

Abstract
Objective
The American Diabetes Association has called for further research on how patient demographics should determine drug choices for individuals with type 2 diabetes mellitus (T2DM). Here, using in-depth physiology studies, we investigate whether obese patients with T2DM are likely to benefit from thiazolidinediones, medications with a known side effect of weight gain.
Materials and Methods
11 obese and 7 non-obese individuals with T2DM participated in this randomized, placebo-controlled, double-blind, crossover study. Each subject underwent a pair of “stepped” pancreatic clamp studies with subcutaneous adipose tissue biopsies following 21 days of pioglitazone (45 mg) or placebo.
Results
Obese subjects demonstrated significant decreases in insulin resistance and many adipose inflammatory parameters with pioglitazone relative to placebo. Specifically, significant improvements in glucose infusion rates, suppression of hepatic glucose production, and whole fat expression of certain inflammatory markers (IL-6, IL-1b, and iNOS) were observed in obese subjects but not in non-obese subjects. Additionally, adipose tissue from obese subjects demonstrated reduced infiltration of macrophages, dendritic cells, and neutrophils as well as increased expression of factors associated with fat “browning” (PGC-1α and UCP-1).
Conclusions
These findings support the efficacy of pioglitazone to improve insulin resistance and reduce adipose tissue inflammation in obese patients with T2DM.
Keywords: Type 2 Diabetes, Obesity, Pioglitazone, Adipose Tissue Inflammation, Insulin Resistance
Introduction
The current global epidemic of obesity has important sequelae of insulin resistance, type 2 diabetes mellitus and other non-communicable diseases. Obesity is associated with chronic low- grade inflammation, insulin resistance, and increased infiltration of macrophages into adipose tissue (1-3). In fact, adipose macrophage content is inversely correlated to insulin action in normal subjects (4). Additionally, adipose tissue itself produces several pro-inflammatory and pro- coagulant factors in proportion to the degree of adiposity (5-8). One such cytokine, IL-6, appears to be an important culprit in obesity-induced insulin resistance, atherosclerosis, and cardiovascular disease (9). Indeed, IL-6 acts on adipocytes, hepatocytes, myocytes, and pancreatic beta cells to influence the body's regulation of glucose metabolism (10). Likewise, IL-1b is purported to be involved in the pathogenesis of both T1DM and T2DM and, interestingly, it is highly expressed in the epicardial adipose tissue of patients with multiple risk factors for coronary atherosclerosis (10, 11). iNOS, too, has been implicated in insulin resistance and obesity and conversely its inhibition has been shown to reduce obesity-induced insulin resistance (12). Further studies have suggested that crosstalk between adipocytes and inflammatory cells results in greater increases in adipose tissue inflammation (4, 13, 14).
Of note, the thiazolidinedione class of anti-diabetic medications works by activating the transcription factor peroxisome proliferator-activated receptor gamma (PPARγ). PPARγ is expressed in both adipocytes (15) and macrophages (16, 17), and is responsible for regulating fatty acid storage, glucose metabolism, adipocyte differentiation, adipose tissue gene expression, and adipose depot formation (15, 18, 19). Its activation with thiazolidinediones has been shown to have both anti-inflammatory and insulin-sensitizing effects (18, 19). Increasing evidence implicates infiltration of adipose tissue with inflammatory cells as an important pathogenetic mechanism in systemic insulin resistance. In both rodents and humans, obesity is associated with increased adipose tissue macrophage, dendritic cell, and neutrophil content (3,20,21). Adipose tissue macrophages produce fat-derived cytokines which generate a pro-inflammatory state that is thought to contribute to insulin resistance (22). We recently reported that after 21 days of treatment with the thiazolidinedione, pioglitazone, subjects with T2DM displayed substantial decreases in macrophage content, improved hepatic and peripheral insulin action, and increased levels of circulating adiponectin. These effects were preceded by decreases in whole fat macrophage chemoattractant factors, which are the signaling molecules that recruit macrophages into fat, and their receptors after only 10 days of treatment (23,24).
Interestingly, thiazolidinedione treatment has been shown to cause significant and seemingly paradoxical weight gain when used clinically (25). It is known that subcutaneous adipose tissue, as opposed to visceral adipose tissue, does not increase the risk for many of the diseases commonly associated with obesity (26). Thus, the benefits of thiazolidedione-associated weight gain have been partially explained by demonstrations that thiazolidinediones induce terminal differentiation of subcutaneous but not visceral preadipocytes and that thiazolidinediones result in the remodeling of fat depots from visceral to subcutaneous sites (27). Furthermore, it has been shown in rodents and rodent cell cultures that thiazolidinediones induce a conversion of white fat, which stores energy and triglycerides, to brown fat, a more metabolically favorable tissue that releases energy as heat (28-31). On the other hand, weight gain associated with thiazolidinedione treatment may indicate a potential detriment to using these agents in obesity. Given the role that thiazolidinediones play in modifying adipose tissue biology, inflammation, and insulin resistance, it is conceivable that thiazolidinediones would have differing effects in obese compared to non-obese patients. Therefore, we examined the effects of pioglitazone separately in obese and non-obese subjects with T2DM.
Materials and Methods
Human subjects
Studies were performed using a randomized, double-blind, placebo- controlled crossover design. All studies and procedures were approved by the Albert Einstein College of Medicine Institutional Review Board. All subjects with a history of heart disease, cerebrovascular disease, seizures, bleeding disorders, muscular disease, smoking, or any disease other than T2DM and well-controlled hypertension were excluded. Women of childbearing age were allowed to participate in the study if a pregnancy test within a week of the clamp study was negative. Prior to enrollment in the study, the purpose, nature, risks, and benefits of the study were explained to subjects and their informed, written consent was obtained. Eighteen volunteers with moderately-to-poorly controlled T2DM were enrolled. Subjects were instructed to maintain their typical diet and activity level during the study period. Prior to clamp studies, sulfonylureas and metformin were discontinued for 3 days and long-acting insulin was discontinued for 24 hours. Subjects took pioglitazone on the morning of their studies but were otherwise directed to fast from the night before each study until the completion of each study.
