

Abstract
Cardiovascular disease is one of the leading causes of death worldwide, and evidence indicates a correlation between the inflammatory process and cardiac dysfunction. Selective inhibitors of cyclooxygenase-2 (COX-2) enzyme are not recommended for long-term use because of potentially severe side effects to the heart. Considering this and the frequent prescribing of commercial celecoxib, the present study analyzed cellular and molecular effects of 1 and 10 µM celecoxib in a cell culture model. After a 24-h incubation, celecoxib reduced cell viability in a dose-dependent manner as also demonstrated in MTT assays. Furthermore, reverse transcription-polymerase chain reaction analysis showed that the drug modulated the expression level of genes related to death pathways, and Western blot analyses demonstrated a modulatory effect of the drug on COX-2 protein levels in cardiac cells. In addition, the results demonstrated a downregulation of prostaglandin E2 production by the cardiac cells incubated with celecoxib, in a dose-specific manner. These results are consistent with the decrease in cell viability and the presence of necrotic processes shown by Fourier transform infrared analysis, suggesting a direct correlation of prostanoids in cellular homeostasis and survival.
Keywords: Cellular and molecular analyses, Cell death, Cyclooxygenase-2 inhibitor, H9c2 cardiac cell line
Introduction
Diseases of the cardiovascular system represent major causes of human morbidity and mortality worldwide, and heart hypertrophy is a characteristic in a substantial proportion of cases (1,2). Many biochemical and physiological reactions help establish hypertrophy in the heart by activating interconnected intracellular signaling pathways that enhance cardiomyocyte size. In addition, accumulating evidence indicates a strict correlation between the inflammatory process and cardiac hypertrophy and failure (3,4).
Several studies have demonstrated that cyclooxygenase-2 (COX-2) is readily detected in cardiac myocytes of failing human hearts (5,6). COX-2 and its isoenzyme COX-1 catalyze the conversion of arachidonic acid to prostaglandin (PG) H2, the key precursor molecule required in the formation of prostanoids. COX-1 is constitutively expressed; on the other hand, COX-2 is highly inducible in response to proinflammatory stimuli such as cytokines and growth factors, among others, which also contribute to PG release. However, in discrete populations of cells, COX-2 is expressed constitutively, suggesting its relevant function in cellular homeostasis (6). The two different COX isozymes have a molecular weight of 70 to 71 kDa and are almost identical in length, with approximately 600 amino acids and 63% identity between them. In addition, the three-dimensional structures of COX-1 and COX-2 share similar structural properties, including a hydrophobic tunnel, which allows arachidonic acid to access the active respective sites. In COX-2, this hydrophobic tunnel has a side pocket (7), and this structural distinction has supported the development of several nonsteroidal anti-inflammatory drugs (NSAIDs) that selectively inhibit COX-2. Such drugs, classified as coxib, were developed to be a safe alternative to avoid serious gastrointestinal complications caused by the traditional NSAIDs (8). In the late 1990's, they dominated the prescription-drug market of the United States, until 2004, when the COX-2 inhibitor rofecoxib (Vioxx¯, Merck & Co., USA) was withdrawn because of the possibility of increased risk for heart attack and stroke in users of this kind of drug (9,10). Despite that, a variety of celecoxibs are still being regularly prescribed to relieve symptoms of arthritis and are also recommended to patients with familial adenomatous polyposis.
The understanding of the exact mechanisms of COX-2 inhibitors in a cell and the entire body (11,12) is still a challenge. Conflicting results were reported on the protective and detrimental activities related to COX-2 activity, making it difficult to clearly understand the risks of specific inhibitors of COX-2 on human health. Based on these observations, we aimed to assess the effects of the selective COX-2 inhibitor, celecoxib, in H9c2, a cardiac myoblast cell that responds to a different set of stimuli, similar to an adult cardiomyocyte (13), and also has been used for drug clinical trials (14,15). This drug was chosen considering its regular prescription worldwide, despite the risks to the cardiovascular system.
In the present study, we hypothesized that cellular and molecular crosstalk could explain the undercover details of celecoxib mechanistic activity in cardiac cells and its biological basis of toxicity. To achieve this, we used the most common concentrations of this class of drug assayed in cell culture models (16-18). We first determined the consistent results of cell death in a dose-dependent manner. Next, the COX-2 protein and its metabolites were verified in H9c2 cells after their incubation with celecoxib; a direct correlation between the level of protein activity and the disruption of cellular homeostasis was verified. To understand the subtle cellular and molecular effects of the drug, Fourier transform infrared spectroscopy (FTIR) was performed. This technique has been broadly used in medical and molecular studies to verify biochemical changes of cells induced by different stimuli (19). In our cardiac cells, the drug was able to induce cell death, represented by the necrotic spectra found. The results presented in this article could be useful to future investigation of the effects of celecoxib using in vivo models to elucidate the effects of the drug on COX-2 activity and its consequence to the heart.
