosome 22q11 and is deleted in patients with velo-cardio-facial syndrome/DiGeorge syndrome (20). The possible biochemical activity of the gene product(s) might be gene regulation. High levels of the DGCR6 protein have been found in ovaries, heart, kidney, adrenal gland, and, to a lesser extent, in liver and spleen (20). Sorting nexin 16 (SNX16) encodes a member of the sorting nexin family, which contain a phox (PX) domain. The Phox (PX) domain, which is a phosphoinositide-binding domain, represents a novel PI-binding module capable of regulating membrane targeting of about 47 mammalian proteins, 30 of which are tentatively referred to as sorting nexins (SNXs). Some SNXs have been implicated in regulating membrane trafficking in the endocytic pathway (21). SNX16 is a novel 343-amino acid protein consisting of a central PX domain followed by a potential coiled-coil domain and a C-terminal region (22). Like other sorting nexins, SNX16 associates with the membrane via the PX domain, which interacts with the phospholipid phosphatidylinositol 3-phosphate. The most documented function of the PX domain is to target proteins to membranes enriched in PtdIns(3)P or other PIs (21). The PX domain may also participate in protein-protein interactions. Many PX domains contain a Pro-rich PXXP motif, suggesting that they may mediate interaction with SH3 domains. The PX domain may also function to regulate the activity of its host protein. Contactin 4 (CNTN4) encodes a member of the contactin family of immunoglobulins. In this study, the expression of CNTN4 was upregulated. This may be associated with disposition of immune-complex involved in MN. The presence of glomerular subepithelial immune deposits is the distinctive pathologic feature of MN, thus supporting the concept of an immunologic origin (23). Deposits of IgG1, IgG2, IgG3, and IgG4 were detected in MN patients. Contactins are axon-associated cell adhesion molecules that function in neuronal network formation and plasticity. The encoded protein is a glycosylphosphatidylinositol-anchored neuronal membrane protein that may play a role in the formation of axon connections in the developing nervous system. Deletion or mutation of this gene may play a role in neurodevelopmental disorders, including autism (24). Alternative splicing results in multiple transcript variants. CNTN4 has been associated with cerebellar degeneration in spinocerebellar ataxia type 16 (25). Disruption of a single copy of CNTN4 is sufficient to confer key aspects of the 3p deletion syndrome phenotype including developmental delay (26). Thus, CNTN4 is one of a relatively small number of autosomal genes identified as contributing to cognitive deficits in a contiguous-gene syndrome.
We also observed that the other two candidate genes BIRC2 and BIRC3 showed a significant decrease in histone H3K9me3 in MN patients compared with healthy subjects. Baculoviral inhibitor of apoptosis (IAP) repeat containing 2 (BIRC2) and baculoviral IAP repeat containing 3 (BIRC3) encode a member of the IAP family of proteins that inhibit apoptosis by binding to tumor necrosis factor receptor (TNFR)-associated factors TRAF1 and TRAF2, probably by interfering with activation of ICE-like proteases. TRAF2 is the main adaptor protein responsible for the signals triggered by TNFR2; its degradation exerts a regulatory role over the biological activity of the receptor (27). The encoded protein inhibits apoptosis induced by serum deprivation but does not affect apoptosis resulting from exposure to menadione, a potent inducer of free radicals. It contains 3 baculovirus IAP repeats and a ring finger domain. IAP proteins are negative regulators of cell death and play pivotal roles in cellular survival by blocking apoptosis, modulating signal transduction, and affecting cellular proliferation (28). BIRC2 is essential for maintaining endothelial cell survival and blood vessel homeostasis during vascular development (29). BIRC2 regulates endothelial cell survival in vivo by modulating TRAF2-dependent signaling and, possibly, by ubiquitinating target proteins in the TNFR-associated complex, such as NEMO and TRAF221. BIRC2 appears to function by modulating death-receptor signaling in endothelial cells, possibly by regulating the formation and function of TNFR complexes. This may contribute to kidney disease progression. BIRC3 plays an important role in promoting estrogen dependent breast cancer cell survival and protecting against TNFα-induced cell death (30). The nature of the BIRC3 estrogen response element is nearly identical to a consensus sequence, and conversion to a perfect consensus sequence does not significantly increase estrogen receptor activity, suggesting that another factor may be preventing estrogen receptor from interacting with this binding site.
