

Abstract
In this study we offer a mechanistic interpretation of the previously known but unexplained substrate inhibition observed for CYP2E1. At low substrate concentrations, p-nitrophenol (pNP) was rapidly turned over (47 min−1) with relatively low Km (24 μM); nevertheless, at concentrations of >100 μM, the rate of pNP oxidation gradually decreased as a second molecule bound to CYP2E1 through an effector site (Kss = 260 μM), which inhibited activity at the catalytic site. 4-Methylpyrazole (4MP) was a potent inhibitor for both sites through a mixed inhibition mechanism. The Ki for the catalytic site was 2.0 μM. Although we were unable to discriminate whether an EIS or ESI complex formed, the respective inhibition constants were far lower than Kss. Bicyclic indazole (IND) inhibited catalysis through a single CYP2E1 site (Ki = 0.12 μM). Similarly, 4MP and IND yielded type II binding spectra that reflected the association of either two 4MP or one IND molecule(s) to CYP2E1, respectively. Based on computational docking studies with a homology model for CYP2E1, the two sites for monocyclic molecules, pNP and 4MP, exist within a narrow channel connecting the active site to the surface of the enzyme. Because of the presence of the heme iron, one site supports catalysis, whereas the other more distal effector site binds molecules that can influence the binding orientation and egress of molecules for the catalytic site. Although IND did not bind these sites simultaneously, the presence of IND at the catalytic site blocked binding at the effector site.
CYP2E1 (P450 or CYP for a particular isoform) is a mammalian cytochrome P450 enzyme, which oxidizes a structurally diverse class of endogenous and exogenous (xenobiotic) compounds (1, 2). A majority of studies have focused on the role of CYP2E1 in phase I metabolism of xenobiotic compounds, e.g. drugs, food additives, and environmental contaminants. Growing evidence also supports an important physiological role for CYP2E1 in gluconeogenesis. CYP2E1 is regulated similarly to enzymes contributing to gluconeogenesis in relation to starvation and diabetes and, in fact, recognizes precursors to gluconeogenesis, acetone, acetol (1-hydroxyacetone), and fatty acids (3) as substrates.
Nevertheless, the selectivity that governs the transformation of molecules by CYP2E1 is poorly understood. A better knowledge of the molecular features that confer specificity of substrates allows predictions to be made as to pharmaco- and toxico-kinetic properties, and such insights are ultimately exploitable in novel drug development and assessment of risk associated with exposure to environmental chemicals. Moreover, the catalytic capacity for CYP2E1 makes the enzyme an excellent target for engineering specific catalytic properties for commercial production of specialty chemicals and remediation activities into plants and other organisms, as reported recently (4, 5).
CYP2E1 has broad substrate specificity toward typically small (molecular weight < 100) and hydrophobic molecules (2, 6). Of the more than 70 different chemicals recognized by this enzyme, a majority of the substrates are short chain alcohols/ketones/aldehydes (e.g. ethanol), nitrosamines, alkanes, halogenated alkanes, and anesthetics. CYP2E1 also recognizes many monocyclic compounds possessing minimal substitutions, e.g. benzene, p-nitrophenol (pNP),2 acetaminophen, isoniazid, and xylenes. Chlorzoxazone, coumarin, quinoline, and caffeine form a smaller class of bicyclic substrates recognized by CYP2E1. Surprisingly, even the long chain fatty acid, arachidonate, is a substrate. Of these compounds, pNP (7) and chlorzoxazone (8) are commonly used in model reactions for CYP2E1 activity. Taken together, these findings implicate a degree of selectivity for the CYP2E1 activity through a restrictive active site.
Although many CYP2E1 substrates conform to a binary kinetic scheme (Scheme 1), Koop (7) reported the first example of substrate inhibition for pNP oxidation. Instead of the hyperbolic curve predicted by Michaelis-Menten kinetics, the increase in pNP concentration led to a rise in the observed rate of turnover until a maximum was reached, and then the rate gradually decreased at higher pNP concentrations. Although the effect was attributed to a classical substrate inhibition mechanism, the data were not fit to the mechanism. The substrate inhibition mechanism predicts that association of a second molecule with the enzyme forms an inactive ternary complex (ES2) (Scheme 1). Higher substrate concentrations favor this latter complex resulting in decreased activity. Further evidence for the possibility of two binding sites for monocyclic molecules was shown through inhibition studies with CYP2E1 (9). The introduction of a variety of 5- and 6-membered heterocyclic molecules to pNP catalytic assays demonstrated competitive, uncompetitive, noncompetitive, and mixed type inhibition. The latter three models require the presence of a secondary site for the inhibitor to bind; however, the preference for these different mechanisms based on ligand structure was not explored. A possible limitation of these studies was the use of only three concentrations of pNP for analyses. These conditions may not have been sufficient to elucidate the mode of interaction between these molecules and CYP2E1.
SCHEME 1. Possible reaction mechanisms for CYP2E1 activity toward pNP.
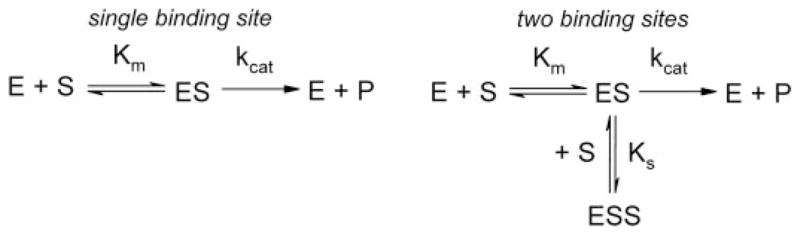
E = CYP2E1; S = pNP.
Here, we explain this kinetic anomaly in terms of both mechanistic and structural views and extend our analysis to include typical representatives of monocyclic and polycyclic aromatic inhibitors. Specifically, we employed 4-methylpyrazole (4MP) and indazole (IND) (Fig. 1) as inhibitors for CYP2E1 oxidation of pNP. The monocyclic 4MP presumably binds competitively to each sub-site occupied by pNP, whereas the larger bicyclic IND would bind to both sub-sites simultaneously. This mode of interaction should yield different inhibition kinetic profiles. In addition to serving as potent inhibitors, the pyrazole ring of these molecules can ligate the heme iron resulting in type II binding spectra (10), and thus provide a useful measure of binding affinity between the monocyclic and bicyclic molecules and CYP2E1. Unfortunately, pNP was not suitable for binding studies because of the spectral overlap between pNP and the heme. To identify the mode of interaction between these molecules and CYP2E1, we proposed multiple possible mechanisms incorporating one or two binding sites for molecules. We fit the resulting data from experimental studies to these models and performed model discrimination analysis based on the second-order Akaike Information Criterion, which properly takes into account the fact that various fitting models contain a different number of adjustable parameters. As a complement to these indirect approaches to identifying complexes, we explored the ability of CYP2E1 to accommodate one or two of these monocyclic and bicyclic compounds through computational docking studies with a homology model developed by our group (11). The location of these potential binding sites would demonstrate whether a second site for a monocyclic compound exists within the active site or distal from the site of oxidation.
FIGURE 1.
Substrate and inhibitors used in this study to probe CYP2E1 binding and activity toward monocyclic and bicyclic compounds.