Clamp studies
Of the 18 subjects enrolled, 16 participated in a pair of crossover clamp studies after receiving either 21 days of treatment with 45 mg/day of pioglitazone or placebo. Study agents were given in a randomized, double-blind fashion with a washout period of at least three weeks. Adipose tissue biopsies were performed under local anesthesia during the final 30 minutes of each clamp study. Six-hour “stepped” euglycemic-hyperinsulinemic clamp studies (Figure 1A) were used to assess the effects of the study agents on hepatic and peripheral insulin action. Euglycemic-hyperinsulinemic clamps are considered the gold standard for evaluating insulin sensitivity (32). The purpose of performing “stepped” clamps is to obtain information on insulin action at both ends of the physiologic insulin concentration range. Specifically, the low-insulin phase of the clamp is designed to measure insulin's ability to suppress hepatic glucose production (HGP) by the liver, while the high-insulin phase is designed to assess insulin's ability to stimulate whole body glucose uptake (33). Subjects were admitted on the evening prior to the studies and intravenous access was obtained. Insulin infusions (Novolin Regular) were started at 3:00 a.m. and insulin infusion rate was adjusted based on hourly serum glucose measurements to reach a euglycemic state. At 7:30 a.m., we obtained intravenous access in the opposite arm for blood sampling. “Stepped” euglycemic-hyperinsulinemic clamps were performed as described previously (24). At t=0, a primed continuous infusion of high-performance liquid chromatography purified [3- 3H] glucose was started (bolus 21.6 μCi for 5 min followed by continuous infusion of 0.15 μCi/min) to measure glucose fluxes. From t=0 to t=120, insulin infusion rate was frequently adjusted to keep plasma glucose levels at 90 mg/dl and to determine the basal insulin replacement rate required to maintain euglycemia. From t=120 to t=240, insulin infusion rates were increased to 20 mU/m2/min above basal corresponding to the “low-insulin step” of the clamp. From t=240 to t=360, infusions were increased to 150 mU/m2/min above basal corresponding to the “high-insulin step” of the clamp. Somatostatin (250 mg/h) was infused for the duration of all clamp studies with replacement of glucoregulatory hormones (glucagon 1 ng/kg/min; growth hormone 3 ng/kg/min) to maintain constant hormone levels throughout studies. Plasma glucose concentrations were measured at 5 to 10 minute intervals during the study and maintained at normal fasting concentrations (∼90 mg/dl) by frequently adjusting a variable infusion of [3-3H] glucose-enriched 20% dextrose. Blood samples were obtained regularly during studies for determinations of plasma insulin, C-peptide, free fatty acids, glycerol, and [3-3H] glucose determinations. At t=360, infusions were stopped and the subject was given a standard meal. To avoid hypoglycemia, dextrose infusion was continued for 30 minutes following the completion of each study with measurements of plasma glucose for another 60 minutes.
Figure 1.
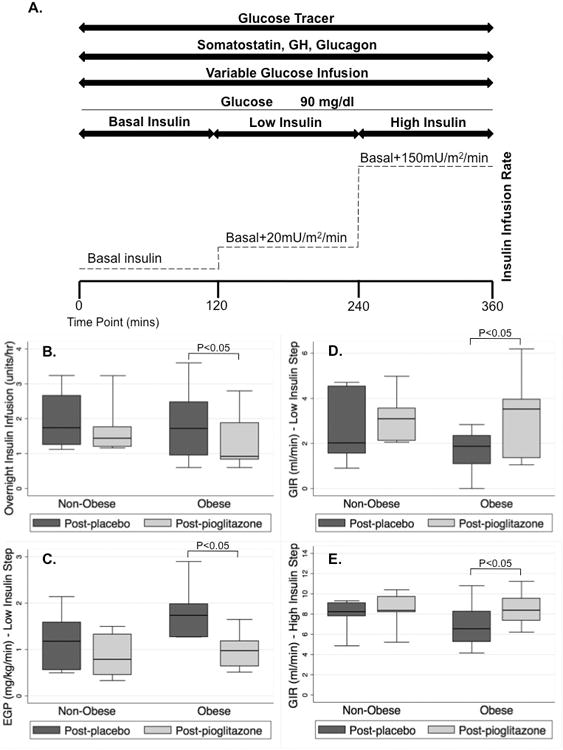
(A) “Stepped” euglycemic-hyperinsulinemic pancreatic clamp study time course. (B) Overnight insulin infusion rate required to maintain euglycemia was significantly lower following pioglitazone in the obese group (p=0.04) but not in the non-obese group (p=0.85). (C) Hepatic glucose production (HGP) during the low-step of the clamp was significantly suppressed in the obese group (p=0.02) while suppression of HGP in the non-obese group following pioglitazone failed to reach significance (p=0.09). (D) Glucose infusion rate (GIR) required to maintain euglycemia during the low-insulin step of the clamp was significantly increased with pioglitazone in the obese group (p=0.02) while the GIR in the non-obese group did not significantly differ (p=0.40). (E) GIR required to maintain euglycemia during the high-insulin step of the clamp was significantly increased in the obese group with pioglitazone (p=0.009) while GIR did not significantly differ in the non-obese group (p=0.13).
Plasma hormone and substrate determinations
Plasma glucose was measured at the bedside using a Beckman glucose analyzer (Fullerton, CA) by use of the glucose oxidase method. Plasma insulin was measured by radioimmunoassay. Plasma FFA levels were measured by an acyl-CoA oxidase-based colorimetric kit (Wako, Osaka, Japan) as previously described (34). Plasma glycerol was measured by colorimetric enzymatic methods and plasma lactate by a previously described fluorometric enzyme technique (23).
Adipose tissue biopsies
All 18 subjects underwent adipose tissue biopsies after receiving either 21 days of pioglitazone or placebo as described above. A 0.25 cm cutaneous incision in the periumbilical region was made under local anesthesia (Lidocaine 1%) and 1-2g of adipose were obtained by aspiration (35). Biopsy specimens were homogenized in Trizol (Invitrogen Technologies, Carlsbad, CA) to inhibit any RNAase activity, and subsequently stored at −80°C.
Adipose tissue separation
Subcutaneous adipose tissue samples were washed and digested with collagenase type 1. Adipocytes and macrophages were separated from the stromal vascular fraction as previously described (23). Separated adipocytes and macrophages were washed with PBS, stored in Trizol, and analyzed by quantitative real time RT-PCR.
Quantitative Real-Time RT-PCR
Total RNA was extracted from the adipose tissue samples using Trizol. cDNA was synthesized using the Superscript First Strand Synthesis System for RT-PCR (Invitrogen Technologies, Carlsbad, CA). Gene expression was studied by quantitative, real-time RT-PCR using the protocol for the LightCycler instrument (Roche Diagnostics, Indianapolis, IN). SYBRGreen1 dye (Roche Diagnostics) was used for detection of double-stranded DNA and housekeeping genes as previously described (24). Results are expressed as relative copy number of a gene following pioglitazone treatment versus following placebo treatment and corrected for the geometric mean of housekeeping genes run with that pair of samples.
