

Abstract
In the effort to produce proteins coded by diverse genomes, structural genomics projects often must express genes containing codons that are rare in the production strain. To address this problem, genes expressing tRNAs corresponding to those codons are typically coexpressed from a second plasmid in the host strain, or from genes incorporated into production plasmids. Here we describe the modification of a series of LIC pMCSG vectors currently used in the high-throughput production of proteins to include crucial tRNA genes covering rare codons for Arg (AGG/AGA) and Ile (AUA). We also present variants of these new vectors that allow analysis of ligand binding or co-expression of multiple proteins introduced through two independent LIC steps. Additionally, to accommodate the cloning of multiple large proteins, the size of the plasmids was reduced by approximately one kilobase through the removal of non-essential DNA from the base vector. Production of proteins from core vectors of this series validated the desired enhanced capabilities: higher yields of proteins expressed from genes with rare codons occurred in most cases, biotinylated derivatives enabled detailed automated ligand binding analysis, and multiple proteins introduced by dual LIC cloning were expressed successfully and in near balanced stoichiometry, allowing tandem purification of interacting proteins.
Keywords: LIC, rare codons, tRNA genes, His-tag, co-expression, biotinylation, ligand binding, high-throughput, structural genomics
Introduction
The extensive genome sequence data made available through systematic sequencing of individual and community genomes allows for global and integrated analysis of complex biological systems. Structural Genomics programs, such as the Protein Structure Initiative (PSI), have used this data to expand our understanding of protein structure/function relationships by determining the structures of numerous novel proteins. Structures of individual members of a protein sequence family provide structural and often functional information for the whole family (1). To achieve this objective efficiently, several groups have developed new technical approaches to accelerate protein structure determination (2-6). While the ‘pipelines’ developed by these groups are not optimized for individual proteins, they work well and can be applied to many related proteins to obtain relevant structural information. Current efforts in PSI:Biology within the established structural genomics centers and collaboratively with Biology Partnerships and the biology community, however, focus more specifically on proteins of known function that participate in important cellular processes related to human health and disease (http://www.nigms.nih.gov/News/Reports/PSIAC_Future_2009.htm). This approach places less emphasis on determining the structures of proteins distantly related to the target. It also forces the use of salvage methods such as alternative construct design, cloning, purification and crystallization strategies in order to obtain the structure of the specifically desired target or its very close variant. That said, modifications to existing high-throughput pipelines can diversify and expand the options for producing these proteins. Target proteins that are large or form functional multicomponent complexes with other proteins require alternative cloning strategies. Several approaches have already been developed to allow coexpression of multiple proteins, either through expression of separate genes or through cloning or construction of operons expressing a set of proteins from a single promoter (7-11). The option to append removable purification affinity tags to selected members of coexpressed proteins at either the N- or C-terminal should improve the chances of creating constructs that fold and interact properly (12,13). Additionally, crystallization can often be enhanced by binding of ligands to the purified proteins, which can add considerable relevance to the determined structure (14-19). For these purposes, proteins appended with appropriate affinity tags can allow for efficient identification of interacting ligands and determination of their binding affinities.
To address the above mentioned challenges, we have constructed a series of new vectors based on a previous vector set used extensively by the Midwest Center for Structural Genomics (MCSG) in the high-throughput production of single proteins (12,13,20). The core modifications of the vectors were the addition of two genes encoding tRNAs that are rare in the expression host, E. coli, and the excision of an approximately 1 kilobase segment not essential for plasmid maintenance or protein expression in order to maintain relatively small overall size of the plasmids (Fig. 1, Table 1). The smaller size is intended to facilitate cloning of larger genes, multiple genes, or operons without the possible effects on cloning efficiency that the vectors’ larger size could cause (21). The added tRNA genes, in addition to the well established value of allowing production of proteins whose genes include rare codons (22,23), eliminates the need to provide those tRNAs on a second plasmid that is resident in the host strain used in the MCSG expression pipeline (5,24). This allows the use of a second plasmid instead to produce one or more companion proteins for production of protein complexes (11). In addition, to aid ligand screening by target proteins, we constructed a vector that appends a biotinylation site to the N-terminus of proteins and that also contains the birA gene (25,26). The coexpression of the birA gene in vivo tags the target protein with biotin. Analysis of ligand binding to the purified biotinylated proteins using biolayer interferometry (BLI) (27,28) allows for rapid, semiautomated screening of many potential ligands, facilitating crystallization and providing functional insights (14,15,17).
Fig. 1.