EXPERIMENTAL PROCEDURES
Reagents
C41 (DE3) cells used in CYP2E1 expression were purchased from Imaxio (France). Top3 cells, which are no longer commercially available, were propagated in the laboratory. Terrific broth modified for genomics was bought from United States Biological (Swampscott, MA). Protein purification resins, 2′,5′-ADP-agarose and Reactive Red 120 type 3000-CL, were obtained from Sigma. SP-Sepharose was purchased from GE Healthcare. Components of the NADPH-regenerating system (NADP+, glucose 6-phosphate, torula yeast glucose-6-phosphate dehydrogenase) were purchased from Sigma. In addition, dilauroyl-L-α-phosphatidylcholine (DLPC), pNP, p-nitrocatechol (pNC), 2-nitroresorcinol, 6-hydroxychlozoxazone, 4MP, bovine erythrocyte superoxide dismutase, catalase, and sodium dithionite (hydrosulfite) were obtained from Sigma. In addition to HPLC-grade acetonitrile (CH3CN) and trifluoroacetic acid, ampicillin, isopropyl β-D-thiogalactopyranoside, lysozyme, diethylaminoethyl cellulose, dithiothreitol, protease inhibitors, and other basic chemicals were purchased from Fisher. Rabbit CPR-K56Q and CYP2E1 were prepared from bacterial expression systems using published protocols (12, 13) with modifications (11, 14). Purified rabbit liver cytochrome b5 was provided as a generous gift from Wayne L. Backes (Louisiana State University Health Science Center, New Orleans).
Steady-state pNP Oxidation Studies
Initial velocities for rabbit P450 2E1 oxidation of pNP to p-nitrocatechol were determined by a high throughput HPLC method developed in our laboratory (11). For reactions, 25 nM CYP2E1 was reconstituted with 100 nM CPR-K56Q and 50 nM cytochrome b5 in a 96-well assay block containing 50 mM potassium phosphate, pH 7.4, 20 μM DLPC, pNP (varied from 5 to 750 μM), 2 units μl−1 catalase, 0.04 μg μl−1 superoxide dismutase, and an NADPH-regenerating system (2 microunits μl−1 glucose-6-phosphate dehydrogenase, 10 mM glucose 6-phosphate, 2 mM MgCl2, 500 μM NADP+). Prior to use, catalase was dialyzed against 20 mM potassium phosphate buffer, pH 7.4, 10% glycerol to remove the thymol preservative.
Specific reactions in the absence or presence of inhibitor were prepared in sets of eight, which facilitated sample manipulation with a multichannel pipette. 4MP prepared in water was added to the reactions to final concentrations of 0, 1, 5, 25, and 125 μM to attempt to saturate a possible second site for 4MP. IND stocks were made in methanol because of solubility concerns. The steady-state kinetics for the reactions in the presence of the methanol alone (final 0.25%) were determined to correct for co-solvent effects. Final concentrations for this inhibitor were 0.1, 0.3, and 1.0 μM. Following addition of all components except NADP+, reactions were incubated at 37 °C for 5 min. Upon addition of NADP+, a reaction aliquot was taken at four time points, transferred to a 96-well microplate, quenched with acetonitrile containing 2-nitroresorcinol (internal standard), and then centrifuged. The supernatant was transferred to a low volume HPLC vial held in a 96-vial rack that matched the 96-well format for the quenched samples. As described previously (11), an autosampler housing 96 vials injected each rack of samples onto a Waters Symmetry C18 3.5 μm 4.6 ×75 mm column with a 75:25 0.1% trifluoroacetic acid/H2O/CH3CN mobile phase at a flow rate of 1.5 ml min−1. The elution of pNC, the internal standard, and pNP were monitored at 320 nm. pNC production during the reaction was quantitated relative to pNC standards. The corresponding concentrations were plotted as a function of time, and the initial rate was determined by linear regression with the software program Graph-Pad Prism (San Diego, CA). Reported initial rates reflect averages of data from 2 to 4 experiments.
Given the observed substrate inhibition during pNP oxidation (7), all possible mechanisms for CYP2E1 included two binding sites for substrate leading to one active complex; nevertheless, there were multiple possible modes of inhibition (Scheme 2). Similar to the traditional mechanism for competitive inhibition, the inhibitor could bind only to free enzyme at the catalytic site to yield single-site inhibition. Alternatively, the inhibitor could bind to both sites occupied by substrate (two-site inhibition). Although substrate was an allosteric effector toward itself, the issue of allosterism may not apply for all binding events. In fact, there were four possible outcomes. Both substrate and inhibitor could alter binding of the other molecule (model 1). For model 2, only substrate acted allosterically, such that substrate affected inhibitor binding (Ki ≠ Ksi) but inhibitor did not affect substrate binding (Ks = Kis). Model 3 described the alternative possibility wherein inhibitor was the only allosteric effector. In this case, the inhibitor altered substrate binding (Ks ≠ Kis), whereas substrate did not affect inhibitor binding (Ki = Ksi). In the absence of allosterism (traditional noncompetitive inhibition, model 4), all inhibition constants were the same and the ESI and EIS complexes were equivalent.
SCHEME 2. Possible inhibition mechanisms for CYP2E1 pNP activity by 4MP and IND.

E = CYP2E1; S = pNP; I = 4MP or IND.
We then identified the most probable mechanism and corresponding parameters for inhibition by these molecules using the advanced tools of numerical analysis and applied statistics, as implemented in the software DynaFit (15, 16). Input files (scripts) for this analysis are included in the Supplemental Material.
Determination of Binding Mechanism for 4MP and IND
Perturbation of the P450 Soret spectra because of the association of ligands is a useful tool for assessing binding events (17). In this study, we monitored the shift from the high spin to the low spin state for the iron upon ligation of nitrogen-bearing molecules, 4MP and IND, to the heme iron. Formation of the Fe–N bond increased the absorbance near 430 nm and decreased the absorbance near 390 nm as a function of ligand concentration. Stocks for 4MP were prepared in H2O and IND in methanol based on the solubility of these compounds. The titrations with these solutions were performed using tandem cuvettes to correct for any solvent effects on CYP2E1 absorbance and possible contributions from titrant to observed changes in absorbance. Specifically, we titrated 0.1 μM CYP2E1 in 50 mM potassium phosphate, pH 7.4, 20 μM DLPC with increasing amounts of ligand at 25 °C. Glycerol was included to stabilize the protein against denaturation. Spectral changes were recorded from 350 to 475 nm using a Jasco V-550 spectrophotometer. In the process, we generated difference spectra by substrating the reference CYP2E1 sample with solvent from the absorbance of the sample with titrant present. Data from 4 to 6 experiments were compiled and averaged for analyses.
Although only one molecule can associate with the heme iron, multiple binding events can be observable if the association of subsequent molecules induces a different spectroscopic species. Based on the observed substrate inhibition for pNP oxidation by CYP2E1, the binding of two monocyclic molecules was conceivable. To determine the stoichiometry of CYP2E1 complexes with small molecules, we employed the software program DynaFit as described for analysis of the catalytic data. For these studies we fit absorbance data to mechanisms incorporating either one or two binding events (Scheme 3). When two molecules were bound to CYP2E1, we included the possibility that EL, EL2, or both complexes yielded a spectroscopic signal as denoted by the asterisk. Input files (scripts) for this analysis are included in Supplemental Material.
SCHEME 3. Possible CYP2E1 binding modes for 4MP and IND.

E = CYP2E1; L = 4MP or IND.