Statistical Analysis
For the purposes of this analysis, subjects were categorized by body mass index (BMI) as either obese (BMI>30) or non-obese (BMI<30). Statistical analysis was performed using STATA software, version 12 (StataCorp, College Station, TX). Given our relatively small sample size, the assumption of normality required to conduct parametric analyses was not met. Therefore, non-parametric analyses were conducted. Comparison of independent medians was assessed using the Mann-Whitney test and comparison of paired medians was assessed using the Wilcoxon matched pairs signed-rank test. All data are presented as median (interquartile range).
Results
Subject Characteristics
In the current crossover study, obese subjects with T2DM (n=11, BMI>30) and non-obese subjects with T2DM (n=7, BMI<30) received 21-days of either pioglitazone or placebo in a randomized, double-blind fashion prior to each clamp study and/or adipose tissue biopsy. Baseline subject characteristics are outlined in Table 1. Of note, the study groups did not significantly differ in age, HbA1c%, or plasma lipids, though, as expected, they did differ in weight and BMI. Though long-term pioglitazone treatment is known to cause significant weight gain, there were no significant changes in mean BMI or weight over this time course in either group.
Table 1.
Subject Characteristics (N=18).
| OBESE (N=11) | NON-OBESE (N=7) | P-value | |
|---|---|---|---|
| Continuous Variables | Median (IQR) | Median (IQR) | |
| Age (years) | 47 (40 – 51) | 53 (42 – 54) | 0.36 |
| BMI (kg/m2) | 35.76 (32.49 – 39.82) | 28.89 (28.09 – 29.35) | <0.001* |
| Weight (kg) | 100 (89.5 – 112.5) | 88 (77.5 – 94.0) | 0.03* |
| HbA1c (%) | 9.1 (8.7 – 11) | 10.2 (9.1 – 12.7) | 0.22 |
| HbA1c (mmol/mol) | 76 (72 – 97) | 88 (76 – 115) | 0.22 |
| Total cholesterol (mg/dl) | 189 (176 – 200) | 187 (169 – 199) | 0.71 |
| LDL (mg/dl) | 116 (95 – 122) | 118.5 (107 – 134) | 0.52 |
| HDL (mg/dl) | 40 (36 – 46) | 40 (37 – 53) | 0.67 |
| Triglycerides (mg/dl) | 156 (93 – 168) | 129 (78 – 270) | 0.79 |
| Categorical Variables | N(%) | N(%) | |
| Sex | |||
| Male | 7 (36.36) | 5 (71.43) | – |
| Female | 4 (63.64) | 2 (28.57) | – |
p<0.05 in obese patients relative to non-obese patients
Responses of the obese and non-obese study groups to pioglitazone versus placebo were assessed. Complete results are tabulated in Table 2 (obese group) and Table 3 (non-obese group), with only p-values included in the body of the text.
Table 2.
Fasting Conditions, Glucose Fluxes, and Adipose Inflammatory Profile in Obese Subjects.
| Post-Placebo | Post-Pioglitazone | P-value | |
|---|---|---|---|
| Median (IQR) | Median (IQR) | ||
| Subject Characteristics | |||
| Weight (kg) | 100.0 (89.5 – 112.5) | 98.1 (89.5 – 110.5) | 0.66 |
| Fasting Conditions | |||
| O/N glucose (mg/dl) | 148 (114 – 166) | 127 (123 – 141) | 0.50 |
| O/N IIR (units/hour) | 1.72 (0.95 – 2.48) | 0.92 (0.84 – 1.88) | 0.04* |
| Glucose Fluxes – Low Insulin Step | |||
| HGP (mg/kg/min) | 1.74 (1.28 – 1.98) | 0.98 (0.64 – 1.18) | 0.02* |
| Rd (mg/kg/min) | 2.84 (2.41 – 3.59) | 3.54 (2.78 – 4.64) | 0.14 |
| GIR (ml/min) | 1.87 (1.09 – 2.34) | 3.52 (1.36 – 3.96) | 0.02* |
| Glucose Fluxes – High Insulin Step | |||
| HGP (mg/kg/min) | 0.46 (-0.23 – 0.87) | 0.05 (-0.34 – 0.40) | 0.50 |
| Rd (mg/kg/min) | 7.58 (6.18 – 7.97) | 7.83 (6.82 – 10.79) | 0.11 |
| GIR (ml/min) | 6.55 (5.29 – 8.24) | 8.38 (7.38 – 9.55) | 0.009* |
| Clamp Conditions – Low Insulin Step | |||
| Free fatty acids | 65 (48 – 103) | 54 (17 – 69) | 0.26 |
| Glycerol | 82 (37 – 111) | 62 (35 – 82) | 0.24 |
| Insulin | 70.85 (54.33 – 107) | 83.29 (55.67 – 105.1) | 0.59 |
| C-peptide | 0.13 (0.10 – 0.17) | 0.12 (0.12 – 0.14) | 0.90 |
| Clamp Conditions – High Insulin Step | |||
| Free fatty acids | 28 (18 – 69) | 15.5 (6 – 25) | 0.29 |
| Glycerol | 60 (40 – 77) | 47 (34 – 56) | 0.15 |
| Insulin | 429.22 (367.00 – 519.32) | 379.75 (340.18 – 479.16) | 0.58 |
| C-peptide | 0.06 (0.06 – 0.08) | 0.08 (0.06 – 0.10) | 0.21 |
| Macrophage chemoattractant factors† | |||
| MCP-1 | 1.24E-1 (6.43E-2 – 1.94E-1) | 8.57E-2 (6.34E-2 – 8.77E-2) | 0.02* |
| CCR-2# | 4.63E-3 (2.08E-3 – 5.36E-3) | 1.42E-3 (1.38E-3 – 1.51E-3) | 0.08 |
| CD44 | 9.04E-2 (6.83E-2 – 1.33E-1) | 4.80E-2 (4.22E-2 – 6.50E-2) | 0.03* |
| Adipose macrophage content† | |||
| CSF-1R | 1.67E-2 (7.23E-3 – 3.22E-2) | 1.00E-2 (6.32E-3 – 1.91E-2) | 0.06 |
| CD14 | 9.93E-2 (6.19E-2 – 1.76E-1) | 6.58E-2 (4.10E-2 – 1.12E-1) | 0.02* |
| CD68 | 3.12E-1 (2.29E-1 – 3.51E-1) | 1.81E-1 (1.47E-1 – 2.58E-1) | 0.02* |
| Inflammatory markers and macrophage activation† | |||