Material and Methods
Cell culture and celecoxib treatment
H9c2 is a rat embryonic cardiac myoblast cell line (ATCC: CRL-1446) obtained from Banco de Células do Rio de Janeiro, Brazil. Cells were routinely grown in Dulbecco's modified Eagle's medium (Gibco, USA), containing 2 mM L-glutamine and 1.5 g/L sodium bicarbonate and supplemented with 10% fetal bovine serum (Gibco) at 37°C in a 5% CO2 atmosphere. Celecoxib (Celebra¯, C17H14F3N3O2S, 381.373 g/mol; Pfizer Inc., USA) was reconstituted in 1% dimethyl sulfoxide (DMSO). In the control assays (0 µM celecoxib), DMSO was also added at the same concentration (1%).
Cell viability assay
The effect of celecoxib on cell viability was determined using the colorimetric 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. This method measures mitochondrial activity based on the reductive cleavage of yellow tetrazolium salt to a purple formazan compound by the dehydrogenase activity of intact mitochondria. The methodology was performed by adapting the protocol described in Mosmann (20). Cells (1.5×104) were grown on 96-well plates and exposed to 0, 1, 10, and 100 µM celecoxib for 24 h. The culture medium was replaced by a fresh one containing 0.5 mg/mL MTT (Sigma-Aldrich, Germany) and cells were incubated for an additional 2 h. The medium was removed and the formazan product was dissolved in 200 µL DMSO for 30 min under gentle agitation. The plates were read with a 570-nm filter in an ELISA Spectracount Reader (Packard Instrument Company Inc., USA).
Fluorescence microscopy and DNA analyses
Fluorescence microscopy analyses were performed with 3.0×104 cells per group, previously grown on sterile coverslips. After treatments (0, 1, 10 µM celecoxib), cells were fixed and permeabilized with 3.8% paraformaldehyde in phosphate-buffered saline (PBS) containing 0.1% Triton X-100 for 7 min. Next, cells were incubated in a PBS solution containing 3.33 ng/µL 4′,6-diamidino-2-phenylindole (DAPI) at room temperature for 5 min for nuclei staining. Finally, cells were washed three times with PBS for 5 min, mounted on slides using 200 mM propyl gallate (Sigma-Aldrich) in 90% glycerol, and subjected to microscopic analysis. Images were taken with a Leica photomicroscope (DMLB) equipped with an HBO 100-W mercury lamp and the corresponding ultraviolet fluorescence microscopy filter. The statistical analyses were performed by counting 100 cells.
RNA extraction and semiquantitative reverse transcription-polymerase chain reaction (RT-PCR)
H9c2 cells were incubated with celecoxib (0, 1, 10 µM) for 24 h and then used for RT-PCR analysis. The total RNA was extracted from 1×106 cells according to the instructions from the supplier of the Trizol reagent (Life Technologies™, USA) and then quantified using NanoDrop_1000 (Thermo Scientific, USA). One microgram of each RNA sample was reverse-transcribed into first-strand cDNA using the cloned AMV Reverse Transcriptase (Life Technologies™) following instructions from the supplier. cDNAs were used as templates in RT-PCR analyses, and specific genes were amplified using the following sets of primers (Life Technologies™): β-actin, 5′-gatcatgtttgagaccttcaacac and 5′-cgtcacacttcatgatggagttga; bcl2, 5′-tttgagttcggtggggtcat and 5′-tgacttcacttgtggcccag; Bax, 5′-tggcagctgacatgttttctgac and 5′-gtcccaaccaccctggtcttgg; caspase, 5′-gcacacattatagctactgg and 5′-gttaaactccgacgacgtatta; caspase-8, 5′-cgatattgctgaacgtctgg and 5′-ctgcaagacaactcgagc; caspase-9, 5′-cgtggtggtcattctctctca and 5′-cttgacactgcgtccagctg; Cox-2, 5′-tccagatcacatttgattgacag and 5′-tctttgactgtgggaggataca; c-Fos, 5′-gacagatcaacttgaagacg and 5′-ggtgaagacaaaggaacg. PCRs were carried out using appropriate annealing temperatures, and the products were analyzed on 1% agarose gels stained with ethidium bromide, and photographed using Kodak Gel Logic 100 Imaging System (USA). The bands were quantified with the Quantity One Software, Bio-Rad (USA).
Western blot analyses and PGE2 immunoassays
Protein samples were extracted from cultured cells according to Sambrook et al. (21). Equal amounts of protein (50 µg) were subjected to 10% polyacrylamide gel and then transferred to a polyvinylidene fluoride membrane. Membranes were blocked with 5% milk for 2 h and then incubated with primary antibodies anti-COX-2 (1:250, Cayman Chemical, USA) or β-actin (1:250, Santa Cruz Biotechnology Inc., USA) and next with secondary antibodies (α-rabbit immunoglobulin G (IgG), Cayman Chemical) at 1:6000 dilution. After extensive washes with saline solution, membranes were developed with enhanced chemiluminescence scintillation fluid (GE Healthcare Life Sciences, USA) and exposed to Hyperfilm (GE Healthcare Life Sciences). The bands were quantified using the Quantity One Software. For the PGE2 immunoassay, the supernatant of H9c2 cultures incubated with celecoxib for 24 h were measured using the Prostaglandin E2 Express EIA kit (Cayman Chemical). The assays were performed in triplicate.