Generation of Liganded Complexes through Computational Docking Efforts
Sybyl7.2 (Tripos, Inc., St. Louis, MO) was utilized to model the interaction between the CYP2E1-binding site and potential substrates. The three-dimensional coordinates of the CYP2E1 enzyme were obtained from previous experimental work that yielded a homology model for the enzyme (11). Essential hydrogen atoms and Gasteiger-Huckel partial charges were assigned to the protein sequence. The three-dimensional coordinates of the substrates (including hydrogen atoms and partial charges) were constructed and energy-minimized using the Tripos force field with the default settings in Sybyl7.2.
Surflex-Dock 2.0 (18) was used to perform flexible docking (with default settings) of the substrates in the binding site of CYP2E1. Surflex-Dock uses an empirical scoring function and a patented search engine to dock the substrates into the CYP2E1-binding site. The scoring function is a weighted sum of nonlinear functions involving van der Waals surface distances between the appropriate pairs of exposed enzyme and substrate atoms (19). Scores are expressed in pKd units to represent binding affinities.
RESULTS
CYP2E1 Steady-state Activity toward pNP
As reported previously (7), CYP2E1 oxidation of pNP led to significant substrate inhibition. The substrate saturation curve displayed a maximum followed by a decrease in activity (open circle, Fig. 2), indicating the traditional Michaelis-Menten kinetic scheme for a single substrate binding site (Scheme 1) could not explain substrate turnover. As an alternative, we fit the data to a two-substrate binding mechanism (Scheme 1) using DynaFit as described (15). CYP2E1 demonstrated a relatively low Kss (24 μM) and the rapid turnover of 47 min−1 for pNP; nevertheless, at higher pNP concentrations (>100 μM), the activity gradually decreased as a second molecule bound to CYP2E1 through an effector site (Ks 260 μM), which inhibited activity at the catalytic site (Table 3). This set of experiments was included in 4MP inhibition studies because of common absence of an organic co-solvent (see below).
FIGURE 2.

Steady-state oxidation of pNP by CYP2E1 in the presence of 4MP (A) or IND (B). For reactions, 25 nM CYP2E1, 100 nM CPR-K56Q, and 50 nM cytochrome b5 were reconstituted in 50 mM potassium phosphate, pH 7.4, 20 μM DLPC, pNP (varied from 5 to 750 μM), 2 units μl−1 catalase, 0.04 μg μl−1 superoxide dismutase, and an NADPH-regenerating system (2 microunits μl−1 glucose-6-phosphate dehydrogenase, 10 mM glucose 6-phosphate, 2 mM MgCl2, 500 μM NADP+) at 37 °C. To determine initial velocities, product p-nitrocatechol was quantitated as a function of time by HPLC as described (11). The reported values reflect the average from 2 to 4 experiments, including the mean ± S.D. A, for 4MP studies, 0, 1, 5, 25, and 125 μM inhibitor was added to reactions. Data were fit to model 2a in Scheme 2. B, IND studies were carried out in the presence of 0.1, 0.3, and 1.0 μM inhibitor at a final concentration of 0.25% methanol. Final data were fit to the single-site competition mechanism (model 1) shown in Scheme 2.
TABLE 3. Inhibition parameters for CYP2E1 pNP activity by 4MP and IND.
Inhibition models are shown in Scheme 2. The nonsymmetrical 99% confidence intervals for the parameters are shown in parentheses.
| Inhibitor | Model | Catalytic parameters
|
Inhibition constants
|
||||
|---|---|---|---|---|---|---|---|
| kcat | Km | Kss | Ki | Kis | Ksi | ||
| min−1 | μM | μM | μM | μM | μM | ||
| 4MP | Two-site inhibition | ||||||
| Model 2a (no ESI) | 47 (40–53) | 24 (17–32) | 260 (210–370) | 2.0 (1.3–3.3) | 120 (58–250) | ||
| Model 2a (no EIS) | 47 (41–53) | 24 (17–32) | 260 (210–370) | 2.0 (1.3-.2) | 10 (6.9–17) | ||
| IND | Single-site inhibition | 37 (27–65) | 51 (27–120) | 130 (64–220) | 0.12 (0.073–0.18) | ||
Inhibition of CYP2E1 Activity toward pNP
To probe binding sites for monocyclic and bicyclic molecules, we identified the mechanism of inhibition by 4MP and IND during steady-state turnover of pNP. Given the observed substrate inhibition during pNP oxidation (7), all possible mechanisms for CYP2E1 inhibition by these molecules included two binding sites for substrate leading to one active complex; nevertheless, there were multiple possible modes of inhibition that depended on whether substrate or inhibitor acted allosterically (Scheme 2). After globally fitting reactions to these mechanisms, we generated a corresponding Akaike information criterion (AICc) to identify statistically the most plausible based on the quality of the fits. The significance rules as outlined by Burnham and Anderson (20) provided a metric to rank models such that the lower AICc values indicated comparatively high support for the mechanism. When comparing the ΔAICc, values between 0 and 2 indicate substantial support, whereas values between 4 and 7 signify considerably less support. A value for ΔAICc greater than 10 indicates essentially no support for the model.
For 4MP, there was a significant preference for model 2a based on ΔAICc values greater than 10 for all other possibilities (Table 1). In this mechanism, 4MP and pNP compete for the same binding sites and alter affinity at the opposing site. Analysis of the parameters revealed extremely large uncertainty in Kis and Ksi, which correspond to the mixed substrate-inhibitor complexes, despite using concentrations of 4MP up to 125 μM or 100-fold greater than the predicted Kd for the second site (see below). This observation suggested that we perform a second round of model discrimination analysis. Data were fit to variations of model 2a whereby either the ESI or EIS complexes did not form (Scheme 2). These simpler mechanisms were preferred over model 2a with equal probability, as shown through model discrimination analysis (Table 2).
TABLE 1. Model discrimination analysis for 4MP and IND inhibition of CYP2E1 pNP activity.
Tested models are shown in Scheme 2. In all cases, the substrate is a homotypic allosteric effector; however, other possible effector roles between substrate (S) and inhibitor (I) are indicated. P indicates the number of adjustable parameters for the model; RSS indicates the relative sum of the squares; ΔAICc shows the increase in the second-order Akaike information criterion relative to the best model (ΔAICc 0); w indicates the Akaike weight such that 0.10 = 10% probability.
| Model | Allosteric effector | P | 4MP
|
IND
|
||||
|---|---|---|---|---|---|---|---|---|
| RSS | ΔAICc | w | RSS | ΔAICc | w | |||
| Single-site inhibition 1 | None | 4 | 0.07145 | 43.3 | 0.0000 | 0.01465 | 0 | 0.9525 |
| Two-site inhibition | ||||||||
| 2a | S, I | 6 | 0.02859 | 0 | 0.9998 | 0.01465 | 6.00 | 0.0473 |
| 2b | S | 5 | 0.04449 | 20.8 | 0.0003 | 0.02471 | 21.7 | 0.0000 |
| 2c | I | 5 | 0.04414 | 20.4 | 0.0004 | 0.04207 | 40.9 | 0.0000 |
| 2d | None | 4 | 0.04404 | 17.7 | 0.0014 | 0.02342 | 16.9 | 0.0002 |
TABLE 2. Second round of model discrimination analysis for 4MP inhibition of CYP2E1 pNP activity.