| IL-6 | 6.28E-3 (3.76E-3 – 1.58E-2) | 3.06E-3 (2.49E-3 – 4.68E-3) | 0.04* |
| IL-1b | 1.84E-2 (7.33E-3 – 3.34E-2) | 6.52E-3 (1.20E-3 – 9.16E-3) | 0.03* |
| TNF-α | 1.14E-2 (6.34E-3 – 2.50E-2) | 5.99E-3 (4.67E-3 – 6.28E-3) | 0.005* |
| PAI-1 | 9.97E-2 (8.77E-3 – 1.56E-2) | 7.89E-3 (4.03E-3 – 1.45E-2) | 0.02* |
| Arginase-1#° | 1.80E-3 (8.42E-4 – 2.23E-3) | 2.46E-3 (1.01E-3 – 3.65E-3) | 0.07 |
| IL-10#° | 2.06E-3 (9.81E-4 – 2.37E-3) | 2.93E-3 (1.28E-3 – 4.09E-3) | 0.07 |
| iNOS | 1.91E-3 (6.37E-4 – 1.98E-3) | 6.25E-4 (5.01E-4 – 1.41E-3) | 0.04* |
| Dendritic cell and neutrophil content† | |||
| MPO-3 | 1.20E-2 (1.04E-2 – 2.09E-2) | 5.06E-3 (4.54E-3 – 7.89E-3) | 0.04* |
| DC-SIGN | 4.00E-3 (3.90E-3 – 4.53E-3) | 2.06E-3 (1.61E-3 – 2.51E-3) | 0.04* |
| DEC-205 | 2.72E-3 (4.69E-4 – 9.82E-3) | 1.54E-3 (4.33E-4 – 6.56E-3) | 0.04* |
| Adipose tissue characteristics† | |||
| Adiponectin | 1.63E0 (1.07E-1 – 1.82E0) | 2.29E0 (1.94E0 – 1.79E0) | <0.05* |
| PGC-1α | 3.94E-4 (1.07E-4 – 8.97E-4) | 1.05E-3 (1.79E-4 – 3.29E-3) | 0.07 |
| UCP-1 | 1.43E-2 (2.90E-3 – 1.03E-1) | 7.74E-2 (3.51E-3 – 1.65E-1) | 0.04* |
p<0.05
All genes are expressed as relative copy number
Gene Expression by macrophages rather than whole fat
Arginase-1 and IL-10 are said to be protective cytokines
O/N=overnight, IIR=insulin infusion rate, HGP=hepatic glucose production, Rd=rate of glucose disappearance, GIR=glucose infusion rate.
Table 3.
Fasting Conditions, Glucose Fluxes, and Adipose Inflammatory Profile in Non-Obese Subjects.
| Post-Placebo | Post-Pioglitazone | P-value | |
|---|---|---|---|
| Median (IQR) | Median (IQR) | ||
| Subject Characteristics | |||
| Weight (kg) | 88.0 (77.5 – 94.0) | 88.0 (81.5 – 94.0) | 0.05 |
| Fasting Conditions | |||
| O/N glucose (mg/dl) | 147.5 (131 – 165) | 131.5 (118 – 146) | 0.99 |
| O/N IIR (units/hour) | 1.74 (1.26 – 2.66) | 1.44 (1.20 – 1.76) | 0.85 |
| Glucose Fluxes – Low Insulin Step | |||
| HGP (mg/kg/min) | 1.18 (0.56 – 1.59) | 0.79 (0.46 – 1.33) | 0.09 |
| Rd (mg/kg/min) | 3.82 (2.66 – 5.70) | 3.93 (3.06 – 4.64) | 0.61 |
| GIR (ml/min) | 2.02 (1.56 – 4.54) | 3.09 (2.12 – 3.56) | 0.40 |
| Glucose Fluxes – High Insulin Step | |||
| HGP (mg/kg/min) | 0.14 (-0.19 – 0.55) | 0.15 (0.09 – 0.33) | 0.99 |
| Rd (mg/kg/min) | 8.86 (5.37 – 10.46) | 9.11 (8.44 – 11.22) | 0.18 |
| GIR (ml/min) | 8.23 (7.80 – 9.08) | 8.36 (8.21 – 9.73) | 0.13 |
| Clamp Conditions – Low Insulin Step | |||
| Free fatty acids | 80 (49 – 125) | 55 (19 – 96) | 0.40 |
| Glycerol | 24 (21 – 72) | 18 (17 – 66) | 0.02* |
| Insulin | 81.23 (50.13 – 107.24) | 44.33 (38.03 – 84.41) | 0.03* |
| C-peptide | 0.18 (0.11 – 0.20) | 0.16 (0.11 – 0.17) | 0.61 |
| Clamp Conditions – High Insulin Step | |||
| Free fatty acids | 26 (24 – 43) | 28.5 (16.5 – 40.5) | 0.74 |
| Glycerol | 31 (17 – 59) | 21 (12 – 51) | 0.09 |
| Insulin | 405.63 (327 – 460.18) | 350.25 (326.47 – 440.06) | 0.60 |
| C-peptide | 0.09 (0.06 – 0.14) | 0.08 (0.06 – 0.12) | 0.80 |
| Macrophage chemoattractant factors† | |||
| MCP-1 | 5.39E-2 (2.90E-2 – 1.08E-1) | 4.20E-2 (1.88E-2 – 1.02E-1) | 0.07 |
| CCR2# | 3.62E-3 (2.29E-3 – 3.66E-3) | 2.06E-3 (9.33E-4 – 3.06E-3) | 0.27 |
| CD44 | 1.24E-1 (7.31E-2 – 1.40E-1) | 3.74E-2 (2.19E-2 – 5.33E-2) | 0.14 |
| Adipose macrophage content† | |||
| CSF-1R | 3.30E-2 (1.64E-2 – 5.70E-2) | 1.39E-2 (8.42E-3 – 2.75E-2) | 0.07 |
| CD14 | 8.26E-2 (5.91E-2 – 2.20E-1) | 6.33E-2 (4.66E-2 – 1.33E-1) | 0.07 |
| CD68 | 5.07E-1 (2.28E-1 – 1.12E0) | 2.42E-1 (1.81E-1 – 5.95E-1) | 0.07 |
| Inflammatory markers and macrophage activation† | |||
| IL-6 | 3.77E-3 (5.37E-4 – 2.87E-2) | 1.68E-3 (3.89E-4 – 2.95E-2) | 0.59 |
| IL-1b | 1.84E-2 (1.32E-2 – 8.51E-2) | 1.69E-2 (4.85E-3 – 2.02E-2) | 0.11 |
| TNF-α | 1.75E-2 (6.89E-3 – 6.43E-2) | 4.95E-3 (2.12E-3 – 1.66E-2) | 0.03* |
| PAI-1 | 9.92E-3 (5.91E-3 – 3.01E-2 | 4.68E-3 (4.22E-3 – 3.12E-2) | 0.07 |
| Arginase-1#° | 2.38E-4 (2.61E-5 – 3.01E-3) | 764E-4 (5.69E-5 – 4.53E-3) | 0.11 |
| IL-10#° | 2.70E-3 (5.05E-5 – 4.31E-3) | 4.89E-3 (1.35E-4 – 5.04E-3) | 0.11 |
| iNOS | 9.30E-4 (4.63E-4 – 1.26E-3) | 3.64E-4 (9.10E-6 – 7.35E-4) | 0.11 |
| Dendritic cell and neutrophil content† | |||
| MPO-3 | 2.34E-2 (2.14E-2 – 3.37E-2) | 8.19E-3 (5.79E-3 – 1.40E-2) | 0.07 |
| DC-SIGN | 4.43E-3 (1.68E-3 – 7.13E-3) | 1.95E-3 (9.22E-4 – 4.13E-3) | 0.07 |
| DEC-205 | 2.85E-3 (4.59E-4 – 6.57E-3) | 1.50E-3 (3.63E-4 – 5.03E-3) | 0.07 |
| Adipose tissue characteristics† | |||
| Adiponectin | 4.62E-1 (9.57E-2 – 1.10E0) | 1.87E0 (9.71E-1 – 2.46E0) | 0.07 |
| PGC-1α | 1.67E-3 (8.78E-4 – 1.27E-1) | 2.00E-2 (5.14E-4 – 4.06E-2) | 0.47 |
| UCP-1 | 8.52E-4 (3.04E-4 – 3.81E-3) | 7.05E-2 (6.03E-4 – 3.97E-1) | 0.07 |