FTIR spectroscopy analysis
FTIR analysis was performed with 1×106 cells per group incubated with celecoxib (0, 1, 10 µM) for 24 h. After treatment, cells were washed with PBS and spread out on a glass slide. After 10 min incubation in a 5% CO2 atmosphere, the cells were dried at 60°C for 3 h. The spectra of the samples were obtained with methods that use KBr pellets on a KBr window. The data were collected via Spectrum GX FTIR (Perkin Elmer, USA). Measurements were made with a 15× objective lens. Thirty-two scans in the region of 4000 to 800 cm-1, with resolution of 4 cm-1, and 20°C were conducted in transmission mode to obtain the infrared spectra. The assays were repeated three times for each group, and the spectra were preprocessed with the Spectrum 5.3 software (Perkin Elmer). The Savitzky-Golay algorithm with nine points was used to reduce the noise level in a spectrum.
Graphs and statistical analyses
Values from independent assays were used for analysis, and graphs were generated using Graph Pad Prism¯ 5. The differences between control and treated groups were also measured using one-way analysis of variance, followed by the Dunnett test. Significance was set at P<0.05.
Results
Cell viability
In H9c2, the addition of celecoxib to the medium reduced its viability in a dose-dependent manner after 24 h incubation. The analyses of the results (data not shown) demonstrated that, in cell cultures where 1 µM concentration of the chemical was used, no significant difference in cell viability was observed compared to the control group of cells where no drug was added (0 µM). With final concentrations of 10 and 100 µM celecoxib, a 45 and 92% decrease in cell viability was observed, respectively.
Fluorescence microscopy and DNA damage in the H9c2 cardiac cell line
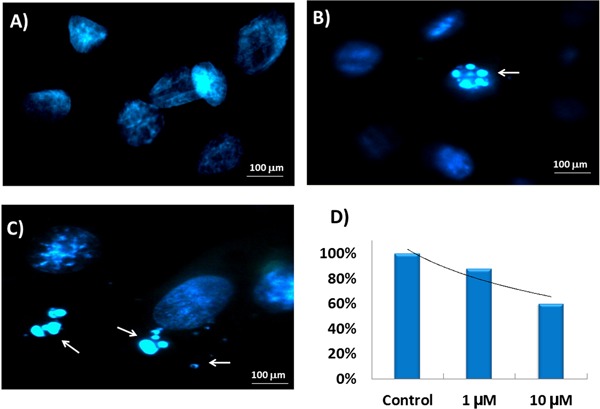
We performed fluorescence microscopy to investigate morphological changes to the DNA of H9c2 cells after celecoxib treatment. For the assays, cells were incubated with 1 and 10 µM celecoxib for 24 h. As shown in Figure 1, DAPI staining detected nuclear condensation, DNA fragmentation, and perinuclear apoptotic/necrotic bodies in cells treated with the pharmaceutical drug. No significant nuclear damage was verified in a control group of cells. The percentage of cells with nuclear morphological alteration in each group was approximately 12 and 40% in the presence of 1 and 10 µM celecoxib, respectively (Figure 1D).
Figure 1. Nuclear morphological damage in H9c2 cells incubated with different concentrations of celecoxib for 24 h. A, 0 µM; B, 1 µM; C, 10 µM celecoxib. Cells were stained with DAPI and the arrows indicate perinuclear apoptotic/necrotic bodies. D, Percentage of nuclear morphological alteration in each group after treatment.
Modulatory effect of selective COX-2 inhibitor in gene expression
To explore gene expression mechanisms that underlie cellular treatment with the selective COX-2 inhibitor, H9c2 cells were incubated for 24 h in the presence of 0, 1, and 10 µM concentrations of the drug. Next, mRNAs were extracted and reverse transcribed, and a specific set of primers for genes involved in death pathways were used in RT-PCR analyses. β-actin was used as a reference gene, and the data obtained were normalized to the expression level of each gene to its own 0-h time point group and were plotted as relative terms. The experiments were performed in triplicate and, with the exception of Bcl2, c-Fos, and Cox-2 transcripts, which changed after treatment, other genes analyzed did not show consistent alterations in their mRNA levels (Figure 2).
Figure 2. Effects of celecoxib (0, 1, and 10 µM) on H9c2 gene expression. Relative expression of Bax, Bcl2, c-Fos, caspases, Cox-2, and β-actin genes were measured after cells were incubated with the drug for 24 h. Data are reported as average of values of assays ±SD. All treatments were conducted in triplicate. ***P<0.001, compared to 0 µM at 24 h (Dunnett test).
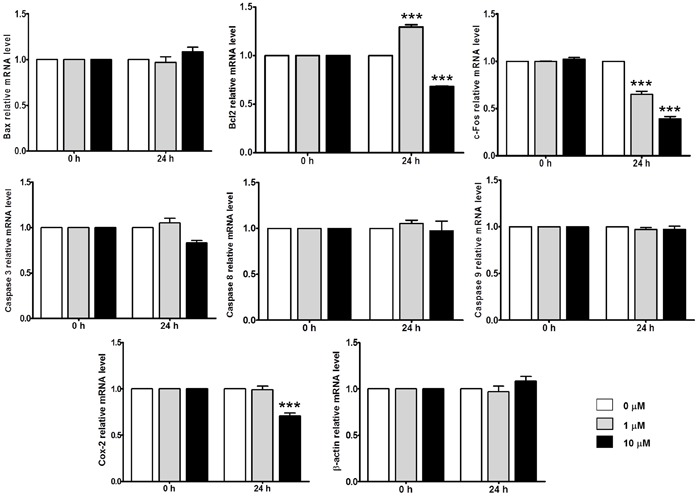
RT-PCR analyses of Bcl2 demonstrated an ∼30% increase in this gene transcript when cells were incubated with 1 µM celecoxib for 24 h (Figure 2). On the contrary, the RT-PCR analyses for the same gene, after cells were incubated with 10 µM concentration of the drug, demonstrated decreased expression of Bcl2. At 24 h, the Bcl2 transcript level decreased ∼31.78% when compared to the control group.