Tested models are variations of model 1 shown in Scheme 2. P indicates the number of adjustable parameters for the model; RSS indicates the relative sum of the squares; ΔAICc shows the increase in the second-order Akaike information criterion relative to the best model (ΔAICc 0); w indicates the Akaike weight such that 0.10 = 10% probability.
| Variation of Model 2a | P | RSS | ΔAICc | w |
|---|---|---|---|---|
| None | 6 | 0.02858 | 2.66 | 0.1166 |
| ESI does not form | 5 | 0.02858 | 0 | 0.4417 |
| EIS does not form | 5 | 0.02858 | 0 | 0.4417 |
Table 3 lists the best fit values for the parameters and non-symmetrical 99% confidence intervals corresponding to the respective mechanisms. The affinity of 4MP for these sites was much higher than that observed for the substrate. The 4MP Ki for the catalytic site was 2.0 μM compared with a Km of 24 μM for pNP. Depending on which mixed complex was present, the second 4MP molecule bound to the effector site with a Kis of 10 μM or a Ksi of 120 μM, whereas the pNP Kss value was 260 μM.
Because of low solubility in aqueous solution, the studies with the inhibitor IND required a co-solvent, methanol, which altered pNP turnover parameters (Table 1). Compared with the absence of co-solvent, kcat deceased slightly from 47 to 37 min−1. More significantly, Km increased from 24 to 51 μM, whereas Kss decreased from 260 to 130 μM. The apparent solvent inhibition as reported by others (21) likely reflects competition or changes in binding sites between methanol and other molecules because of the ability of the methanol to also serve as a substrate (22). To normalize these effects among experiments, we used a constant concentration of methanol (0.25%) in experiments from 0 to 1 μM IND.
Unlike 4MP, IND inhibited pNP oxidation through simple competition for a single site on the enzyme as described by model 1 (Table 1). The presence of bound IND effectively blocks binding of pNP to either catalytic or effector sites resulting in an inactive complex. As shown in Table 3, IND was a potent inhibitor of CYP2E1 activity with a Ki of 0.12 μM. In fact, studies were limited to a maximum of 1 μM IND because of significance inhibition of activity under those conditions.
Binding of 4MP and IND by CYP2E1
In addition to specific inhibition of CYP2E1 activity, 4MP and IND were excellent candidates for yielding spectroscopic signals correlated with binding events. Nitrogen heterocycles and anilines shift the high-low spin equilibrium for heme iron by replacing a water molecule ligated to the iron (if present), which stabilizes the low spin form. By shifting the spin state from high to low for CYP2E1, 4MP and IND induce a type II difference binding spectra (Fig. 3A). For both molecules the peak in absorbance occurred at 430 nm and a trough developed at a minimal absorbance of 392 nm.
FIGURE 3. Type II difference spectra for CYP2E1 binding 4MP and IND.

Titrations were performed using tandem cuvettes to correct for any solvent effects on CYP2E1 absorbance and possible contributions from titrant to observed changes in absorbance. In the process, we generated difference spectra by substrating the reference CYP2E1 sample with solvent from the absorbance of the sample with titrant present. Specifically, 0.1 μM CYP2E1 in 50 mM potassium phosphate, pH 7.4, 20 μM DLPC was titrated with increasing amounts of ligand at 25 °C. The reported values reflect the average from 6 experiments, including the mean ± S.D. A, typical type II binding spectra for these heterocyclic molecules. B, 4MP titration data were fit to the two-site model in Scheme 3. C, IND titrations were performed in the presence of 0.25% methanol. Data were fit to the single-site model in Scheme 3.
As observed from our catalytic studies, the most plausible binding model for CYP2E1 and 4MP was the presence of two binding sites (ΔAICc 0 versus ≥19.5 for other models, see Table 4). The fit of the data to this model is shown in Fig. 3B. The resulting parameters were defined within the 95% confidence intervals. To ensure saturation of these sites, the titration was carried out to 35 μM 4MP. The fit of the data to a two-site model in Scheme 3 resulted in two relatively high affinity sites. Following binding of the first 4MP molecule (Kd1 = 0.67 μM), a second molecule rapidly binds with 2-fold weaker affinity (Kd2 = 1.3 μM). The second binding event led to a slight decrease in the absorbance of the type II complex, as indicated by the respective extinction coefficients. This effect suggests a slight perturbation of the Fe–N bond responsible for the original absorbance signal.
TABLE 4. Model discrimination analysis for 4MP and IND binding to CYP2E1.
Tested models are shown in Scheme 3. The asterisk denotes complexes contributing to the observed signal. P indicates the number of adjustable parameters for the model; RSS indicates the relative sum of the squares; ΔAICc shows the increase in the second-order Akaike information criterion relative to the best model (ΔAICc 0); w indicates the Akaike weight such that 0.10 = 10% probability. Minimal sum of squares for 4MP studies was 0.0000015 and the value for IND studies was 0.00000123.
| Model | P | 4MP
|
IND
|
||||
|---|---|---|---|---|---|---|---|
| RSS | ΔAICc | w | RSS | ΔAICc | w | ||
| EL* | 3 | 1.595 | 27.4 | 0.000 | 1.000 | 0.0 | 0.725 |
| EL*, EL2 | 4 | 1.367 | 19.5 | 0.000 | 1.000 | 2.4 | 0.218 |
| EL, EL2* | 4 | 1.749 | 36.7 | 0.000 | 5.219 | 99.9 | 0.000 |
| EL*, EL2* | 5 | 1.000 | 0.0 | 1.000 | 1.003 | 5.1 | 0.057 |
As observed for the catalytic studies, IND bound to CYP2E1 through a single high affinity binding site as shown by Table 4 (ΔAICc 0). Fig. 3C shows the formation of the type II complex with a Kd of 0.0052 μM (Table 5). Of note, this value is much lower than the enzyme concentration (0.1 μM) used in the assay, and thus the accuracy of this value is not clear. To ensure no other complexes were present, these studies were carried out to 3 μM IND, which was almost 1000-fold greater than the predicted dissociation constant for the binary complex. Moreover, the potency of IND inhibition limited catalytic studies to 1 μM IND. Two of the models incorporating two binding sites were statistically probable; however, for both mechanisms the predicted Kd2 value had open confidence intervals, and thus could not be determined.
TABLE 5. CYP2E1 binding parameters for 4MP and IND.
4MP was fit to a two binding site model, whereas IND was fit to the single site model (Scheme 3). The nonsymmetrical 95% confidence intervals for the parameters are shown in parentheses.
| Ligand | Kd1 | ε1(430–392) | Kd2 | ε2(430–392) |
|---|---|---|---|---|
| μM | cm−1 μM−1 | μM | cm−1 μM−1 | |
| 4MP | 0.67 (0.50–0.95) | 0.045 (0.034–0.052) | 1.3 (0.53–3.9) | 0.039 (0.038–0.41) |
| IND | 0.0052 (0.0028–0.0084) | 0.030 (0.029–0.030) |
Docking pNP to Generate Homotypic Complexes with CYP2E1
Based on computational docking studies between a homology model for CYP2E1 (11) and pNP, the substrate bound to proximal and distal sites relative to the active site heme. The surface of these binding sites is shown in Fig. 4 to highlight the docking of these molecules within the substrate access channel. At the proximal, or catalytic, site, pNP adopted an orientation productive for hydroxylation. Of the possible sites for oxidation, the closest one was 4.76 Å from the heme iron indicating some structural dynamics would be necessary to position the substrate for catalysis. Multiple CYP2E1 residues mediated van der Waals contacts with pNP at <3.0 Å, including Ile-115, Phe-207, Ala-299, methyl group from Thr-303, Val-364, Leu-368, and Phe-478. The orientation of pNP was possibly stabilized by an unusual interaction between the oxygen from the nitro group on pNP and Phe-478 (2.6 Å).