p<0.05
All genes are expressed as relative copy number
Expression by macrophages rather than whole fat
Arginase-1 and IL-10 are said to be protective cytokines
O/N=overnight, IIR=insulin infusion rate, HGP=hepatic glucose production, Rd=rate of glucose disappearance, GIR=glucose infusion rate.
Fasting Conditions
Overnight plasma glucose levels did not differ in pioglitazone-treated versus placebo-treated subjects in either the obese group (p=0.50) or the non-obese group (p>0.99). The insulin infusion rate required to maintain plasma glucose in the euglycemic range overnight was significantly lower following pioglitazone treatment versus placebo treatment in the obese group (p=0.04) but not in the non-obese group (p=0.85) (Figure 1B).
Glucose Fluxes
Pioglitazone treatment resulted in significantly greater suppression of hepatic glucose production (HGP) by insulin during the low-insulin step of the clamp (t=180-240) in the obese group (p=0.02) while suppression of HGP in the non-obese group following pioglitazone treatment failed to reach significance (p=0.09) (Figure 1C). The rate of glucose disappearance (Rd) during the low-insulin step of the clamp did not significantly differ in either the obese group (p=0.14) or the non-obese group (p=0.61). However, the glucose infusion rate (GIR) required to maintain euglycemia during the low-insulin step of the clamp was significantly increased following treatment with pioglitazone relative to following treatment with placebo in the obese group (p=0.02) but not in the non-obese group (p=0.40) (Figure 1D).
During the high-insulin phase of the clamp, the HGP in both groups and following both treatments was highly suppressed. There were no significant differences in HGP following pioglitazone treatment versus following placebo treatment in either the obese group (p=0.50) or the non-obese group (p>0.99). As expected, the Rd in both groups and following both treatments was elevated during the high-insulin step of the clamp. No differences were seen in Rd in either the obese group (p=0.11) or the non-obese group (p=0.18) during the high-insulin phase, indicating no differences in insulin-mediated glucose uptake. The GIR required to maintain euglycemia during the high-insulin step of the clamp was again significantly increased in the obese group following treatment with pioglitazone relative to following treatment with placebo (p=0.009) but not in the non-obese group (p=0.13) (Figure 1E).
Clamp Conditions
Plasma levels of free fatty acids, glycerol, insulin, and C-peptide are included in tables 2 and 3. Glucose specific activity remained stable throughout the studies as previously described (23).
Macrophage chemoattractant factors
To assess the degree to which pioglitazone reduces the inflammatory environment of whole fat with respect to macrophage chemoattraction, we assessed the expression of two macrophage chemoattractant factors, Macrophage Chemoattractant Protein-1 (MCP-1) and CD44. Additionally, we examined the expression of the receptor for MCP-1, chemokine (C-C motif) receptor 2 (CCR2), in macrophages. Pioglitazone treatment significantly reduced the expression of MCP-1 in whole fat in obese patients relative to administration of placebo (p=0.02). This reduction in MCP-1 expression in whole fat was of borderline significance in the non-obese group (p=0.07). Neither the obese group nor the non- obese group had significantly differing expression of CCR2 in macrophages (p=0.08 in the obese group; p=0.27 in the non-obese group) with pioglitazone relative to placebo. However, the expression of the macrophage chemoattractant factor, CD44, in whole fat was significantly decreased following pioglitazone relative to placebo in the obese group (p=0.03) but not in the non- obese group (p=0.14).
Adipose macrophage content
To measure the adipose macrophage content of fat, we measured expression of three macrophage-specific markers. Decreased expression of colony- stimulating factor 1 receptor (CSF-1R) following pioglitazone treatment trended toward significance in both groups relative to placebo (p=0.06 in the obese group; p=0.07 in the non-obese group). The expression of CD14 was significantly reduced following administration of pioglitazone relative to placebo in the obese group (p=0.02) with borderline significant reduction of CD14 expression in the non-obese group (p=0.07). Similarly, the expression of CD68 was significantly reduced with pioglitazone relative to placebo in the obese group (p=0.02) and borderline significantly reduced in the non-obese group (p=0.07).