The analyses were also performed for c-Fos. Our assays demonstrated a decreased transcript level of gene expression after 24 h incubation with celecoxib (Figure 2). Using 1 µM concentration of the drug, the c-Fos level decreased ∼35.01%, and a 10 µM concentration caused an ∼61.04% reduction in transcription, compared to the control groups. In addition, the effect of celecoxib on Cox-2 mRNA levels in H9c2 was investigated. The results demonstrated no significant changes in Cox-2 transcripts when cells were incubated with 1 µM celecoxib, and an ∼22.67% reduction in transcripts analyzed was observed after cells were treated with 10 µM celecoxib for 24 h.
Effects of celecoxib on COX-2 protein synthesis and PGE2 cellular production
The levels of COX-2 protein following celecoxib treatment for 24 h were examined by Western blotting to determine whether the chemical modulates COX-2 translation. As shown in Figure 3A and B, the chemical caused cellular changes in the low basal levels of COX-2 protein. The analyses for cells incubated with 1 µM celecoxib demonstrated an ∼70% increase in COX-2 compared to untreated cells. For cells incubated with 10 µM celecoxib, an ∼30.5% increase in COX-2 protein levels was observed.
Figure 3. Celecoxib modulates COX-2 protein translational rates in cardiac H9c2 cells. A, Western blot analyses of COX-2 protein level after celecoxib-specific treatments. The results are from one representative experiment of three performed assays that showed similar patterns. B, Quantitative analysis of the Western blots. C, Supernatant of treated cells was used for prostaglandin E2 (PGE2) measurement. ***P<0.001 compared to control (B) or control and 1 µM (C) (Dunnett test).
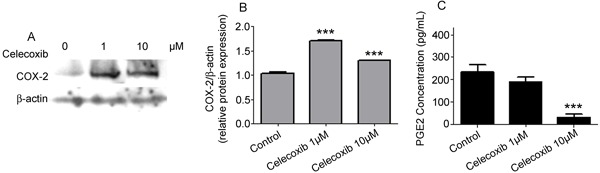
To further determine celecoxib-mediated inhibition of PGE2 synthesis in H9c2 cells, the supernatant of cellular cultures treated or untreated with the chemical was collected, and PGE2 was measured. The results presented in Figure 3C show that, even at 1 µM, celecoxib decreased PGE2 production after a 24-h incubation compared to control levels. PGE2 secretion decreased from 235 pg/mL in uninduced cells to 189.33 and 32.66 pg/mL when H9c2 cells were incubated with 1 and 10 µM concentrations of the drug, respectively.
Infrared spectroscopy analyses of cellular damage
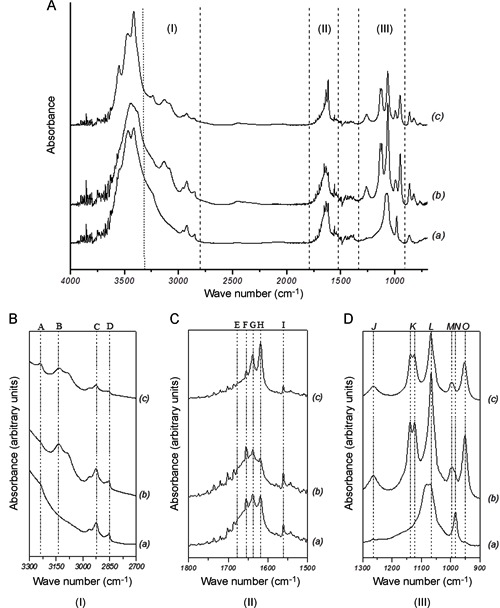
FTIR spectral profiles obtained from cells treated or not with celecoxib were analyzed. The results are reported in Figure 4A. Three spectral regions (3300-2700, 1800-1500, and 1300-900 cm-1) are detailed in Figure 4B, C, and D, and Table 1 summarizes vibrational assignments of the molecules in the spectra.
Figure 4. FTIR spectra between 4000 and 800 cm-1 of H9c2 cells after a 24-h incubation with celecoxib. A, Spectra overview: (a) 0 µM, (b) 1 µM, (c) 10 µM celecoxib. Spectra intervals: B, 3300 to 2700 cm-1 (I); C, 1800 to 1500 cm-1 (II); D, 1300 to 900 cm-1 (III). A-N were assigned as bands in those spectra. Bands A, B, J-N represent changes in DNA; bands assigned from E to I represent structural changes of proteins; C and D represent general changes in cellular structures; O represents P-O stretching in phosphorylated protein.