FIGURE 4. Surface depiction of catalytic and effector binding sites for molecules.
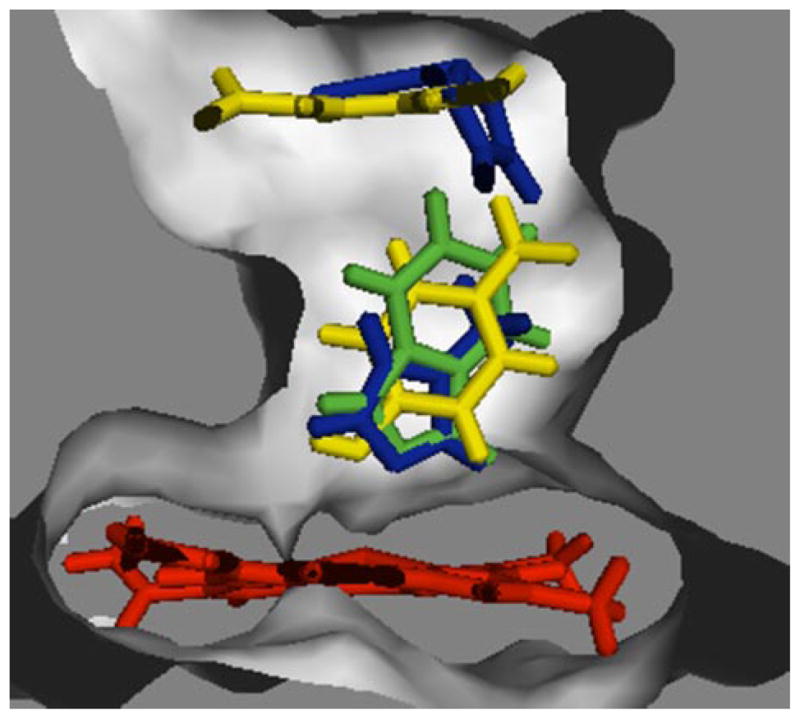
Overlaid ligands are pNP (yellow), 4MP (blue), and IND (green). Molecular graphics were generated by PYMOL.
The second pNP molecule bound to a site distal from the heme, referred to as the effector site, because catalysis cannot occur at this site. In the absence of substrate at the catalytic site, the pNP molecule at the effector site formed close van der Waals contacts (<3.0 Å) with Ile-115, Phe-116, Phe-207, Leu-210, Leu-368, and Phe-478, as well as a π-stacking interaction between the pNP ring and Phe-478 (3.03 Å). Hydrogen bonds also formed between the phenolic hydrogen of pNP and the peptide backbone carbonyls from residues Phe-207 (2.09 Å) and Leu-210 (2.41 Å). The binding of pNP to the catalytic site induced a rotation of pNP at the effector site, leading to changes in certain protein-ligand interactions. Although van der Waals contacts were broken with Ile-115, new contacts were formed with Phe-298. The π-stacking interaction was also lost between pNP and Phe-478. Finally, occupancy of the distal effector site induced a slight ~0.42 Å shift of the proximally bound pNP molecule, but no changes in interactions with amino acids.
Docking 4MP to Generate Homotypic Complexes with CYP2E1
4MP binding at the catalytic site resulted in similar interactions as observed for pNP. Like the oxygen from the hydroxyl group of pNP, the 4MP nitrogen at position 1 was 3.05 Å from the heme iron. CYP2E1 residues (Ile-115, Ala-299, the methyl group from Thr-303, Val-364, and Leu-368) provided van der Waals contacts to 4MP (<3.0 Å). Despite occupancy of the effector site, the second 4MP molecule could bind to the catalytic site indicating 4MP did not block access through the channel (Fig. 4). Side chains from Phe-207 and Leu-210 stabilized 4MP binding through close van der Waals contacts. Unlike pNP bound to this site, Phe-478 formed π-stacking interactions with 4MP. The formation of the homotypic 4MP complex did not significantly perturb binding of 4MP initially bound to CYP2E1. In addition to contacts with the enzyme, the bound 4MP molecules formed van der Waals contacts through their respective methyl groups.
Docking 4MP and pNP to Form a Heterotypic Complex with CYP2E1
For our inhibition studies, the presence of two binding sites for pNP and 4MP in the reaction suggests heterotypic complexes likely form and contribute to reaction kinetics. For the ESI complex (Scheme 2), pNP was bound at the catalytic site and 4MP at the effector site. The docking solutions for this complex were dependent on the order of association leading to ambiguity for the pNP binding mode. The presence of pNP at the catalytic site forced 4MP binding at the effector site at 110° relative to 4MP in the homotypic complex. The different binding orientation for 4MP in the CYP2E1·pNP·4MP complex obviated formation of π-stacking between 4MP and Phe-478, as observed for the CYP2E1·4MP complex. Phe-207, Leu-210, and Phe-478 formed van der Waals contacts with 4MP (<3.0 Å). The introduction of 4MP did not alter pNP binding. In contrast, docking pNP to the CYP2E1 complex with 4MP bound to the effector site led to pNP adopting a nonproductive orientation for oxidation to occur. The nitro group was nearest to the heme iron (2.97 Å) rather than the hydroxyl group and potential sites of oxidation (5.17 Å at the closest point). The amino acid contacts with pNP were the same as those observed for the CYP2E1·pNP complex. The minimization of this mixed ternary complex caused a slight change in 4MP binding. Ligand-ligand interactions were also observed. The methyl group of 4MP formed van der Waals contacts with the phenyl ring of pNP, and a hydrogen bond (1.7 Å) was observed between the N-H group of 4MP and the oxygen of the pNP hydroxyl group.
The docking solution for the EIS complex (Scheme 2) indicated 4MP binding at the catalytic site was the same as observed in the 4MP homotypic complex, which was not the case for pNP. Unlike the pNP homotypic complex, pNP bound to the effector site in a similar orientation as observed in the absence of ligand at the catalytic site. Close van der Waals contacts formed with Ile-115, Phe-116, Phe-207, Leu-210, Leu-368, and Phe-478. Further stabilization of the complex derived from hydrogen bonds formed between the phenolic hydrogen from pNP and peptide backbone carbonyls from residues Phe-207 (2.36 Å) and Leu-210 (2.34 Å). As observed in the previous complex, the methyl group from 4MP formed van der Waals contacts (2.75 Å) with the phenyl ring of pNP.
Docking IND to Generate Homotypic Complexes with CYP2E1
Despite the larger size, IND mediated contacts with the same residues as observed for pNP and 4MP (Fig. 4). The catalytic site residues, Ile-115, Phe-207, Ala-299, Thr-303 (methyl group), Val-364, and Leu-368, and Phe-478, mediated van der Waals contacts at a distance of less than 3.2 Å from IND. In support of the observed type II binding spectra, the nitrogen at position 1 for the IND pyrazole ring was poised for formation of a bond with the heme iron at 3.12 Å. There were no hydrogen bonds or π-stacking interactions for this complex. Because IND binding at the catalytic site did not completely occlude the effector site, we attempted to dock either pNP or another IND molecule. These molecules bound to a different pocket composed of the B-C and F-G loops, which was distal (~10 Å) from the active site heme.