Design of tRNA producing LIC vectors.
Table 1.
Truncated vectors expressing tRNA genes1.
| Parental Vector3 | Protein Product2 | Final Vector |
|---|---|---|
| pMCSG7 | His6-TEV-PROTEIN | pMCSG53 |
| pMCSG19 | MBP -TVMV- His6-TEV- PROTEIN | pMCSG69 |
| pMCSG26 | PROTEIN -His6 | pMCSG58 |
| pMCSG28 | PROTEIN -TEV- His6 | pMCSG59 |
| pMCSG29 | PROTEIN -TEV-His10TVMV-MBP | pMCSG70 |
| pMCSG32 | MBP -TVMV- PROTEIN -TEV-His6 | pMCSG71 |
| pMCSG50 | His6-biotin-TEV- PROTEIN | pMCSG62 |
| pMCSG60 | His6-TEV- PROTEIN 1, PROTEIN 2 | pMCSG63 |
| pMCSG63 | His6-TEV- PROTEIN 1, PROTEIN 2 (pCDF ori) | pMCSG76 |
| pMCSG63 | His6-TEV- PROTEIN 1, PROTEIN 2 (p15A ori) | pMCSG77 |
Nonessential region of approximately 1 kb excised and genes and promotors encoding ileX and argU tRNAs added to parental vector (see materials and methods).
Target proteins (PROTEIN) encoded into the vector are made with appended sequences to aid in purification or analysis. Tag, listed from N- to C- terminus, were His6, hexahistidine; TEV, tobacco etch virus protease recognition sequence (34); MBP, maltose binding protein; TVMV, tobacco vein mottling virus protease recognition sequence (35); biotin, biotinylation sequence, which in the presence of coexpressed BirA ligase adds covalently linked biotin. For vectors 63, 76, and 77 PROTEIN 1 is the protein produced after cloning into LIC site 1, PROTEIN 2 the tag-less protein expressed on cloning into LIC site 2 (see text). All vectors are pBR322-based (AmpR) except pMCSG76 (Clo DF13, SpecR) and pMCSG77 (p15A, KanR).
Ten new pMCSG LIC vectors were constructed. LIC vectors expressing rare tRNAs were created by the introduction of the genes ileX and argU from E. coli BL21 DE3, encoding tRNAs for arginine and isoleucine, into the Sph I and SgrA I restriction sites, respectively, of the parental vector pMCSG7. These tRNAs cover three rare codons in E. coli for Arg (AGG/AGA) and Ile (AUA). Subsequent excision of approximately 1 kb of DNA by digestion with PshA I and Tth111 I completed the construction of pMCSG53. Replacement of the region between Xba I and Bgl I of pMCSG53 with expression cassettes from established production vectors allowed production of proteins with a variety of tags and cleavage sites (Table 1). Addition of a biotinylation site to the pMCSG7 LIC region and the birA gene outside the expression region allowed for construction of pMCSG62 through a similar truncation and tRNA gene addition. For coexpression of multiple proteins, a second different LIC site was introduced at a Sma I site to give pMCSG63. Variants of pMCSG63 with different origins of replication were constructed by insertion of the tRNA and LIC regions from pMCSG63 into plasmids with the p15A and pCDF origins (Materials and Methods).
Materials and Methods
Truncated LIC vector
A smaller version of our standard LIC vector was constructed from pMCSG7 (20). Vector pMCSG7 was digested with the restriction enzymes BsaA I and PshA I, removing a fragment of DNA about 1 kb in length containing the rop repressor coding sequence and flanking sequences from the pBR322 origin of replication. The plasmid fragments were separated by agarose gel electrophoresis and the larger fragment was extracted with the QIAEX II Gel Extraction Kit (Qiagen, Inc., Valencia, CA) following the manufacturer’s instructions. The purified linear plasmid was re-circularized by ligation with T4 DNA Ligase (Invitrogen Life Technologies, Grand Island, NY). The resulting plasmid was designated pMCSG49 and is 4278 bp in length.
LIC vector containing rare tRNAs
The ileX tRNA gene that encodes the tRNA recognizing the AUA codon for Ile, along with the endogenous promoter and terminator sequences (22) was synthesized by PCR of E. coli BL21 genomic DNA using Platinum Pfx DNA Polymerase (Invitrogen) with primers that incorporated the Sph I restriction site at each end. The purified PCR product was ligated into the Sph I site of vector pMCSG7. The E. coli argU tRNA gene that encodes the tRNA recognizing AGA and AGG for Arg with its endogenous promoter and terminator (23,29) was synthesized by PCR of E. coli BL21 genomic DNA with primers containing the SgrA I restriction site. The purified fragment was ligated into the SgrA I site of the pMCSG7 plasmid already containing the ileX gene. The resulting vector was digested with PshA I and Tth111 I to remove the rop repressor and flanking sequences, followed by treatment with the Klenow fragment of DNA polymerase and dNTPs to create flush ends. The re-circularized plasmid (pMCSG53) is 4808 bp in length and contains both tRNA genes in the counter-clockwise orientation (Fig. 1).
Dual LIC vector
An expression vector containing two LIC sites with associated ribosome-binding sites (rbs) and controlled by a single T7 promoter was constructed from pMCSG7. Two 71-mer synthetic oligonucleotides that contain the single-stranded Ssp I LIC overhangs when annealed were cloned into pMCSG7 by the standard LIC procedure (24). The resulting plasmid (pMCSG60) contained the original pMCSG7 LIC region followed by the rbs and LIC region from pMCSG26 without the complete 3′ His-tag. (12). This allows the cloning of a second protein coding sequence using the standard pMCSG26 primers with the inclusion of a termination codon. This gene will be expressed without an affinity tag as a part of an artificial operon. A shortened version of pMCSG60 containing the two rare tRNA genes was also constructed. The LIC region and a portion of the β-lactamase coding region from pMCSG60 were removed by digestion with Bgl I and Xba I. This fragment was ligated into pMCSG53 between the same restriction sites. The resulting 4864 bp plasmid (pMCSG63) contains the dual LIC cloning region along with the two rare tRNA genes. Variants of pMCSG63 with different origins of replication (30) were also constructed to allow co-transformation with our standard pBR322-based plasmids. The tRNA and dual LIC region from pMCSG63 was synthesized by PCR with primers that added the restriction sites EcoN I and Tth111 I at opposite ends. After digesting with these enzymes, the PCR product was ligated into the plasmid pMCSG21 (10) digested with the same enzymes. The resulting plasmid (pMCSG76) contains the Clo DF13 origin of replication and streptomycin/spectinomycin resistance. A related plasmid (pMCSG77) was constructed with the pACYC177-based vector pMGK (5,24) as the vector backbone. The dual LIC region with tRNA genes from pMCSG63 was synthesized by PCR with primers that added the restriction site BsrG I at the 3′ end. After digestion with BsrG I the DNA was ligated into pMGK digested with BsrG I and Sca I. It was first necessary to remove a Sma I site and an Ssp I site from the Tn903 kanamycin resistance gene in pMGK. This was done by introducing two point mutations through a two-step PCR. The PCR product was digested with Xho I and Hind III, then ligated into pMGK between the same two sites. The resulting vectors, pMCSG76 and pMCSG77, are compatible with each other and pMCSG53.
Vector for producing biotinylated proteins