Inflammatory markers and macrophage activation
To assess the degree to which pioglitazone treatment affects the inflammatory milieu of adipose tissue, we measured changes in the expression of the proinflammatory cytokines IL-6, IL-1B, and TNF-α, the protective cytokines arginase-1 and IL-10, the serine protease inhibitor PAI-1 which is associated with obesity and insulin resistance, and iNOS which is a marker of adipose tissue macrophage activation. Following pioglitazone treatment, obese subjects had significantly reduced whole fat expression of the inflammatory cytokines IL-6 (p=0.04), IL-1b (p=0.03), and TNF-α (p = 0.005) relative to placebo. In contrast, in non-obese subjects the whole fat expression of IL-6 (p=0.59) and IL-1B (p=0.11) was not significantly different between the two treatment arms with only TNF-α expression undergoing a significant reduction in expression level with pioglitazone treatment (p=0.03). Macrophage expression of the protective cytokine, arginase-1, after treatment with pioglitazone relative to placebo demonstrated an upward trend in obese subjects (p=0.07) but a downward trend in non- obese subjects (p=0.11). The expression of another protective cytokine, IL-10, by macrophages was increased in both groups but not significantly in either group (p=0.07 in the obese group and p=0.11 in non-obese group). Whole fat expression of PAI-1 was significantly reduced following pioglitazone relative to placebo in obese subjects (p=0.02) and borderline significantly reduced in non-obese subjects (p=0.07). iNOS expression, another measure of adipose tissue macrophage activation, was significantly reduced with pioglitazone in obese subjects (p=0.04) but not in non- obese subjects (p=0.11).
Dendritic cell and neutrophil content
Following pioglitazone treatment, obese subjects demonstrated significantly reduced whole fat expression of the dendritic cell markers DEC-205 (p=0.04) and DC-SIGN (p=0.04) while non-obese subjects displayed borderline significant reductions in DEC-205 (p=0.07) and DC-SIGN (p=0.07). Similarly, the expression of the neutrophil marker myeloperoxidase-3 (MPO-3) was significantly reduced in obese subjects with pioglitazone relative to placebo (p=0.04) with borderline significant reduction in the expression of MPO-3 in non- obese subjects (p=0.07).
Adipose tissue characteristics
Following pioglitazone treatment, whole fat expression of adiponectin, a marker of fatty acid oxidation and an insulin-sensitizing effector, were significantly increased in obese subjects relative to administration of placebo (p=0.04) with a borderline significant increase in the non-obese group (p=0.07). Whole fat expression of peroxisome proliferator activated receptor gamma coactivator-1 alpha (PGC-1α), a master regulator of mitochondrial biogenesis and fat browning, was not significantly increased in either group following pioglitazone. However, it did approach significance in the obese group (p=0.07) whereas it did not do so in the non-obese group (p=0.47). Expression of uncoupling protein-1 (UCP-1), also known as thermogenin, a downstream component of the fat browning process, was significantly increased in the whole fat of obese subjects following pioglitazone (p=0.03) and borderline significantly increased in non-obese subjects (p=0.07).
Discussion
The need for “personalized medicine” is increasingly recognized in light of the growing complexities of managing T2DM and other chronic diseases (36). Indeed, despite the availability of multiple pharmacologic options, management of T2DM frequently remains sub-optimal. While metformin is universally recommended as a first-line agent for the treatment of T2DM, several additional drug classes are recommended as second-line options, to be selected on a case-by- case basis (37). One of these medication classes, thiazolidinediones, activate PPARγ which is a nuclear receptor that mediates a range of processes including adipocyte differentiation, adipose tissue gene expression, adipose depot formation, and glucose metabolism (15,18,19). Additionally, thiazolidinediones have been shown to have both anti-inflammatory and insulin-sensitizing effects (18,19).
Given these findings, it is interesting that pioglitazone, a drug approved for the treatment of T2DM, has been shown to cause significant weight gain (25). This may cause clinicians to be hesitant to use pioglitazone to treat their obese patients with diabetes. However, additional data is necessary for clinicians to determine which patient sub-populations are likely to benefit from thiazolidinedione treatment. In fact, since none of the second-line anti-diabetic medication classes has proven to be superior, the American Diabetes Association position statement on the management of T2DM calls for further research “on how phenotype and other patient/disease characteristics should drive drug choices (37).”
The current study demonstrated that treatment of obese subjects with pioglitazone resulted in substantial improvements in insulin sensitivity and adipose tissue inflammatory and secretory profiles. Furthermore, major differences between the responses of the obese and non-obese groups were observed with regard to improvement in glucose fluxes (specifically insulin sensitivity, glucose infusion rate, and hepatic glucose production) and expression of certain inflammatory markers (IL-6, IL-1b, and iNOS) with pioglitazone treatment relative to placebo. These three inflammatory markers were noted earlier to be associated with diabetes, obesity, insulin resistance, and cardiovascular disease (9-12). The pioglitazone-associated reduction in the expression of these markers in obese subjects is likely to be beneficial. These findings are supported by prior studies suggesting that thiazolidinedione-associated weight gain results in the remodeling of adipose tissue to a more metabolically active form (27).
It is also striking that in this cohort of obese subjects, pioglitazone resulted in a near- significant increase in expression of an upstream regulator of fat browning and a significant increase in expression of a downstream factor associated with fat browning (PGC-1α and UCP-1). Such white-to-brown adipose differentiation, which was previously demonstrated in rodents and rodent cell cultures following PPARγ agonist treatment (28-31,38), may prove to be a benefit of thiazolidinedione-treatment resulting in protective metabolic effects. Of course, in terms of assessing pioglitazone's effects on adipose tissue browning, our findings are early ones that will need to be supported by further studies designed to assess adipose tissue structure, mitochondrial content, and energy expenditure, with an extensive look at adipose-browning-associated gene expression.
Since the effort of conducting such in-depth physiology studies in human subjects with diabetes limited the current sample size, non-parametric statistical methods were used to reduce the influence that outliers would typically have on smaller studies. Of note, although the majority of measured parameters did not reach significance in the non-obese group, we cannot rule out the possibility that this was due to its slightly smaller sample size. However, this ought not to detract from the important findings demonstrated in obese subjects. Interestingly, though pioglitazone is known to cause weight gain when used long-term, the weights of these subjects were not affected by this short-term exposure to pioglitazone.