DISCUSSION
In this study, we explain CYP2E1 substrate inhibition in terms of both mechanistic and structural views and extend our analysis to include typical representatives of monocyclic and polycyclic aromatic inhibitors. Under steady-state conditions, both substrate pNP and inhibitor 4MP competed for catalytic and effector sites. In contrast, the larger IND molecule inhibited catalysis through the occupancy of a single CYP2E1 site associated with substrate turnover. Similarly, 4MP and IND yielded type II binding spectra that reflected the association of either two 4MP or one IND molecule(s) to CYP2E1, respectively. Based on computational docking studies, the two sites for monocyclic molecules, pNP and 4MP, exist within a narrow channel connecting the active site to the surface of the enzyme. Because of the presence of the heme iron, one site supports catalysis, whereas the other, more distal effector site binds molecules that can influence the binding orientation and egress of molecules for the catalytic site. Although IND did not bind these sites simultaneously, the presence of IND at the catalytic site blocked binding of a second molecule at the effector site.
Despite early catalytic evidence for multiple binding sites (7, 9), the kinetic mechanism for the metabolism of monocyclic molecules by CYP2E1 has been relatively unexplored. In the original study, a maximal rate of pNP turnover was observed at 100 μM followed by a decrease in rate by 70% at 500 μM. Despite the evidence for substrate inhibition, the observed data were not fit to a particular kinetic mechanism. Subsequent steady-state studies by others have been typically limited to a maximal concentration of 100 μM pNP and relied on a simple Michaelis-Menten scheme to analyze data (23–25). For the rabbit CYP2E1 system, kcat and Km were reported to be 30 min−1 and 38 μM, respectively (23). In contrast, we measured a kcat of 47 min−1 and Km of 24 μM. As measured by kcat/Km, the differences in kinetic parameters translated to a 2.5-fold higher catalytic efficiency observed in our study when compared with the results from the previous study. Although the reaction conditions were not identical between the studies, the use of a lower reaction pH (6.8 versus physiological pH 7.4) and higher ratios of CPR and cytochrome b5 by the authors should have yielded more efficient turnover than we observed (7). The most likely explanation for the discrepancy is our use of a wider substrate concentration range (5–750 μM) to saturate a second substrate binding site and analysis of the resulting rates with the substrate inhibition model (Scheme 1). Taken together, the simplicity and convenience of the Michaelis-Menten scheme can lead to significant erroneous predictions of the true kinetic parameters for substrate turnover by CYP2E1.
Similar to the predictions by other homology models (26–29), our structure for pNP bound to the catalytic site consisted of a binding pocket derived solely from contacts with hydrophobic residues. Ile-115, Phe-207, Ala-299, methyl group from Thr-303, Val-364, Leu-368, and Phe-478 all mediated van der Waals contacts with pNP. The significance of the Thr-303 methyl group was shown by changes in the regioselectivity of fatty acid oxidation by CYP2E1 when the residue was substituted with serine (30). V364L and L368V substitutions led to~2-fold decrease in kcat (originally reported as Vmax) and Km, respectively, although the data were analyzed with the Michaelis-Menten scheme (31). Interestingly, Leu-210 was not observed to contact pNP, yet substitution of this residue with isoleucine decreased kcat but not Km for pNP (31), which could be explained by binding at the effector site (see below). Despite the presence of multiple phenylalanines, there was no observable π-stacking between residues and pNP as predicted by others (27). The orientation of pNP reflected the conformation of the active site pocket formed by hydrophobic residues and possibly through an unusual interaction between the oxygen from the nitro group on pNP and Phe-478, as described previously (32). Substitution of Phe-478 with valine actually led to complete loss of pNP activity (31). The ortho position was poised for hydroxylation, although the site of oxidation was 4.76 Å from the heme iron indicating conformational dynamics would be necessary for catalysis to occur.
Unlike previous reports for CYP2E1 homology models (26, 27), we observed no contacts between charged or polar groups and pNP. Thr-303 provided only van der Waals contacts through the methyl group of the side chain rather than through the hydroxyl group. Moreover, neither Glu-302 nor Arg-100 was even observed in the active site. Although these former models assigned critical roles for charged and polar residues in binding and orienting substrates, we argue that restrictions of the active site geometry and reliance on multiple weak interactions dominate the selectivity and productivity of enzyme-substrate interactions.
The potent CYP2E1 inhibitor 4MP competed with pNP for the CYP2E1 catalytic site as shown through catalytic, binding, and docking studies. The inhibition constant (Ki) during pNP turnover was 2.0 μM, which was 12-fold less than the apparent Km for substrate (24 μM) (Table 3). Direct evidence for the high affinity interaction between the catalytic site and 4MP was shown through the generation of the type II binding spectra between CYP2E1 and the inhibitor (Fig. 3A). The spectral shift required the ligation of the lone pair of electrons from 4MP to ligate the heme iron in the active site, thereby stabilizing the low spin state for CYP2E1. During catalysis, CYP2E1 cycles through different spin states and possibly conformations (33). These processes could explain the lower 4MP dissociation constant for this site (Kd1 = 0.67 μM, see Table 5) when compared with the corresponding Ki value. When bound to the catalytic site, 4MP mediated van der Waals contacts with the same residues as observed for pNP. An additional interaction with Leu-210 and formation of the Fe–N bond likely played a role in the higher selectivity for 4MP over pNP. Other than the hydrophobic side chains in the binding pocket, there were no obvious contacts contributing to the orientation of 4MP.
The mechanism of substrate inhibition incorporates a second binding event by the substrate to produce a dead end complex, which explains the decrease in activity at higher substrate concentrations. Substrate then acts as an effector during turnover through the occupancy of this second binding site. The binding of pNP to the effector site was 10-fold weaker than to the catalytic site, i.e. Km of 24 μM versus Kss of 260 μM. Nevertheless, the contribution of the effector site to pNP catalysis is significant. If we assume CYP2E1 obeyed the Michaelis-Menten scheme (Scheme 1), i.e. no effector site, then the Vmax for pNP turnover would have been 1.2 μM min−1 based on a kcat of 47 min−1 and CYP2E1 concentration of 0.025 μM. Instead, we observed a maximal rate of 0.76 μM min−1 at 100 μM pNP indicating only 64% of the maximal possible rate was observed. Moreover, the observed maximal rate from our study is the same value as reported previously by others (0.025 μM CYP2E1 × 30 min−1 = 0.76 μM min−1) (23); however, in their study, initial rates at pNP concentrations greater than 100 μM were excluded from the analysis of the data. This observation illustrates how designing experiments to conform to Michaelis-Menten kinetics can mask the “true” catalytic properties of an enzyme, e.g. CYP2E1.