A vector containing a 15 residue biotinylation site (31) between the His-tag and the TEV protease recognition sequence along with the E. coli birA biotin ligase gene was constructed by modifying the dual LIC plasmid pMCSG60. The birA gene was synthesized by a two-step PCR of E. coli BL21 genomic DNA in order to remove an Ssp I restriction site by point mutation. The first round of PCR produced two DNA fragments with overlapping sequences at one end. After gel purification the fragments were combined in a second PCR reaction and amplified. The DNA was purified by agarose gel electrophoresis, treated with T4 DNA Polymerase (EMD Millipore, Billerica, MA) and dTTP, then inserted into the second LIC site of pMCSG60 by the standard LIC procedure (12). A complementary pair of synthetic oligonucleotides encoding the His6-tag and the biotinylation site were annealed, resulting in single-strand overhangs for the Nde I and Acc65 I restriction sites. This double-stranded oligonucleotide was ligated into the birA gene-containing vector that had been digested with the same two enzymes. The resulting plasmid was designated pMCSG50. An approximately 1 kb region of pMCSG50 was removed by digestion with PshA I and Tth111 I followed by treatment with the Klenow fragment of DNA polymerase and dNTPs, and re-circularization with T4 DNA ligase. The LIC region, BirA coding region and a portion of the β-lactamase gene was removed by digestion with Xba I and Bgl I and then ligated into pMCSG53 that had been digested with the same two enzymes. The resulting vector is a shortened version of pMCSG50 containing the two rare tRNA genes from pMCSG53. This new plasmid was designated pMCSG62.
Analytical procedures
Plasmids were purified by a modification of the alkaline lysis method (32) and analyzed by agarose gel electrophoresis after digestion with restriction endonucleases. Potential successful constructs were verified by partial sequencing using primers that spanned the modified portion of the vectors at the DNA Sequencing & Genotyping Facility, University of Chicago. All restriction enzymes and their respective 10× buffers were obtained from New England BioLabs, Inc., Ipswich, MA. Function of the vectors was evaluated by high-throughput, automated LIC or manual LIC of target genes into the vectors using previously described procedures (10,33). For the dual LIC vectors, a gene or genes were first introduced into LIC1, followed by addition of a gene or genes to the LIC2 site in the resulting construct by standard LIC protocols (10,12). Following induction of constructs introduced into BL21(DE3) hosts, expressed proteins were analyzed by polyacrylamide gel electrophoresis (PAGE) under denaturing conditions.
Purification of biotinylated protein
Inosine 5′-monophosphate dehydrogenase from B. anthracis (BaIMPDH) was chosen as a test protein for the biotinylated vector system. BaIMPDH was purified according to a standard protocol (15) that was modified to include only a single IMAC purification step. Specifically, lysis was performed as described previously (15), the lysate was clarified by centrifugation at 36,000g for 1 h and filtered through a 0.44 μm membrane. Clarified lysate was applied to a 5 mL HiTrap Ni-NTA column (GE Healthcare Life Sciences, Piscataway, NJ) on an ÄKTAxpress system (GE Healthcare Life Sciences). The column was washed with lysis buffer (50 mM HEPES pH 8.0, 500 mM KCl, 5% glycerol, 10 mM β-mercaptoethanol) containing 20 mM imidazole, and the protein was eluted with the same buffer containing 250 mM imidazole. Fractions containing target protein were pooled, concentrated, and loaded onto a Superdex 300 16/60 size exclusion chromatography column equilibrated with buffer containing 50 mM Tris-HCl (pH 8.0), 100 mM KCl, 1 mM DTT, 3 mM EDTA, and 10% glycerol.
Analysis of biotinylated protein-inhibitor interactions using biolayer interferometry
Analysis of the binding of inhibitors to IMPDH proteins was performed using BLI (27,28) on the Octet RED (ForteBio, Menlo Park, CA). Assays were performed in 96-well black microplates (Fisher Scientific, Pittsburgh, PA) at 25 °C. All volumes were 300 μL. Biotinylated IMPDH was loaded onto Super Streptavidin (SSA) Biosensors (ForteBio, Menlo Park, CA) at a concentration of 50 μg/mL in phosphate-buffered saline (PBS). Reference SSA Biosensors were blocked with biocytin at 10 μg/mL in PBS buffer. IMPDH inhibitors A110 and C91 (obtained as a gift from Dr. Lizbeth Hedstrom) (16) were titrated in triplicates from 0.2 nM to 10 μM in running buffer containing 50 mM Tris-HCl (pH 8), 100 mM KCl, 1 mM DTT, 3 mM EDTA, 0.1 mg/mL BSA, 5% DMSO, 1 mM IMP, and 1.2 mM NAD+. Inhibitor association and dissociation events were measured for 240 and 120 seconds, respectively. Assays were run first using the protein biosensors, followed by the reference biosensors using the same protocol to remove system artifacts and minor buffer inconsistencies. To correct for signal drift associated with the target protein, one or two reference biosensors exposed to running buffer were analyzed in parallel with the biosensors undergoing sample analysis. The reference data were then subtracted from the protein-inhibitor data. The association and dissociation curves were fit using a single-exponential fitting model using analysis software provided by the manufacturer.
Results
Truncated vectors encoding rare tRNAs
Ten new vectors based on the production vector pMCSG7 and its derivatives were constructed by removal of approximately 1 kb of non-essential DNA and addition of genes encoding two tRNAs (Table 1). The first six vectors listed express individual proteins in various configurations to allow high-throughput purification by appending polyhistidine tags of various lengths (His6 or His10), with or without maltose-binding protein (MBP), either cleavable or not, to either the N- or C-termini of target proteins. Vector pMCSG62 and its precursor pMSCG50 produce proteins labeled with the affinity tag biotin (in addition to the polyhistidine tag) for automated evaluation of ligand binding (27,28). The final three vectors combine elements of both the N- and C-terminal tagging vectors, which use different LIC sites, to allow coexpression of multiple proteins introduced by sequential LIC reactions into the two sites. The presence of this expression region in three vectors with different origins of replications in principle allows expression of up to six transcripts simultaneously (11), each potentially encoding one or more proteins.
Production of single proteins for his-tag purification
Expression vectors designed for the high-throughput production of single proteins, pMCSG53, pMCSG58, pMCSG59, pMCSG69, pMCSG70, and pMCSG71, were verified by DNA sequence analysis to contain all designed modifications. To evaluate the effect of the introduction of the tRNA genes, six protein genes containing a high proportion of rare codons and one with few rare codons were selected (Table 2) and introduced into pMCSG49, the truncated intermediate vector lacking tRNA genes, and pMCSG53, which contained the rare tRNA genes. Following induction, protein expression level was analyzed by PAGE (Table 2). The gels were loaded with protein extracted from a fixed volume of culture to insure protein bands reflect the total productivity of the construct. As a consequence, bands from highly expressed genes are overloaded slightly. The gene encoding protein APC100724 was included as a control because it contained only three rare codons (less than 8% of the total Arg and Ile codons). Its expression was not improved by expression in the tRNA vector; in fact, less protein was observed than in the parental vector. However, expression of all six genes containing rare tRNA codons (range 10-34 rare codons, corresponding to 40-64% of Arg and Ile codons) clearly improved with inclusion of the tRNA genes.
Table 2.
Expression of genes containing rare tRNA codons.
| Protein1 | Size (kDa)2 | Arg Codons3 | Ile Codons3 | pMCSG49 | pMCSG53 |
|---|---|---|---|---|---|
| 100724 | 40.5 | 3/17 | 0/22 |