In addition to weight gain, pioglitazone has been linked to bone loss, fluid retention, and, perhaps, bladder cancer (39). On the other hand, pioglitazone has been shown to have particular benefits in certain populations, such as improving liver function in individuals with nonalcoholic fatty liver disease and reducing proteinuria in patients with both T2DM and reduced glomerular filtration rate (40). The sample size and duration of the current study were not designed to address the safety of pioglitazone, though it was reassuring that none of the subjects in this study experienced any adverse effects.
Of note, the prevalence of obesity in patients with diabetes is greater than 50% (41). This underscores the importance of identifying therapeutic agents whose mechanisms of action would make them likely to benefit obese patients. In particular, adipose tissue inflammatory cells appear to be an important unifying mechanism linking nutrient excess, inflammation, and insulin resistance. In the absence of targeted therapeutic approaches to decrease adipose inflammation, demonstrating beneficial effects of thiazolidinediones on adipose inflammatory cells and their activation, especially in an obese population, has important clinical implications. Though subsequent larger studies will be helpful to confirm the findings of this mechanistic study, from the standpoint of efficacy our work suggests that pioglitazone treatment favorably impacts insulin resistance and adipose biology in obese patients with T2DM.
Acknowledgments
The authors thank Dr. Aileen McGinn of Albert Einstein College of Medicine for her critical reading of the manuscript and for her statistical advice. The project described was supported by the National Center for Advancing Translational Sciences (NCATS), a component of the National Institutes of Health (NIH), through grant numbers UL1TR000086, TL1RR000087, KL2TR000088 as well as by an Investigator-Initiated Research Grant from Takeda Pharmaceuticals North America and by research grants from the NIH (DK-069861, and DK-48321) and the American Diabetes Association. Meredith Hawkins is a Beeson Scholar of the American Federation for Aging Research and an investigator in the Diabetes Research and Training Center (DK-20541) and the Clinical Research Center (RR 12248). Y.B.E. researched data, performed experimental studies, and wrote the manuscript. K.Z., S.Ko., and S.Ke. performed experimental studies. P.K. developed the study and performed or supervised experimental studies. P.R., M.C., and S.R.M. reviewed/edited the manuscript. M.H. developed the study, researched data, edited the manuscript, and is the guarantor of this study.
Sources of Support: This project was supported by the National Center for Advancing Translational Sciences (NCATS), a component of the National Institutes of Health (NIH), through grant numbers UL1TR000086, TL1RR000087, KL2TR000088 as well as by an Investigator-Initiated Research Grant from Takeda Pharmaceuticals North America and by research grants from the NIH (DK-069861, and DK-48321) and the American Diabetes Association.
References
- 1.Alessi MC, Bastelica D, Morange P, et al. Plasminogen activator inhibitor 1, transforming growth factor-beta1, and BMI are closely associated in human adipose tissue during morbid obesity. Diabetes. 2000;49:1374–1380. doi: 10.2337/diabetes.49.8.1374. [DOI] [PubMed] [Google Scholar]
- 2.Coppack SW. Pro-inflammatory cytokines and adipose tissue. The Proceedings of the Nutrition Society. 2001;60:349–356. doi: 10.1079/pns2001110. [DOI] [PubMed] [Google Scholar]
- 3.Weisberg SP, McCann D, Desai M, et al. Obesity is associated with macrophage accumulation in adipose tissue. The Journal of clinical investigation. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hotamisligil GS, Spiegelman BM. Tumor necrosis factor alpha: a key component of the obesity- diabetes link. Diabetes. 1994;43:1271–1278. doi: 10.2337/diab.43.11.1271. [DOI] [PubMed] [Google Scholar]
- 5.Griffin ME, Marcucci MJ, Cline GW, et al. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes. 1999;48:1270–1274. doi: 10.2337/diabetes.48.6.1270. [DOI] [PubMed] [Google Scholar]
- 6.Kim JK, Fillmore JJ, Sunshine MJ, et al. PKC-theta knockout mice are protected from fat- induced insulin resistance. The Journal of clinical investigation. 2004;114:823–827. doi: 10.1172/JCI22230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loskutoff DJ, Samad F. The adipocyte and hemostatic balance in obesity: studies of PAI-1. Arteriosclerosis, thrombosis, and vascular biology. 1998;18:1–6. doi: 10.1161/01.atv.18.1.1. [DOI] [PubMed] [Google Scholar]
- 8.Trayhurn P, Beattie JH. Physiological role of adipose tissue: white adipose tissue as an endocrine and secretory organ. The Proceedings of the Nutrition Society. 2001;60:329–339. doi: 10.1079/pns200194. [DOI] [PubMed] [Google Scholar]
- 9.Abeywardena MY, Leifert WR, Warnes KE, et al. Cardiovascular biology of interleukin-6. Current pharmaceutical design. 2009;15:1809–1821. doi: 10.2174/138161209788186290. [DOI] [PubMed] [Google Scholar]
- 10.Kristiansen OP, Mandrup-Poulsen T. Interleukin-6 and diabetes: the good, the bad, or the indifferent? Diabetes. 2005;54(2):S114–124. doi: 10.2337/diabetes.54.suppl_2.s114. [DOI] [PubMed] [Google Scholar]
- 11.Sacks HS, Fain JN, Cheema P, et al. Inflammatory genes in epicardial fat contiguous with coronary atherosclerosis in the metabolic syndrome and type 2 diabetes: changes associated with pioglitazone. Diabetes care. 2011;34:730–733. doi: 10.2337/dc10-2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaneki M, Shimizu N, Yamada D, et al. Nitrosative stress and pathogenesis of insulin resistance. Antioxidants & redox signaling. 2007;9:319–329. doi: 10.1089/ars.2006.1464. [DOI] [PubMed] [Google Scholar]
- 13.Kern PA, Ranganathan S, Li C, et al. Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. American journal of physiology Endocrinology and metabolism. 2001;280:E745–751. doi: 10.1152/ajpendo.2001.280.5.E745. [DOI] [PubMed] [Google Scholar]
- 14.Wu H, Ghosh S, Perrard XD, et al. T-cell accumulation and regulated on activation, normal T cell expressed and secreted upregulation in adipose tissue in obesity. Circulation. 2007;115:1029–1038. doi: 10.1161/CIRCULATIONAHA.106.638379. [DOI] [PubMed] [Google Scholar]