If the effector site can accommodate pNP, then other monocyclic compounds would likely be able to bind and alter reaction kinetics. Hargreaves et al. (9) assayed the effect of a series of 5- and 6-membered cyclic molecules on pNP oxidation by CYP2E1. Studies were limited to four pNP concentrations, and the resulting data were fit to the typical reversible inhibition mechanisms: competitive (single site), uncompetitive, non-competitive, and mixed type. Although there was no clear correlation between the structures of the inhibitors and corresponding types of inhibition mechanisms, the observation of uncompetitive, noncompetitive, and mixed type inhibition supports an effector site accessible to monocyclic molecules. In our study, 4MP displayed inhibition toward catalytic and effector sites recognized by pNP (Table 3), which is mechanistically similar to mixed type inhibition. Like the catalytic site, 4MP demonstrated a much higher affinity for the effector site than pNP. Although we were not able to determine whether EIS or ESI were present, the corresponding dissociation constants Kis and Ksi were 120 and 10 μM, respectively. Regardless of the mechanism of inhibition, these values are well below the dissociation constant for pNP at the effector site (260 μM) and thus could impact the significance of 4MP inhibition of CYP2E1 activity. Although indicative of a homotypic complex, we were able to detect two binding events using 4MP as a titrant, which is consistent with this model.
To provide structural explanations for these binding and catalytic results, we docked a second monocyclic molecule to binary CYP2E1 complexes for pNP and 4MP. In both cases, the second pNP or 4MP molecule bound within the substrate access channel created by the B-C loop and F helix, as well as sheets 1 and 4 (Fig. 4). As a site distal from the active site heme, the corresponding binding pocket likely reflects the effector site because of the inability to carry out substrate oxidation. For pNP, binding at the effector site resulted in close van der Waals contacts with Phe-116, Phe-207, Leu-210, Leu-368, Phe-298, and Phe-478. A role for Leu-210 in pNP oxidation was shown by the decrease in Vmax when the residue was substituted with isoleucine (31). The extension of the side chain by a methylene group may alter the steps limiting catalysis, e.g. product release. From the same study, an L368V substitution may have perturbed the effector site resulting in a 2-fold decrease in both Vmax and Km. Nevertheless, these experiments were designed to enable the use of the Michaelis-Menten kinetic scheme at pNP concentrations ≤100 μM, and thus the impact of these substitutions on specific pNP sites is not known. Based on our docking studies, the aromatic rings at the effector site mediated multiple weak polar interactions to the pNP oxygens (32), rather than π-stacking interactions with the benzyl ring of pNP. The orientation of pNP was further determined by hydrogen bonding between the phenolic hydrogen of pNP and the peptide backbone carbonyls from residues Phe-207 and Leu-210. Additional van der Waals contacts between the two pNP molecules contributed to the stability of the complex.
Although contacts between pNP and the CYP2E1 homology model did not change, 4MP binding at the effector site resulted in two different pNP orientations that depended on the order of docking for the molecules. Substrate pNP bound with either the hydroxyl or nitro moiety directed toward the heme iron reflecting orientations productive or nonproductive for oxidation. Ligand-ligand interactions further stabilized the structure. The methyl group of 4MP formed van der Waals contacts with the phenyl ring of pNP, and a hydrogen bond was observed between the N-H group of 4MP and the oxygen of the pNP hydroxyl group. In contrast, occupancy of the effector site by either molecule did not significantly affect 4MP binding at the catalytic site.
The catalytic, binding, and docking studies with IND further supported the juxtaposition of two sites for monocyclic molecules. The bicyclic heterocycle inhibited pNP oxidation (Table 3) and bound to CYP2E1 (Table 5) through a single binding event. IND was a potent competitive inhibitor during pNP catalysis demonstrating a Ki of 0.12 μM. In fact, the magnitude of the inhibition at 1 μM IND decreased the observed initial velocities to the limit of detection for the assay. Similarly, the IND binding isotherm was hyperbolic up to 3 μM resulting in a bimolecular Kd of 0.0052 μM. In both cases, there were no binding events following the association of IND with CYP2E1. Although bound to the catalytic site, IND likely occluded a binding pocket present in previous studies with monocyclic molecules. In support of this conclusion, the docking studies with IND confirmed the presence of IND at the catalytic site prevented subsequent binding of pNP or another IND molecule to the effector site (Fig. 4). Rather, these molecules bound to a pocket composed of the B-C and F-G loops, which likely reflects the entrance to the substrate access channel at the protein-membrane interface as predicted by progesterone-bound structure of CYP3A4 (34).
Taken together, these complexes for cyclic molecules may provide an explanation for the contributions of the effector site to CYP2E1 catalysis. As a sequential binding event within a single substrate access channel, occupancy of the effector site could block egress of products, e.g. p-nitrocatechol following pNP oxidation. Evidence for a narrow channel between the active site and solvent was shown through CO flash photolysis studies with CYP2E1 (35). The binding of arachidonic acid, a large 20-carbon fatty acid, blocked the release of dissociated CO, whereas the smaller ethanol substrate had no effect. More analogous to our studies with monocyclic and bicyclic molecules, isotopic experiments with deuterated substrates p-xylene and dimethylnaphthalene yielded similar isotope effects by CYP2E1 (36). One explanation preferred by the authors was the presence of two binding sites for the monocyclic p-xylene molecule. Occupancy of these sites would render the isotope effect for this molecule similar to that for the bicyclic dimethylnaphthalene molecule. In the case of heterotypic complexes, the effector could further stabilize nonproductive complexes, even orientations of the substrate. With respect to binding at the effector site, 4MP was a more potent inhibitor (Kis = 120 μM or Ksi = 10 μM) than pNP (260 μM). As a consequence of protein-ligand and ligand-ligand interactions generating these dead-end complexes, the selectivity of the effector site ultimately determines the impact of molecules on reaction kinetics. Variations in these contacts may explain why 5- and 6-membered cyclic molecules inhibited pNP oxidation through different mechanisms (9).
Concluding Remarks
Numerous studies have focused on the prospect of multiple binding sites for major drug metabolizing P450s (37–39); nevertheless, there has been no investigation of this phenomenon with CYP2E1 aside from the initial report by Koop (7). We are the first to explore the impact of multiple binding sites on CYP2E1 activity. The distinctive inhibition mechanisms for 4MP and IND highlighted the significance of understanding the underlying kinetic mechanism for substrate metabolism. These heterocyclic molecules displayed similar high affinities toward the catalytic site, yet IND was far more potent inhibitor of CYP2E1. To achieve the same degree of inhibition, much higher 4MP concentrations were necessary to saturate both binding sites. Because of the high preference for monocyclic molecules, these findings could significantly alter the interpretation and prediction of many CYP2E1 reactions, considering that the prospect of allosterism has not been explored. Ultimately, the specificities of these molecules for both sites will determine the contribution of heterotypic complexes to the metabolism of CYP2E1 substrates. Elucidating the details of these interactions will require suitable modeling and discrimination among possible reaction mechanisms. Moreover, the design of experiments to favor Michaelis-Menten kinetics may mask critical structure-function relationships between CYP2E1 and small molecules, e.g. substrates and inhibitors.
Supplementary Material
Acknowledgments
We thank Arvind P. Jamakhandi for helpful discussion and the preparation of CYP2E1 and CPR-K56Q. We also thank Wayne L. Backes (Louisiana Health Science Center, New Orleans) for providing the rabbit cytochrome b5 used in this study.
Footnotes
The on-line version of this article (available at http://www.jbc.org) contains the scripts (input data) for the DynaFit software (15, 16).
The abbreviations used are: pNP, p-nitrophenol; 4MP, 4-methylpyrazole; IND, bicyclic indazole; HPLC, high pressure liquid chromatography; DLPC, dilauroyl-L-α-phosphatidylcholine; pNC, p-nitrocatechol.