|

|
| 101279 | 20.7 | 7/16 | 3/9 |

|

|
| 100441 | 29.6 | 9/19 | 4/10 |

|

|
| 100385 | 36.6 | 17/17 | 9/28 |

|

|
| 100395 | 40.8 | 17/17 | 12/40 |

|

|
| 100375 | 45.5 | 22/22 | 17/39 |
|

|
| 100369 | 29.6 | 12/12 | 14/25 |

|

|
Seven genes containing varying numbers of rare codons for isoleucine and arginine were cloned into the LIC site of pMCSG49 and pMCSG53, expressed and analyzed by standard procedures (33). Extracts of total protein were separated on polyacrylamide gels. Individual bands corresponding to the overexpressed proteins from the scanned gel image are displayed.
Protein number is the APC number in the MCSG database (http://olenka.med.virginia.edu/mcsg/) and corresponds to the following proteins (100724 - subunit II of cytochrome C oxidase from Sphaerobacter thermophilus, 101279 - response regulator receiver domain-containing protein from Planctomyces limnophilus, 100441 - CobB/CobQ domain protein glutamine amidotransferase from Desulfotomaculum acetoxidans, 100385 - Ppx/GppA phosphatase from Anaerococcus prevotii, 100395 - heat-inducible transcription repressor HrcA from A. prevotii , 100369 - 4-diphosphocytidyl-2C-methyl-Derythritolsynthase from A. prevotii and 100375 - glycogen biosynthesis protein from A. prevotii).
Predicted molecular weight based on expressed protein plus N-terminal His6-tag.
Rare/total codons. For Arg, values are AGA + AGG over total; for Ile, AUA over total.
Biotinylation of target proteins and analysis of ligand binding
To allow analysis of ligand binding, sequences appending a biotinylation peptide and the gene encoding the biotinylating enzyme, BirA, were introduced into pMCSG7 to give the vectors, pMSCG50 and pMCSG62. The changes were verified by sequence analysis (Fig. 2).
Figure 2.
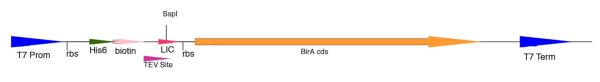
Vectors producing a biotinylation site in the leader sequence of proteins.
The birA gene from E. coli BL21 was synthesized by PCR and cloned into the second LIC site of pMCSG60 by the standard procedure. A pair of synthetic oligonucleotides encoding the 15-amino acid biotinylation site was inserted upstream of the TEV recognition sequence by ligation. T7 Prom, T7 promoter; rbs, ribosome binding site: His6, N-terminal his6-tag; TEV Site, tobacco etch virus protease cleavage site; LIC, ligation independent cloning site; BirA cds, coding sequence for BirA protein; T7 Term, T7 terminator.
The utility of this construct was established by introduction of the gene encoding IMPDH from Bacillus anthracis and analysis of the interaction of the protein and two known IMPDH noncompetitive inhibitors, compounds A110 and C91 (16). Binding of the inhibitors was monitored utilizing BLI (27,28) on an Octet RED instrument (ForteBio). The BLI technique generates an interference pattern by monitoring visible light reflected from two surfaces within the fiber-optic biosensor. When a biological molecule binds to the biosensor tip surface, a shift in the interference pattern can be measured and reported as a change in wavelength as a function of time. This wavelength shift is reported in relative intensity units, nm. Biotinylated BaIMPDH was overexpressed from vector pMSCG50 and purified as described in Materials and Methods. Manufacturer recommended amounts (5-8 nM) of biotinylated BaIMPDH were loaded onto super-streptavidin (SSA) biosensors (Fig. 3).
Figure 3.