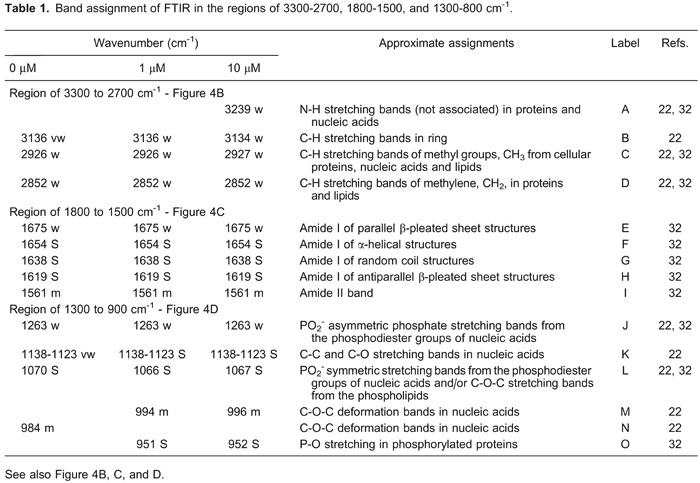
In the spectra, proteins are represented by the amide I and amide II bands (Figure 4C). The amide I bands represented ∼80% of C=O stretching vibrations of the amide groups connected to N-H bending and C-N stretching. Figure 5 shows a varied pattern of secondary structures in proteins of cells incubated with the selective COX-2 inhibitor. At 1 µM celecoxib, parallel β-sheets and α-helixes increased their intensity; at 10 µM, these levels of intensity decreased compared to the control group. In addition, antiparallel β-sheet structures decreased in intensity in cells treated with 1 µM celecoxib and increased with 10 µM celecoxib.
Figure 5. Protein secondary structure distribution in cells treated with 0, 1, or 10 µM celecoxib. Irregular distribution of β-sheets, α-helixes, and random coils are shown.
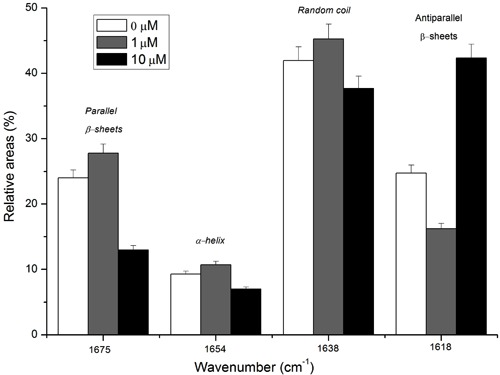
The spectra (Figure 4 and Table 1) also demonstrated important changes in DNA structure. Bands assigned as A, B, J-N show symmetric and asymmetric stretching vibrations of the PO2 − central group, and C-C and C-O stretching vibrations and C-O-C deformation modes in the sugar groups (22). Band J, corresponding to PO2 − asymmetric stretching modes in nucleic acids, increased in intensity in spectra (b) and (c). In the band assigned as K, the intensity and band shape of C-C and C-O stretching modes were modified with celecoxib treatment, and overlapping bands with two peaks in (b) and (c) could be associated with changes in nucleic acids. Band N corresponding to C-O-C deformation modes at 984 cm−1 in (a) shifted to 994-996 cm−1 in (b) and (c), respectively, which is correlated to phosphodiester groups presented in nucleic acids and chromatin structure change. To analyze subtle differences in nucleic acid vibration, the ratio of integrated areas of absorption between nucleic acids and the amide II band at 1561 cm−1 was calculated. The ratio AX/A1561 (where X represents the wave number of the nucleic acid analyzed) was used to compare the relative amounts of nucleic acid and proteins in a sample. The results are summarized in Figure 6. The overall intensity of the region assigned to nucleic acid increased gradually when compared to amide II in a cell incubated with celecoxib. Major differences on relative areas of nucleic acid were demonstrated between the control experiment (0 µM) and the other two experimental conditions (1 and 10 µM), instead of between the two conditions analyzed.
Figure 6. Nucleic acid vibrational state. A, Ratio of the integrated areas of absorption between nucleic acids and amide II band at 1561 cm-1 after H9c2 were incubated with 0, 1, and 10 µM celecoxib for 24 h. B, Relative areas of nucleic acid bands of H9c2 after incubation with celecoxib for 24 h.
Discussion
Cardiovascular disease is one of the leading causes of death worldwide, and accumulating evidence indicates a strict correlation between the inflammatory process and cardiac dysfunction (3,4). However, understanding the fine-tuning of cellular and molecular aspects of cardiac metabolism in controlling inflammatory pathways and the development of safe prescription drugs for inflammatory processes without the risk of cardiac injury is still considered a challenge. Despite their adverse side effects, selective COX-2 inhibitors, such as celecoxibs, are usually prescribed, because the benefits may outweigh the risks for some patients, which generates impasses worldwide. Based on such controversy, in the present investigation, we demonstrated cellular and molecular studies of a commercial celecoxib on a cardiac cell line.