Taken together, we systematically evaluated the status of H3K9me3 in PBMCs of MN patients and gained new insights into the links between key genes and histone methylation in the context of MN. However, as our study relies on only 10 patients and 10 controls, there are some limitations. Our results may indicate that H3K9 trimethylation is involved in nonphysiological MN environment and these novel candidate genes may become potential biomarkers or future therapeutic targets. Further investigations with a large sample are needed to clarify and validate the roles of identified H3K9me3 candidate genes in the pathogenesis of MN.
Supplementary Material
Acknowledgments
The authors are deeply grateful to all the volunteers who donated blood. Research supported by grants from the Key Project for Science and Technology of Guangxi (#2012GXNSFDA053017).
Footnotes
First published online December 12, 2013.
References
- 1.Makker SP, Tramontano A. Idiopathic membranous nephropathy: an autoimmune disease. Semin Nephrol. 2011;31:333–340. doi: 10.1016/j.semnephrol.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Elie V, Fakhoury M, Deschenes G, Jacqz-Aigrain E. Physiopathology of idiopathic nephrotic syndrome: lessons from glucocorticoids and epigenetic perspectives. Pediatr Nephrol. 2012;27:1249–1256. doi: 10.1007/s00467-011-1947-1. [DOI] [PubMed] [Google Scholar]
- 3.Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005;6:838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- 4.Ciccone DN, Chen T. Histone lysine methylation in genomic imprinting. Epigenetics. 2009;4:216–220. doi: 10.4161/epi.8974. [DOI] [PubMed] [Google Scholar]
- 5.Park JA, Kim AJ, Kang Y, Jung YJ, Kim HK, Kim KC. Deacetylation and methylation at histone H3 lysine 9 (H3K9) coordinate chromosome condensation during cell cycle progression. Mol Cells. 2011;31:343–349. doi: 10.1007/s10059-011-0044-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shinkai Y, Tachibana M. H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev. 2011;25:781–788. doi: 10.1101/gad.2027411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu CC, Chen JS, Huang CF, Chen CC, Lu KC, Chu P, et al. Approaching biomarkers of membranous nephropathy from a murine model to human disease. J Biomed Biotechnol. 2011;2011:581928. doi: 10.1155/2011/581928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raha D, Wang Z, Moqtaderi Z, Wu L, Zhong G, Gerstein M, et al. Close association of RNA polymerase II and many transcription factors with Pol III genes. Proc Natl Acad Sci U S A. 2010;107:3639–3644. doi: 10.1073/pnas.0911315106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wacker DA, Kim TH. From sextant to GPS: twenty-five years of mapping the genome with ChIP. J Cell Biochem. 2009;107:6–10. doi: 10.1002/jcb.22060. [DOI] [PubMed] [Google Scholar]
- 10.Euskirchen GM, Rozowsky JS, Wei CL, Lee WH, Zhang ZD, Hartman S, et al. Mapping of transcription factor binding regions in mammalian cells by ChIP: comparison of array- and sequencing-based technologies. Genome Res. 2007;17:898–909. doi: 10.1101/gr.5583007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cybulsky AV. Membranous nephropathy. Contrib Nephrol. 2011;169:107–125. doi: 10.1159/000313948. [DOI] [PubMed] [Google Scholar]
- 12.Lin B, Wang J, Hong X, Yan X, Hwang D, Cho JH, et al. Integrated expression profiling and ChIP-seq analyses of the growth inhibition response program of the androgen receptor. PLoS One. 2009;4: doi: 10.1371/journal.pone.0006589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Langmead B, Schatz MC, Lin J, Pop M, Salzberg SL. Searching for SNPs with cloud computing. Genome Biol. 2009;10:R134. doi: 10.1186/gb-2009-10-11-r134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feng J, Liu T, Zhang Y. Using MACS to identify peaks from ChIP-Seq data. Curr Protoc Bioinformatics. 2011;Chapter 2:Unit 214. doi: 10.1002/0471250953.bi0214s34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shin JH, Li RW, Gao Y, Baldwin R, Li CJ. Genome-wide ChIP-seq mapping and analysis reveal butyrate-induced acetylation of H3K9 and H3K27 correlated with transcription activity in bovine cells. Funct Integr Genomics. 2012;12:119–130. doi: 10.1007/s10142-012-0263-6. [DOI] [PubMed] [Google Scholar]