Real-time binding of biotinylated BaIMPDH to super-streptavidin biosensors. Biosensors A1-H1 were loaded with the BaIMPDH enzyme. Biosensor H2 was loaded with biocytin and was used as a negative control. Region 0-900 seconds indicates protein loading in PBS buffer, region 901-1020 seconds shows blocking of the unused streptavidin sites with biocytin, and region 1021-2021 seconds show equilibration of loaded biosensors in the running buffer.
Compounds A110 and C91 were titrated from 0.2 nM to 10 μM in the running buffer. Loaded protein biosensors were then used to obtain binding responses from the prepared inhibitor dilution series (Fig. 4A). The association and dissociation curves were fitted using a single-exponential fitting model to determine the apparent dissociation constant, KD. Binding of A110 and C91 to BaIMPDH produced KD values of 150 ±20 and 150 ±40 nM, respectively (Fig. 4B). The KD values obtained using BLI agree within three-fold with the IC50 values obtained spectrometrically (57 ±7 and 57 ±1 nM for A110 and C91, respectively, personal information). The IC50 value can be used as an approximation of the KD value for a noncompetitive inhibition (37), the type of inhibition observed for the case presented here. Confirmation of A110 and C91 binding allowed for successful co-crystallization experiments.
Figure 4.

Binding of inhibitors to biotinylated BaIMPDH. A. Association and dissociation responses for serial diltions of inhibitors A110 and C91. Values associated with lines are concentrations of the inhibitors in μM. B. Steady-state analysis plots used to determine KD.
Coexpression of multiple proteins via dual LIC sites
For coexpression of multiple protein genes, a second LIC site was introduced into pMCSG7 to give the vector pMCSG60. The expression region of this vector combines the distinct LIC regions of pMCSG7 and pMCSG26 (Fig. 5). Sequencing indicated the expected changes. The entire LIC region with the tRNA genes was transferred into vectors with alternative replication origins to give pMCSG76 and pMCSG77 (Materials and Methods).
Figure 5.
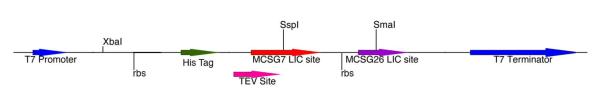
Diagrammatic representation of the LIC region of dual-expression vectors.
Insertion of the rbs and LIC region from pMCSG26 into pMCSG7 generated the dual-expression vector pMCSG60. Abbreviations are as in Fig. 2. The pMCSG7 LIC site appends an N-terminal his6-tag, whereas the pMCSG26 LIC region generates an untagged product (12). Insertion of the pMCSG60 LIC region into pMCSG53 generated the dual-expression plus rare tRNA vector pMCSG63. Dual-expression plus rare tRNA co-transformation vectors pMCSG76 and pMCSG77 were constructed by inserting the LIC and tRNA regions from pMCSG63 into vectors containing the Clo DF13 and p15A origin of replication, respectively, to generate the family of vectors, pMCSG63 (pBR322, AmpR), pMCSG76 (Clo DF13, SpecR), and pMCSG77 (p15A, KanR).
Multi-gene expression from the dual LIC sites was demonstrated in pMCSG60. The GroES/GroEL operon from 72 bacterial species was inserted into the second LIC site of pMCSG60 by high-throughput cloning, and demonstrated to produce the chaperone pair (manuscript in preparation). To the resulting clone carrying the operon GroES/GroEL from B. cereus, the approximately 7.1 kbp six-gene petrobactin operon from B. cereus was synthesized by PCR and inserted into the first LIC. After induction of protein synthesis in E. coli BL21 DE3, total protein and Nickel NTI bound and unbound fractions (Maxwell 16, Promega Corporation, Madison, WI) were analyzed by polyacrylamide gel electrophoresis. Bands corresponding to six of the eight B. cereus proteins were identified by Mass Spectrometry (Fig. 6). GroES and AsbD, the two smallest proteins, were not identified. The presence of the other six proteins demonstrates protein synthesis from both LIC sites as well as protein synthesis from the native B. cereus ribosome binding sites.
Figure 6.