In a dose-dependent use of the COX-2 inhibitor, cell viability decreases considerably. In our experiments, cells treated with 1 µM concentration of the drug for 24 h presented a similar viability pattern compared to the control culture. This preliminary result was relevant, considering that higher concentrations of the drug, such as 10 and 100 µM, are not achieved in the serum of patients that take the drug at low levels, although they are largely used in cell culture models (3,10). However, we only excluded the 100 µM drug concentration in future assays because of cell viability (∼100% death). The 10 µM concentration was kept to investigate its effect in cardiac cells, as in patients under high doses of celecoxib administration (23).
Next, to understand cellular and molecular concerns about the behavior of cardiac cells when treated with celecoxib, we investigated nuclear morphological changes. In general, cardiac myocytes exposed to chronic stress undergo a sequence of phenotypic changes, which eventually lead to heart failure. In H9c2 cells, microscopy analyses demonstrated 12% morphological chromatin damage in cells incubated with 1 µM celecoxib, which diverge from data obtained in MTT assays, where no significant differences in cell viability were found when compared to the control group. The conflicting results could be explained by the cellular division rate (24). Cells incubated with the lowest drug concentration kept their regular cellular metabolism in the majority of cells, which could explain the viability rates found (Figure 1).
Different laboratories have addressed the question of how cells decide whether to undergo apoptosis or necrosis. Some evidence suggests that different cell death mechanisms crosstalk with each other and substantiate the notion that necrosis constitutes a default that is unmasked when essential effectors of apoptosis are inhibited such as changes in expression of pro-apoptotic, anti-apoptotic, and caspase genes (25,26). Based on these observations, the transcriptional level of genes involved in death processes was investigated in H9c2 cells treated with the COX-2 selective inhibitor. The results showed no major changes in Bax and caspase genes along with a modulatory effect at the transcriptional level for Bcl2 and c-fos genes, which suggests the involvement of those two genes in molecular mechanisms that are probably facilitating cell death. Bcl2 is classically described as a protein that provides survival advantages to cells (27,28). In our model, the quite stressful environment created by 1 µM celecoxib upregulated the Bcl2 transcript level, which helps avoid cell death as detected in previous assays (Figure 1). In addition, the downregulation of Bcl2 transcripts when cells were incubated with 10 µM concentration of the drug could contribute to the considerable decrease in cell viability, in a molecular pathway that is probably independent of caspase genes (Figure 2). In addition, the modulatory effects of celecoxib on transcription levels of c-Fos, a proto-oncogene that turns on a cascade of events that lead to cell division and/or programmed cell death (29,30), disrupt cellular homeostasis. A correlation between low levels of c-Fos transcripts and cell death is reinforced by recent studies using chicken embryos submitted to electromagnetic fields (31). That study reported a strong connection between c-Fos downregulation and cell death in early stages of chicken embryonic development. In general, a cell's commitment to death or ability to evade it involves the integration of a complex network of signals. In addition, the analysis of COX-2 protein and PGE2 levels in cells treated with the pharmaceutical drug reinforces the relevant function of the protein in cellular homeostasis (Figure 3). PGE2 at basal levels assures an optimal environment for metabolic reactions in a cell and is an important factor in the maintenance of the cardiovascular system (6). In H9c2 cells, inhibition of COX-2 activity decreased PG levels. As a consequence, cells increased their COX-2 levels trying to reestablish basal levels of PGE2 and other correlated metabolites, which were not reached considering the presence of celecoxib. At this point, it is possible to correlate PGE2 production with safeguarding cells, and it is probably able to reduce death rates in cells where 1 µM COX-2 inhibitor is present. On the other hand, at 10 µM concentrations of the drug, the reduced level of PGE2 present in the culture was not able to control cell death. The detection of necrotic death mechanisms in cells incubated with celecoxib in FTIR assays reinforces these observations.
FTIR spectroscopy analyses were performed to understand the fine-tuning of H9c2 cellular metabolism when cells were treated with celecoxib. This technique has been broadly used in medical and molecular studies to verify biochemical changes in cells induced by different stimuli (19). It is based on the vibration of chemical bonds within molecules and is extremely sensitive to any molecular structural changes, and certain functional groups of atoms produce the same vibrational spectrum even in different molecules, which makes it a powerful tool to identify molecular structures (32). The spectra of H9c2 cells incubated with celecoxib demonstrated irregular changes in the components of amide I bands, and considerable variability at the DNA structural level (Figures 4-6 and Table 1), which suggests the presence of necrotic mechanisms of cell death as described in Jamin et al. (33) and Holman et al. (34).
It is known that, in most of the cell cycle, cells keep their DNA tightly packed in chromosomes, which appear opaque to infrared (IR) radiation. However, in the S phase of the cell cycle, or in the presence of cellular necrotic death mechanisms, the chromatin structure becomes uncondensed, which allows better detection of DNA content by IR radiation (22). On the other hand, in apoptosis, nucleic acids decrease absorption of IR radiation (25). In this study, the results suggested that, in cardiac H9c2 cells, celecoxib treatments were able to induce biochemical changes and chromatin unpacking, which made it possible to analyze DNA structural changes in the spectra, increasing the evidence that the necrotic death process was present. In addition, FTIR spectra showed that damage to the DNA structure of cells occurred at 1 and 10 µM concentrations; however, the higher concentrations of the drug were not able to cause more DNA damage than the 1 µM treatment compared to the group of cells used as control (Figure 6), which reinforces the activity of PGE2 in the control of cell death processes.