- 16.Carbon S, Ireland A, Mungall CJ, Shu S, Marshall B, Lewis S. AmiGO: online access to ontology and annotation data. Bioinformatics. 2009;25:288–289. doi: 10.1093/bioinformatics/btn615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Georgiev S, Boyle AP, Jayasurya K, Ding X, Mukherjee S, Ohler U. Evidence-ranked motif identification. Genome Biol. 2010;11:R19. doi: 10.1186/gb-2010-11-2-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi J, Dawe RK. Partitioning of the maize epigenome by the number of methyl groups on histone H3 lysines 9 and 27. Genetics. 2006;173:1571–1583. doi: 10.1534/genetics.106.056853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pfuhl T, Durr M, Spurk A, Schwalbert B, Nord R, Mysliwietz J, et al. Biochemical characterisation of the proteins encoded by the DiGeorge critical region 6 (DGCR6) genes. Hum Genet. 2005;117:70–80. doi: 10.1007/s00439-005-1267-2. [DOI] [PubMed] [Google Scholar]
- 21.Seet LF, Hong W. The Phox (PX) domain proteins and membrane traffic. Biochim Biophys Acta. 2006;1761:878–896. doi: 10.1016/j.bbalip.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 22.Hanson BJ, Hong W. Evidence for a role of SNX16 in regulating traffic between the early and later endosomal compartments. J Biol Chem. 2003;278:34617–34630. doi: 10.1074/jbc.M300143200. [DOI] [PubMed] [Google Scholar]
- 23.Prunotto M, Carnevali ML, Candiano G, Murtas C, Bruschi M, Corradini E, et al. Autoimmunity in membranous nephropathy targets aldose reductase and SOD2. J Am Soc Nephrol. 2010;21:507–519. doi: 10.1681/ASN.2008121259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cottrell CE, Bir N, Varga E, Alvarez CE, Bouyain S, Zernzach R, et al. Contactin 4 as an autism susceptibility locus. Autism Res. 2011;4:189–199. doi: 10.1002/aur.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manderson EN, Birch AH, Shen Z, Mes-Masson AM, Provencher D, Tonin PN. Molecular genetic analysis of a cell adhesion molecule with homology to L1CAM, contactin 6, and contactin 4 candidate chromosome 3p26pter tumor suppressor genes in ovarian cancer. Int J Gynecol Cancer. 2009;19:513–525. doi: 10.1111/IGC.0b013e3181a3cd38. [DOI] [PubMed] [Google Scholar]
- 26.Fernandez T, Morgan T, Davis N, Klin A, Morris A, Farhi A, et al. Disruption of contactin 4 (CNTN4) results in developmental delay and other features of 3p deletion syndrome. Am J Hum Genet. 2004;74:1286–1293. doi: 10.1086/421474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cabal-Hierro L, Lazo PS. Signal transduction by tumor necrosis factor receptors. Cell Signal. 2012;24:1297–1305. doi: 10.1016/j.cellsig.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 28.Dueber EC, Schoeffler AJ, Lingel A, Elliott JM, Fedorova AV, Giannetti AM, et al. Antagonists induce a conformational change in cIAP1 that promotes autoubiquitination. Science. 2011;334:376–380. doi: 10.1126/science.1207862. [DOI] [PubMed] [Google Scholar]
- 29.Santoro MM, Samuel T, Mitchell T, Reed JC, Stainier DY. Birc2 (cIap1) regulates endothelial cell integrity and blood vessel homeostasis. Nat Genet. 2007;39:1397–1402. doi: 10.1038/ng.2007.8. [DOI] [PubMed] [Google Scholar]
- 30.Pradhan M, Baumgarten SC, Bembinster LA, Frasor J. CBP mediates NF-kappaB-dependent histone acetylation and estrogen receptor recruitment to an estrogen response element in the BIRC3 promoter. Mol Cell Biol. 2012;32:569–575. doi: 10.1128/MCB.05869-11. [DOI] [PMC free article] [PubMed] [Google Scholar]