Coexpression of the asb and groES/groEL operons from B. cereus in pMCSG60. A. Graphical map of the pMCSG60 plasmid with the ~2 kbp B. cereus groES/groEL operon inserted into the second LIC site and the ~7.1 kbp six-gene B. cereus petrobactin operon inserted into the first LIC site. B. PAGE analysis of synthesized protein. Total: total protein synthesized. Unbound: protein not bound to Nickel NTI. Eluted: protein eluted from Nickel NTI beads. The identities of the labeled proteins (AsbA, AsbB, AsbC, AsbE and AsbF and GroEL) were confirmed by mass spectrometry of bands excised from the gel.
Discussion
In PSI:Biology, often desired target proteins are encoded by genes which employ codons that are rare in E. coli, the standard host for HTP protein production. Expression of such genes is well known to lead to translational problems in high-level production of the proteins (38,39). Typically, addition of a helper plasmid encoding two or three rare tRNAs alleviates this problem (22,38,40). However, the presence of an additional plasmid places restrictions on cloning and expression strategies, in particular where a decreased metabolic burden on the host cells is desired or when coexpression of proteins from multiple plasmids is required. These restrictions can be removed by adding tRNA encoding genes into the expression plasmid, thereby providing the needed tRNAs and the message for the target protein from a single plasmid (41,42). To this end, we added the native E. coli tRNA genes, ileX and argU, under control of their native promoter and terminator, to the standard production plasmid pMCSG7. To address the potential detrimental effect of the increased size of the basic plasmid (43), we also eliminated 1 kb of non-essential DNA from the plasmid so that the resulting vector, pMCSG53, was approximately the same size as pMCSG7. Analysis of the expression of genes containing rare codons showed that the increased copy number of these genes resulting from their presence on the resulting plasmid provided sufficient rare tRNAs to improve the production of proteins encoded by rare codons (Table 2). The new vectors are fully compatible with the well-established HTP protocols used at the MCSG and accept the same PCR products (33).
Many protein targets of PSI:Biology, selected based on their importance to fundamental biological studies or their known roles in health-related processes, function in the cell as heterooligomers composed of different proteins. Here we use the asb operon as an example (44). This operon is responsible for biosynthesis of the bacterial siderophore, petrobactin, an important virulence factor in B. anthracis (45,46). Whereas it is sometimes possible to mix proteins overexpressed separately to achieve the native heteroligomer, proper folding and association of the proteins may require coexpression (7-10). Commercially available expression vectors allow cloning of multiple genes into single plasmids, as well as introduction of multiple expression vectors with different origins of replication into a single host cell (11). However, these vectors are not compatible with HTP protocols, in particular those using LIC to introduce the genes. Here we describe a set of vectors containing two independent LIC sites compatible with established HTP protocols. The number of genes expressed by a single vector can be increased by introducing operons instead of single genes, and we have demonstrated this approach by sequential introduction of the B. cereus groEL/ES operon and the B. cereus asb operon into the dual LIC vector pMCSG60. Expression of these genes resulted in the production of at least six proteins from this single vector (Fig. 6). In this particular configuration, only one of these proteins was tagged with an N-terminal his6-tag. Following automated purification by HTP protocols, the six proteins were partially purified and their identities verified by MS. Two companion vectors to pMCSG60 have the identical cloning regions in vectors with different origins of replication, allowing the introduction of up to six fragments of DNA cloned by LIC into a single host. The sequential introduction of multiple compatible expression vectors and coexpression of multiple proteins have been demonstrated previously (10,11).
Analysis of the functional role of proteins often entails characterization of the binding parameters of known ligands, such as substrates, products and effectors, and also includes the identification of novel ligands. Addition of peptide sequences containing a biotinylation site to recombinant proteins can allow semi-automated analysis of ligand binding by biolayer interferometry (18,19). In addition to providing functional insights, the characterization of ligand binding provides an additional tool for improving protein crystallization by guiding the crystallization of alternative forms of a protein or reducing conformational heterogeneity by locking the protein into a single conformation, thereby both increasing the success rate and providing higher resolution data (14,17). Accordingly, the base vector of the MCSG was modified to allow LIC of standard PCR products to link the cloned genes to a leader sequence encoding a His6-tag followed by a biotinylation site and the TEV protease recognition sequence. Following purification by HTP methods, proteins were analyzed by biolayer interferometry (27,28) to provide binding constants (Figs. 3 and 4). The two approaches coupled in this vector, HTP cloning/purification and semiautomated binding analysis, allowed for rapid comparison of the binding of a set of inhibitors to a series of IMPDH enzymes from different bacteria to generate detailed insight into the function of this important enzyme (16).