Altogether, the results showed biochemical changes that were able to lead to cell necrosis. In our assays, the evidence points to a direct involvement of the prostanoid in cell survival or death by necrotic process. Furthermore, we also speculate that in vivo the increased risk of heart attack and stroke in regular users of selective COX-2 inhibitors is due to unbalanced production of PGE2 by cardiomyocytes. The prostanoid could be responsible for direct biochemical mechanisms that protect the cell and assure its homeostatic survival. Further in vivo studies are needed.
Acknowledgments
We thank Alene Alder-Rangel for English revision. Research supported by CNPq (#475586/2009-3 and #506991/2010-5), FAPEMIG (CBB-APQ #02351-10), and Fundação Valeparaibana de Ensino.
Footnotes
First published online November 11, 2013.
References
- 1.Ritter O, Neyses L. The molecular basis of myocardial hypertrophy and heart failure. Trends Mol Med. 2003;9:313–321. doi: 10.1016/S1471-4914(03)00114-X. [DOI] [PubMed] [Google Scholar]
- 2.Latronico MV, Elia L, Condorelli G, Catalucci D. Heart failure: targeting transcriptional and post-transcriptional control mechanisms of hypertrophy for treatment. Int J Biochem Cell Biol. 2008;40:1643–1648. doi: 10.1016/j.biocel.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 3.Smeets PJ, Teunissen BE, Planavila A, Bosch H de Vogel-van den, Willemsen PH, Vusse GJ van der, et al. Inflammatory pathways are activated during cardiomyocyte hypertrophy and attenuated by peroxisome proliferator-activated receptors PPARalpha and PPARdelta. J Biol Chem. 2008;283:29109–29118. doi: 10.1074/jbc.M802143200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Velten M, Duerr GD, Pessies T, Schild J, Lohner R, Mersmann J, et al. Priming with synthetic oligonucleotides attenuates pressure overload-induced inflammation and cardiac hypertrophy in mice. Cardiovasc Res. 2012;96:422–432. doi: 10.1093/cvr/cvs280. [DOI] [PubMed] [Google Scholar]
- 5.Wong SC, Fukuchi M, Melnyk P, Rodger I, Giaid A. Induction of cyclooxygenase-2 and activation of nuclear factor-kappaB in myocardium of patients with congestive heart failure. Circulation. 1998;98:100–103. doi: 10.1161/01.CIR.98.2.100. [DOI] [PubMed] [Google Scholar]
- 6.Curtis-Prior P. The eicosanoids. 1st edn. England: Wiley John & Sons, Inc.; 2004. [Google Scholar]
- 7.Gierse JK, McDonald JJ, Hauser SD, Rangwala SH, Koboldt CM, Seibert K. A single amino acid difference between cyclooxygenase-1 (COX-1) and -2 (COX-2) reverses the selectivity of COX-2 specific inhibitors. J Biol Chem. 1996;271:15810–15814. doi: 10.1074/jbc.271.26.15810. [DOI] [PubMed] [Google Scholar]
- 8.Simon LS, Lanza FL, Lipsky PE, Hubbard RC, Talwalker S, Schwartz BD, et al. Preliminary study of the safety and efficacy of SC-58635, a novel cyclooxygenase 2 inhibitor: efficacy and safety in two placebo-controlled trials in osteoarthritis and rheumatoid arthritis, and studies of gastrointestinal and platelet effects. Arthritis Rheum. 1998;41:1591–1602. doi: 10.1002/1529-0131(199809)41:9<1591::AID-ART9>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 9.Karha J, Topol EJ. The sad story of Vioxx, and what we should learn from it. Cleve Clin J Med. 2004;71:933–939. doi: 10.3949/ccjm.71.12.933. [DOI] [PubMed] [Google Scholar]
- 10.Dajani EZ, Islam K. Cardiovascular and gastrointestinal toxicity of selective cyclo-oxygenase-2 inhibitors in man. J Physiol Pharmacol. 2008;59((Suppl 2)):117–133. [PubMed] [Google Scholar]
- 11.Graham DJ, Campen D, Hui R, Spence M, Cheetham C, Levy G, et al. Risk of acute myocardial infarction and sudden cardiac death in patients treated with cyclo-oxygenase 2 selective and non-selective non-steroidal anti-inflammatory drugs: nested case-control study. Lancet. 2005;365:475–481. doi: 10.1016/S0140-6736(05)17864-7. [DOI] [PubMed] [Google Scholar]
- 12.Fosbol EL, Gislason GH, Jacobsen S, Folke F, Hansen ML, Schramm TK, et al. Risk of myocardial infarction and death associated with the use of nonsteroidal anti-inflammatory drugs (NSAIDs) among healthy individuals: a nationwide cohort study. Clin Pharmacol Ther. 2009;85:190–197. doi: 10.1038/clpt.2008.204. [DOI] [PubMed] [Google Scholar]
- 13.Kimes BW, Brandt BL. Properties of a clonal muscle cell line from rat heart. Exp Cell Res. 1976;98:367–381. doi: 10.1016/0014-4827(76)90447-X. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Zhao X, Gao X, Nie X, Yang Y, Fan X. Development of fluorescence imaging-based assay for screening cardioprotective compounds from medicinal plants. Anal Chim Acta. 2011;702:87–94. doi: 10.1016/j.aca.2011.06.020. [DOI] [PubMed] [Google Scholar]