References
- 1.Guengerich FP. Toxicol Lett. 1994;70:133–138. doi: 10.1016/0378-4274(94)90156-2. [DOI] [PubMed] [Google Scholar]
- 2.Ronis M, Lindros KO, Ingelman-Sundberg M. In: Cytochromes P450, Metabolic and Toxicological Aspects. Ioannides C, editor. CRC Press, Inc; Boca Raton, FL: 1996. pp. 211–239. [Google Scholar]
- 3.Robertson G, Leclercq I, Farrell GC. Am J Physiol. 2001;281:G1135–G1139. doi: 10.1152/ajpgi.2001.281.5.G1135. [DOI] [PubMed] [Google Scholar]
- 4.Doty SL, Shang TQ, Wilson AM, Tangen J, Westergreen AD, Newman LA, Strand SE, Gordon MP. Proc Natl Acad Sci U S A. 2000;97:6287–6291. doi: 10.1073/pnas.97.12.6287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banerjee S, Shang TQ, Wilson AM, Moore AL, Strand SE, Gordon MP, Doty SL. Biotechnol Bioeng. 2002;77:462–466. doi: 10.1002/bit.10151. [DOI] [PubMed] [Google Scholar]
- 6.Guengerich FP, Kim DH, Iwasaki M. Chem Res Toxicol. 1991;4:168–179. doi: 10.1021/tx00020a008. [DOI] [PubMed] [Google Scholar]
- 7.Koop DR. Mol Pharmacol. 1986;29:399–404. [PubMed] [Google Scholar]
- 8.Yamazaki H, Guo Z, Guengerich FP. Drug Metab Dispos. 1995;23:438–440. [PubMed] [Google Scholar]
- 9.Hargreaves MB, Jones BC, Smith DA, Gescher A. Drug Metab Dispos. 1994;22:806–810. [PubMed] [Google Scholar]
- 10.Jefcoate C. Methods Enzymol. 1978;52:258–279. doi: 10.1016/s0076-6879(78)52029-6. [DOI] [PubMed] [Google Scholar]
- 11.Collom SL, Jamakhandi AP, Tackett AJ, Radominska-Pandya A, Miller GP. Arch Biochem Biophys. 2007;459:59–69. doi: 10.1016/j.abb.2006.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanna IH, Teiber JF, Kokones KL, Hollenberg PF. Arch Biochem Biophys. 1998;350:324–332. doi: 10.1006/abbi.1997.0534. [DOI] [PubMed] [Google Scholar]
- 13.Cheng D, Kelley RW, Cawley GF, Backes WL. Protein Expression Purif. 2004;33:66–71. doi: 10.1016/j.pep.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 14.Jamakhandi A, Jeffus BC, Dass VR, Miller GP. Arch Biochem Biophys. 2005;439:165–174. doi: 10.1016/j.abb.2005.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuzmic P. Anal Biochem. 1996;237:260–273. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- 16.Jamakhandi A, Kuzmic P, Sanders DE, Miller GP. Biochemistry. 2007;46:10192–10201. doi: 10.1021/bi7003476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schenkman JB, Remmer H, Estabrook RW. Mol Pharmacol. 1967;3:113–123. [PubMed] [Google Scholar]
- 18.Jain A. J Med Chem. 2003;46:499–511. doi: 10.1021/jm020406h. [DOI] [PubMed] [Google Scholar]
- 19.Jain A. J Comput Aided Mol Des. 1996;10:427–440. doi: 10.1007/BF00124474. [DOI] [PubMed] [Google Scholar]
- 20.Burnham KP, Anderson DR. Model Selection and Multimodel Inference: A Practical Information-Theoretic Approach. 2. Springer-Verlag Inc; New York: 2002. p. 70. [Google Scholar]
- 21.Easterbrook J, Lu C, Sakai Y, Li AP. Drug Metab Dispos. 2001;29:141–144. [PubMed] [Google Scholar]
- 22.Coon MJ, Koop DR, Morgan ET. Pharmacol Biochem Behav. 1983;18:177–180. doi: 10.1016/0091-3057(83)90168-5. [DOI] [PubMed] [Google Scholar]
- 23.Larson JR, Coon MJ, Porter TD. Proc Natl Acad Sci U S A. 1991;88:9141–9145. doi: 10.1073/pnas.88.20.9141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tassaneeyakul W, Veronese ME, Birkett DJ, Gonzalez FJ, Miners JO. Biochem Pharmacol. 1993;46:1975–1981. doi: 10.1016/0006-2952(93)90639-e. [DOI] [PubMed] [Google Scholar]
- 25.Chen W, Peter RM, McArdle S, Thummel KE, Sigle RO, Nelson SD. Arch Biochem Biophys. 1996;335:123–130. doi: 10.1006/abbi.1996.0489. [DOI] [PubMed] [Google Scholar]
- 26.Tan Y, White SP, Paranawithana SR, Yang CS. Xenobiotica. 1997;27:287–299. doi: 10.1080/004982597240613. [DOI] [PubMed] [Google Scholar]
- 27.Lewis DFV. Xenobiotica. 2002;32:305–323. doi: 10.1080/00498250110112015. [DOI] [PubMed] [Google Scholar]
- 28.Lewis D, Sams C, Loizou GD. J Biochem Mol Toxicol. 2003;17:47–52. doi: 10.1002/jbt.10055. [DOI] [PubMed] [Google Scholar]
- 29.Park JY, Harris D. J Med Chem. 2003;46:1645–1660. doi: 10.1021/jm020538a. [DOI] [PubMed] [Google Scholar]
- 30.Fukuda T, Imai Y, Komori M, Nakamura M, Kusunose E, Satouchi K, Kusunose M. J Biochem (Tokyo) 1993;113:7–12. doi: 10.1093/oxfordjournals.jbchem.a124006. [DOI] [PubMed] [Google Scholar]
- 31.Spatzenegger M, Liu H, Wang Q, Debarber A, Koop DR, Halpert JR. J Pharmacol Exp Ther. 2003;304:477–487. doi: 10.1124/jpet.102.043323. [DOI] [PubMed] [Google Scholar]
- 32.Burley S, Petsko GA. Adv Protein Chem. 1988;39:125–189. doi: 10.1016/s0065-3233(08)60376-9. [DOI] [PubMed] [Google Scholar]
- 33.Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet BM, Ringe D, Petsko GA, Sligar SG. Science. 2000;287:1615–1622. doi: 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]
- 34.Williams PA, Cosme J, Vinkovic DM, Ward A, Angove HC, Day PJ, Vonrhein C, Tickle IJ, Jhoti H. Science. 2004;305:683–686. doi: 10.1126/science.1099736. [DOI] [PubMed] [Google Scholar]
- 35.Smith S, Robinson RC, Smith TG, Burks SM, Friedman FK. Biochemistry. 2006;45:15617–15623. doi: 10.1021/bi061873u. [DOI] [PubMed] [Google Scholar]
- 36.Harrelson J, Henne KR, Alonso DOV, Nelson SD. Biochem Biophys Res Commun. 2007;352:843–849. doi: 10.1016/j.bbrc.2006.11.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Houston J, Galetin A. Arch Biochem Biophys. 2005;433:351–360. doi: 10.1016/j.abb.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 38.Atkins W. Annu Rev Pharmacol Toxicol. 2005;45:291–310. doi: 10.1146/annurev.pharmtox.45.120403.100004. [DOI] [PubMed] [Google Scholar]
- 39.Miller GP, Guengerich FP. Biochemistry. 2001;40:7262–7272. doi: 10.1021/bi010402z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.