All new pMCSG expression vectors described in this manuscript are available via PSI Material Repository (http://psimr.asu.edu/).
Acknowledgements
This work was supported by the National Institute of Health Grant GM094585, the National Institute of Allergy and Infectious Diseases Contracts HHSN27200700058C and HHSN272201200026C, and by the US Department of Energy office of Biological and Environmental Research under Contract No. DE-AC02-06CH11357.
References
- 1.Fox BG, Goulding C, Malkowski MG, Stewart L, Deacon A. Nat Methods. 2008;5:129–132. doi: 10.1038/nmeth0208-129. [DOI] [PubMed] [Google Scholar]
- 2.Elsliger MA, Deacon AM, Godzik A, Lesley SA, Wooley J, Wuthrich K, Wilson IA. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2010;66:1137–1142. doi: 10.1107/S1744309110038212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Graslund S, Nordlund P, Weigelt J, Hallberg BM, Bray J, Gileadi O, Knapp S, Oppermann U, Arrowsmith C, Hui R, Ming J, dhe-Paganon S, Park HW, Savchenko A, Yee A, Edwards A, Vincentelli R, Cambillau C, Kim R, Kim SH, Rao Z, Shi Y, Terwilliger TC, Kim CY, Hung LW, Waldo GS, Peleg Y, Albeck S, Unger T, Dym O, Prilusky J, Sussman JL, Stevens RC, Lesley SA, Wilson IA, Joachimiak A, Collart F, Dementieva I, Donnelly MI, Eschenfeldt WH, Kim Y, Stols L, Wu R, Zhou M, Burley SK, Emtage JS, Sauder JM, Thompson D, Bain K, Luz J, Gheyi T, Zhang F, Atwell S, Almo SC, Bonanno JB, Fiser A, Swaminathan S, Studier FW, Chance MR, Sali A, Acton TB, Xiao R, Zhao L, Ma LC, Hunt JF, Tong L, Cunningham K, Inouye M, Anderson S, Janjua H, Shastry R, Ho CK, Wang D, Wang H, Jiang M, Montelione GT, Stuart DI, Owens RJ, Daenke S, Schutz A, Heinemann U, Yokoyama S, Bussow K, Gunsalus KC. Nat Methods. 2008;5:135–146. [Google Scholar]
- 4.Peti W, Page R, Moy K, O’Neil-Johnson M, Wilson IA, Stevens RC, Wuthrich K. J Struct Funct Genomics. 2005;6:259–267. doi: 10.1007/s10969-005-9000-x. [DOI] [PubMed] [Google Scholar]
- 5.Price WN, 2nd, Handelman SK, Everett JK, Tong SN, Bracic A, Luff JD, Naumov V, Acton T, Manor P, Xiao R, Rost B, Montelione GT, Hunt JF. Microb Inform Exp. 2011;1:6. doi: 10.1186/2042-5783-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xiao R, Anderson S, Aramini J, Belote R, Buchwald WA, Ciccosanti C, Conover K, Everett JK, Hamilton K, Huang YJ, Janjua H, Jiang M, Kornhaber GJ, Lee DY, Locke JY, Ma LC, Maglaqui M, Mao L, Mitra S, Patel D, Rossi P, Sahdev S, Sharma S, Shastry R, Swapna GV, Tong SN, Wang D, Wang H, Zhao L, Montelione GT, Acton TB. J Struct Biol. 2010;172:21–33. doi: 10.1016/j.jsb.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kerrigan JJ, Xie Q, Ames RS, Lu Q. Protein expression and purification. 2011;75:1–14. doi: 10.1016/j.pep.2010.07.015. [DOI] [PubMed] [Google Scholar]
- 8.Perrakis A, Romier C. Methods Mol Biol. 2008;426:247–256. doi: 10.1007/978-1-60327-058-8_15. [DOI] [PubMed] [Google Scholar]
- 9.Scheich C, Kummel D, Soumailakakis D, Heinemann U, Bussow K. Nucleic Acids Res. 2007;35:e43. doi: 10.1093/nar/gkm067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stols L, Zhou M, Eschenfeldt WH, Millard CS, Abdullah J, Collart FR, Kim Y, Donnelly MI. Protein expression and purification. 2007;53:396–403. doi: 10.1016/j.pep.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 11.Tolia NH, Joshua-Tor L. Nat Methods. 2006;3:55–64. doi: 10.1038/nmeth0106-55. [DOI] [PubMed] [Google Scholar]
- 12.Eschenfeldt WH, Maltseva N, Stols L, Donnelly MI, Gu M, Nocek B, Tan K, Kim Y, Joachimiak A. J Struct Funct Genomics. 2010;11:31–39. doi: 10.1007/s10969-010-9082-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eschenfeldt WH, Stols L, Millard CS, Joachimiak A, Donnelly MI. Methods Mol Biol. 2009;498:105–115. doi: 10.1007/978-1-59745-196-3_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hassell AM, An G, Bledsoe RK, Bynum JM, Carter HL, 3rd, Deng SJ, Gampe RT, Grisard TE, Madauss KP, Nolte RT, Rocque WJ, Wang L, Weaver KL, Williams SP, Wisely GB, Xu R, Shewchuk LM. Acta Crystallogr D Biol Crystallogr. 2007;63:72–79. doi: 10.1107/S0907444906047020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim Y, Babnigg G, Jedrzejczak R, Eschenfeldt WH, Li H, Maltseva N, Hatzos-Skintges C, Gu M, Makowska-Grzyska M, Wu R, An H, Chhor G, Joachimiak A. Methods. 2011;55:12–28. doi: 10.1016/j.ymeth.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Makowska-Grzyska M, Kim Y, Wu R, Wilton R, Gollapalli DR, Wang XK, Zhang R, Jedrzejczak R, Mack JC, Maltseva N, Mulligan R, Binkowski TA, Gornicki P, Kuhn ML, Anderson WF, Hedstrom L, Joachimiak A. Biochemistry. 