- 15.Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annual review of biochemistry. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- 16.Castrillo A, Tontonoz P. Nuclear receptors in macrophage biology: at the crossroads of lipid metabolism and inflammation. Annual review of cell and developmental biology. 2004;20:455–480. doi: 10.1146/annurev.cellbio.20.012103.134432. [DOI] [PubMed] [Google Scholar]
- 17.Ricote M, Li AC, Willson TM, et al. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- 18.Klappacher GW, Glass CK. Roles of peroxisome proliferator-activated receptor gamma in lipid homeostasis and inflammatory responses of macrophages. Current opinion in lipidology. 2002;13:305–312. doi: 10.1097/00041433-200206000-00011. [DOI] [PubMed] [Google Scholar]
- 19.Vidal-Puig AJ, Considine RV, Jimenez-Linan M, et al. Peroxisome proliferator-activated receptor gene expression in human tissues. Effects of obesity, weight loss, and regulation by insulin and glucocorticoids. The Journal of clinical investigation. 1997;99:2416–2422. doi: 10.1172/JCI119424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chmelar J, Chung KJ, Chavakis T. The role of innate immune cells in obese adipose tissue inflammation and development of insulin resistance. Thrombosis and haemostasis. 2013;109:399–406. doi: 10.1160/TH12-09-0703. [DOI] [PubMed] [Google Scholar]
- 21.Lumeng CN. Innate immune activation in obesity. Molecular aspects of medicine. 2013;34:12–29. doi: 10.1016/j.mam.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maury E, Brichard SM. Adipokine dysregulation, adipose tissue inflammation and metabolic syndrome. Molecular and cellular endocrinology. 2010;314:1–16. doi: 10.1016/j.mce.2009.07.031. [DOI] [PubMed] [Google Scholar]
- 23.Koppaka S, Kehlenbrink S, Carey M, et al. Reduced Adipose Tissue Macrophage Content Is Associated With Improved Insulin Sensitivity in Thiazolidinedione-Treated Diabetic Humans. Diabetes. 2013 doi: 10.2337/db12-0868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tonelli J, Li W, Kishore P, et al. Mechanisms of early insulin-sensitizing effects of thiazolidinediones in type 2 diabetes. Diabetes. 2004;53:1621–1629. doi: 10.2337/diabetes.53.6.1621. [DOI] [PubMed] [Google Scholar]
- 25.Patel J, Anderson RJ, Rappaport EB. Rosiglitazone monotherapy improves glycaemic control in patients with type 2 diabetes: a twelve-week, randomized, placebo-controlled study. Diabetes, obesity & metabolism. 1999;1:165–172. doi: 10.1046/j.1463-1326.1999.00020.x. [DOI] [PubMed] [Google Scholar]
- 26.Porter SA, Massaro JM, Hoffmann U, et al. Abdominal subcutaneous adipose tissue: a protective fat depot? Diabetes care. 2009;32:1068–1075. doi: 10.2337/dc08-2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stumvoll M, Haring HU. Glitazones: clinical effects and molecular mechanisms. Annals of medicine. 2002;34:217–224. [PubMed] [Google Scholar]
- 28.Ohno H, Shinoda K, Spiegelman BM, et al. PPARgamma agonists induce a white-to-brown fat conversion through stabilization of PRDM16 protein. Cell metabolism. 2012;15:395–404. doi: 10.1016/j.cmet.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sears IB, MacGinnitie MA, Kovacs LG, et al. Differentiation-dependent expression of the brown adipocyte uncoupling protein gene: regulation by peroxisome proliferator-activated receptor gamma. Molecular and cellular biology. 1996;16:3410–3419. doi: 10.1128/mcb.16.7.3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tai TA, Jennermann C, Brown KK, et al. Activation of the nuclear receptor peroxisome proliferator-activated receptor gamma promotes brown adipocyte differentiation. The Journal of biological chemistry. 1996;271:29909–29914. doi: 10.1074/jbc.271.47.29909. [DOI] [PubMed] [Google Scholar]
- 31.Wilson-Fritch L, Nicoloro S, Chouinard M, et al. Mitochondrial remodeling in adipose tissue associated with obesity and treatment with rosiglitazone. The Journal of clinical investigation. 2004;114:1281–1289. doi: 10.1172/JCI21752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tam CS, Xie W, Johnson WD, et al. Defining insulin resistance from hyperinsulinemic- euglycemic clamps. Diabetes care. 2012;35:1605–1610. doi: 10.2337/dc11-2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watts GF. Clinical Diabetes Research: Methods and Techniques. Edited by Michael Roden © 2007, John Wiley & Sons Ltd ISBN: 978 0 470 01728 9. Practical Diabetes International. 2008;25:277–277. [Google Scholar]
- 34.Esterson YB, Kishore P, Koppaka S, et al. Fatty acid-induced production of plasminogen activator inhibitor-1 by adipose macrophages is greater in middle-aged versus younger adult participants. The journals of gerontology Series A, Biological sciences and medical sciences. 2012;67:1321–1328. doi: 10.1093/gerona/gls200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mauriege P, Imbeault P, Langin D, et al. Regional and gender variations in adipose tissue lipolysis in response to weight loss. Journal of lipid research. 1999;40:1559–1571. [PubMed] [Google Scholar]
- 36.Spiegel AM, Hawkins M. ‘Personalized medicine’ to identify genetic risks for type 2 diabetes and focus prevention: can it fulfill its promise? Health affairs (Project Hope) 2012;31:43–49. doi: 10.1377/hlthaff.2011.1054. [DOI] [PubMed] [Google Scholar]
- 37.Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD) Diabetes care. 2012;35:1364–1379. doi: 10.2337/dc12-0413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smorlesi A, Frontini A, Giordano A, et al. The adipose organ: white-brown adipocyte plasticity and metabolic inflammation. Obesity reviews: an official journal of the International Association for the Study of Obesity. 2012;13(2):83–96. doi: 10.1111/j.1467-789X.2012.01039.x. [DOI] [PubMed] [Google Scholar]
- 39.Ahmadian M, Suh JM, Hah N, et al. PPARgamma signaling and metabolism: the good, the bad and the future. Nature medicine. 2013;19:557–566. doi: 10.1038/nm.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Defronzo RA, Mehta RJ, Schnure JJ. Pleiotropic effects of thiazolidinediones: implications for the treatment of patients with type 2 diabetes mellitus. Hospital practice (1995) 2013;41:132–147. doi: 10.3810/hp.2013.04.1062. [DOI] [PubMed] [Google Scholar]
- 41.Prevalence of overweight and obesity among adults with diagnosed diabetes--United States, 1988-1994 and 1999-2002. MMWR Morbidity and mortality weekly report. 2004;53:1066–1068. [PubMed] [Google Scholar]