- 15.Lin Z, Will Y. Evaluation of drugs with specific organ toxicities in organ-specific cell lines. Toxicol Sci. 2012;126:114–127. doi: 10.1093/toxsci/kfr339. [DOI] [PubMed] [Google Scholar]
- 16.Pyrko P, Kardosh A, Schonthal AH. Celecoxib transiently inhibits cellular protein synthesis. Biochem Pharmacol. 2008;75:395–404. doi: 10.1016/j.bcp.2007.08.029. [DOI] [PubMed] [Google Scholar]
- 17.Jacobshagen C, Gruber M, Teucher N, Schmidt AG, Unsold BW, Toischer K, et al. Celecoxib modulates hypertrophic signalling and prevents load-induced cardiac dysfunction. Eur J Heart Fail. 2008;10:334–342. doi: 10.1016/j.ejheart.2008.02.013. [DOI] [PubMed] [Google Scholar]
- 18.Yamakawa N, Suemasu S, Kimoto A, Arai Y, Ishihara T, Yokomizo K, et al. Low direct cytotoxicity of loxoprofen on gastric mucosal cells. Biol Pharm Bull. 2010;33:398–403. doi: 10.1248/bpb.33.398. [DOI] [PubMed] [Google Scholar]
- 19.Diem M, Griffiths PR, Chalmers JM. Vibrational spectroscopy for medical diagnosis. 1st edn. England: Wiley John & Sons, Inc.; 2008. [Google Scholar]
- 20.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 21.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. 3rd edn. Vol. 3. New York: Cold Spring Harbor Laboratory Inc; 2001. [Google Scholar]
- 22.Dovbeshko GI, Gridina NY, Kruglova EB, Pashchuk OP. FTIR spectroscopy studies of nucleic acid damage. Talanta. 2000;53:233–246. doi: 10.1016/S0039-9140(00)00462-8. [DOI] [PubMed] [Google Scholar]
- 23.Qin W, Zhu W, Hewett JE, Rottinghaus G, Chen YC, Flynn JT, et al. uPA is upregulated by high dose celecoxib in women at increased risk of developing breast cancer. BMC Cancer. 2008;8:298. doi: 10.1186/1471-2407-8-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sheikh F, Jin Y, Pasumarthi KB, Kardami E, Cattini PA. Expression of fibroblast growth factor receptor-1 in rat heart H9c2 myoblasts increases cell proliferation. Mol Cell Biochem. 1997;176:89–97. doi: 10.1023/A:1006879029333. [DOI] [PubMed] [Google Scholar]
- 25.Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zitvogel L, et al. Cell death modalities: classification and pathophysiological implications. Cell Death Differ. 2007;14:1237–1243. doi: 10.1038/sj.cdd.4402148. [DOI] [PubMed] [Google Scholar]
- 26.Golstein P, Kroemer G. Cell death by necrosis: towards a molecular definition. Trends Biochem Sci. 2007;32:37–43. doi: 10.1016/j.tibs.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 27.Zhang W, Shi HY, Zhang M. Maspin overexpression modulates tumor cell apoptosis through the regulation of Bcl-2 family proteins. BMC Cancer. 2005;5:50. doi: 10.1186/1471-2407-5-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang B, Liu M, Tang HK, Ma HB, Wang C, Chen X, et al. The expression and significance of MRP1, LRP, TOPOIIbeta, and BCL2 in tongue squamous cell carcinoma. J Oral Pathol Med. 2012;41:141–148. doi: 10.1111/j.1600-0714.2011.01066.x. [DOI] [PubMed] [Google Scholar]
- 29.Greenberg ME, Ziff EB. Stimulation of 3T3 cells induces transcription of the c-fos proto-oncogene. Nature. 1984;311:433–438. doi: 10.1038/311433a0. [DOI] [PubMed] [Google Scholar]
- 30.Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol. 2002;4:E131–E136. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- 31.Lahijani MS, Farivar S, Khodaeian M. Effects of 50 Hz electromagnetic fields on the histology, apoptosis, and expression of c-Fos and beta-catenin on the livers of preincubated white Leghorn chicken embryos. Electromagn Biol Med. 2011;30:158–169. doi: 10.3109/15368378.2011.596603. [DOI] [PubMed] [Google Scholar]
- 32.Stuart BH. Biological applications of infrared spectroscopy. 1st edn. New York: John Wiley & Sons, Inc.; 1997. [Google Scholar]
- 33.Jamin N, Miller L, Moncuit J, Fridman WH, Dumas P, Teillaud JL. Chemical heterogeneity in cell death: combined synchrotron IR and fluorescence microscopy studies of single apoptotic and necrotic cells. Biopolymers. 2003;72:366–373. doi: 10.1002/bip.10435. [DOI] [PubMed] [Google Scholar]
- 34.Holman HY, Martin MC, Blakely EA, Bjornstad K, McKinney WR. IR spectroscopic characteristics of cell cycle and cell death probed by synchrotron radiation based Fourier-transform IR spectromicroscopy. Biopolymers. 2000;57:329–335. doi: 10.1002/1097-0282(2000)57:6<329::AID-BIP20>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]