2012;51:6148–6163. doi: 10.1021/bi300511w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vedadi M, Niesen FH, Allali-Hassani A, Fedorov OY, Finerty PJ, Jr., Wasney GA, Yeung R, Arrowsmith C, Ball LJ, Berglund H, Hui R, Marsden BD, Nordlund P, Sundstrom M, Weigelt J, Edwards AM. Proc Natl Acad Sci U S A. 2006;103:15835–15840. doi: 10.1073/pnas.0605224103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morris DP, Roush ED, Thompson JW, Moseley MA, Murphy JW, McMurry JL. Biochemistry. 2010;49:6386–6393. doi: 10.1021/bi100487p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rothbard JB, Zhao X, Sharpe O, Strohman MJ, Kurnellas M, Mellins ED, Robinson WH, Steinman L. J Immunol. 2011;186:4263–4268. doi: 10.4049/jimmunol.1003934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stols L, Gu M, Dieckman L, Raffen R, Collart FR, Donnelly MI. Protein expression and purification. 2002;25:8–15. doi: 10.1006/prep.2001.1603. [DOI] [PubMed] [Google Scholar]
- 21.Godiska R, Mead D, Dhodda V, Wu C, Hochstein R, Karsi A, Usdin K, Entezam A, Ravin N. Nucleic Acids Res. 2010;38:e88. doi: 10.1093/nar/gkp1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Del Tito BJ, Jr., Ward JM, Hodgson J, Gershater CJ, Edwards H, Wysocki LA, Watson FA, Sathe G, Kane JF. J Bacteriol. 1995;177:7086–7091. doi: 10.1128/jb.177.24.7086-7091.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garcia OL, Gonzalez B, Menendez A, Sosa AE, Fernandez JR, Santana H, Meneses N. Ann N Y Acad Sci. 1996;782:79–86. doi: 10.1111/j.1749-6632.1996.tb40549.x. [DOI] [PubMed] [Google Scholar]
- 24.Acton TB, Gunsalus KC, Xiao R, Ma LC, Aramini J, Baran MC, Chiang YW, Climent T, Cooper B, Denissova NG, Douglas SM, Everett JK, Ho CK, Macapagal D, Rajan PK, Shastry R, Shih LY, Swapna GV, Wilson M, Wu M, Gerstein M, Inouye M, Hunt JF, Montelione GT. Methods Enzymol. 2005;394:210–243. doi: 10.1016/S0076-6879(05)94008-1. [DOI] [PubMed] [Google Scholar]
- 25.Kay BK, Thai S, Volgina VV. Methods Mol Biol. 2009;498:185–196. doi: 10.1007/978-1-59745-196-3_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scholle MD, Collart FR, Kay BK. Protein expression and purification. 2004;37:243–252. doi: 10.1016/j.pep.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 27.Cooper MA. Drug Discovery Today. 2006;11:1061–1067. doi: 10.1016/j.drudis.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 28.Schmitt H-M, Brecht A, Piehler J, Gauglitz G. Biosensors and Bioelectronics. 1997;12:809–816. doi: 10.1016/s0956-5663(97)00010-9. [DOI] [PubMed] [Google Scholar]
- 29.Garcia GM, Mar PK, Mullin DA, Walker JR, Prather NE. Cell. 1986;45:453–459. doi: 10.1016/0092-8674(86)90331-4. [DOI] [PubMed] [Google Scholar]
- 30.Selzer G, Som T, Itoh T, Tomizawa J. Cell. 1983;32:119–129. doi: 10.1016/0092-8674(83)90502-0. [DOI] [PubMed] [Google Scholar]
- 31.Beckett D, Kovaleva E, Schatz PJ. Protein Sci. 1999;8:921–929. doi: 10.1110/ps.8.4.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Birnboim HC, Doly J. Nucleic Acids Res. 1979;7:1513–1523. doi: 10.1093/nar/7.6.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim Y, Dementieva I, Zhou M, Wu R, Lezondra L, Quartey P, Joachimiak G, Korolev O, Li H, Joachimiak A. J Struct Funct Genomics. 2004;5:111–118. doi: 10.1023/B:JSFG.0000029206.07778.fc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kapust RB, Tozser J, Fox JD, Anderson DE, Cherry S, Copeland TD, Waugh DS. Protein Eng. 2001;14:993–1000. doi: 10.1093/protein/14.12.993. [DOI] [PubMed] [Google Scholar]
- 35.Nallamsetty S, Kapust RB, Tozser J, Cherry S, Tropea JE, Copeland TD, Waugh DS. Protein expression and purification. 2004;38:108–115. doi: 10.1016/j.pep.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 36.Donnelly MI, Zhou M, Millard CS, Clancy S, Stols L, Eschenfeldt WH, Collart FR, Joachimiak A. Protein expression and purification. 2006;47:446–454. doi: 10.1016/j.pep.2005.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burlingham BT, Widlanski TS. J. Chem. Education. 2003:214–218. [Google Scholar]
- 38.Kane JF. Curr Opin Biotechnol. 1995;6:494–500. doi: 10.1016/0958-1669(95)80082-4. [DOI] [PubMed] [Google Scholar]
- 39.Rosenberg AH, Goldman E, Dunn JJ, Studier FW, Zubay G. J Bacteriol. 1993;175:716–722. doi: 10.1128/jb.175.3.716-722.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spanjaard RA, Chen K, Walker JR, van Duin J. Nucleic Acids Res. 1990;18:5031–5036. doi: 10.1093/nar/18.17.5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Novy R, Drott D, Yaeger K, Mierendorf R. inNovations. 2001;12:1. [Google Scholar]
- 42.Lee SF, Li YJ, Halperin SA. Microbiology. 2009;155:3581–3588. doi: 10.1099/mic.0.030064-0. [DOI] [PubMed] [Google Scholar]
- 43.Brown TA. Gene Cloning and DNA Analysis: An Introduction. Blackwell Pubishing; 2010. [Google Scholar]
- 44.Lee JY, Janes BK, Passalacqua KD, Pfleger BF, Bergman NH, Liu H, Hakansson K, Somu RV, Aldrich CC, Cendrowski S, Hanna PC, Sherman DH. J Bacteriol. 2007;189:1698–1710. doi: 10.1128/JB.01526-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hotta K, Kim CY, Fox DT, Koppisch AT. Microbiology. 2010;156:1918–1925. doi: 10.1099/mic.0.039404-0. [DOI] [PubMed] [Google Scholar]
- 46.Miethke M, Marahiel MA. Microbiol Mol Biol Rev. 2007;71:413–451. doi: 10.1128/MMBR.00012-07. [DOI] [PMC free article] [PubMed] [Google Scholar]