

Abstract
Background
Next-generation sequencing has provided a wealth of plastid genome sequence data from an increasingly diverse set of green plants (Viridiplantae). Although these data have helped resolve the phylogeny of numerous clades (e.g., green algae, angiosperms, and gymnosperms), their utility for inferring relationships across all green plants is uncertain. Viridiplantae originated 700-1500 million years ago and may comprise as many as 500,000 species. This clade represents a major source of photosynthetic carbon and contains an immense diversity of life forms, including some of the smallest and largest eukaryotes. Here we explore the limits and challenges of inferring a comprehensive green plant phylogeny from available complete or nearly complete plastid genome sequence data.
Results
We assembled protein-coding sequence data for 78 genes from 360 diverse green plant taxa with complete or nearly complete plastid genome sequences available from GenBank. Phylogenetic analyses of the plastid data recovered well-supported backbone relationships and strong support for relationships that were not observed in previous analyses of major subclades within Viridiplantae. However, there also is evidence of systematic error in some analyses. In several instances we obtained strongly supported but conflicting topologies from analyses of nucleotides versus amino acid characters, and the considerable variation in GC content among lineages and within single genomes affected the phylogenetic placement of several taxa.
Conclusions
Analyses of the plastid sequence data recovered a strongly supported framework of relationships for green plants. This framework includes: i) the placement of Zygnematophyceace as sister to land plants (Embryophyta), ii) a clade of extant gymnosperms (Acrogymnospermae) with cycads + Ginkgo sister to remaining extant gymnosperms and with gnetophytes (Gnetophyta) sister to non-Pinaceae conifers (Gnecup trees), and iii) within the monilophyte clade (Monilophyta), Equisetales + Psilotales are sister to Marattiales + leptosporangiate ferns. Our analyses also highlight the challenges of using plastid genome sequences in deep-level phylogenomic analyses, and we provide suggestions for future analyses that will likely incorporate plastid genome sequence data for thousands of species. We particularly emphasize the importance of exploring the effects of different partitioning and character coding strategies.
Keywords: Composition bias, Phylogenomics, Plastid genome sequences, Plastomes, RY-coding, Viridiplantae
Background
Viridiplantae, or green plants, are a clade of perhaps 500,000 species [1-6] that exhibit an astounding diversity of life forms, including some of the smallest and largest eukaryotes [3,7]. Fossil evidence suggests the clade is at least 750 million years old [8-10], while divergence time estimates from molecular data suggest it may be more than one billion years old [11-14]. Reconstructing the phylogenetic relationships across green plants is challenging because of the age of the clade, the extinction of major lineages [15-17], and extreme molecular rate and compositional heterogeneity [18-22]. Most phylogenetic analyses of Viridiplantae have recovered two well-supported subclades, Chlorophyta and Streptophyta[23,24]. Chlorophyta contain most of the traditionally recognized “green algae,” and Streptophyta contain the land plants (Embryophyta), as well as several other lineages also considered “green algae”. Land plants include the seed plants (gymnosperms and angiosperms; Spermatophyta), which consist of ~270,000 to ~450,000 species [1,3].
While many of the major green plant clades are well defined, questions remain regarding the relationships among them. For example, the closest relatives of land plants have varied among analyses [23,25-29], as have the relationships among the three bryophyte lineages (mosses, liverworts, and hornworts) [29-35]. The relationships among extant gymnosperms also remain contentious, particularly with respect to the placement of Gnetophyta[20,36-43].
Most broad analyses of green plant relationships based on nuclear gene sequence data have relied largely on 18S/26S rDNA sequences [30,37,44,45], although recent analyses have employed numerous nuclear genes [40,46]. Some studies have used mitochondrial gene sequence data, often in combination with other data [29,47,48]. However, investigations of green plant phylogeny typically have either largely or exclusively employed chloroplast genes (e.g., [29,49-52]). Sequence data from the plastid genome have transformed plant systematics and contributed greatly to the current view of plant relationships. With the plastid genome present in high copy numbers in each cell in most plants, and with relatively little variation in gene content and order [53], as well as few reported instances of gene duplication or horizontal gene transfer [54,55], the plastid genome provides a wealth of phylogenetically informative data that are relatively easy to obtain and use [56,57]. Although early phylogenetic studies using one or a few chloroplast loci provided fundamental insights into relationships within and among green plant clades, these analyses failed to resolve some backbone relationships [56-59]. These remaining enigmatic portions of the green plant tree of life ultimately motivated the use of entire, or nearly entire, plastid genome sequences for phylogenetic inference.
Complete sequencing of the relatively small (~150 kb) plastid genome has been technically feasible since the mid-1980s [60,61], although few plastid genomes were sequenced prior to 2000 (see [62,63]). Next-generation sequencing (NGS) technologies, such as 454 [62] and Illumina [64-67], greatly reduced the cost and difficulty of sequencing plastid genomes, and consequently, the number of plastid genomes available on GenBank increased nearly six-fold from 2006 to 2012 [68]. Phylogenetic analyses based on complete plastid genome sequences have provided valuable insights into relationships among and within subclades across the green plant tree of life (recently reviewed in [26,35,68,69]). Still, studies employing complete plastid genomes generally have either focused on subclades of green plants or have had relatively low taxon sampling. Thus, they have not addressed the major relationships across all green plants simultaneously.
We assembled available plastid genome sequences to build a phylogenetic framework for Viridiplantae that reflects the wealth of new plastid genome sequence data. Furthermore, we highlight analytical challenges for resolving the green plant tree of life with this type of data. We performed phylogenetic analyses of protein-coding data on 78 genes from 360 taxa, exploring the effects of different partitioning and character-coding protocols for the entire data set as well as subsets of the data. While our analyses recover many well-supported relationships and reveal strong support for some contentious relationships, several factors, including base composition biases, can affect the results. We also highlight the challenges of using plastid genome data in deep-level phylogenomic analyses and provide suggestions for future analyses that will incorporate plastid genome data for thousands of species.
Results
Data set
We assembled plastid protein-coding sequences from 360 species (Additional file 1) for which complete or nearly complete plastid genome sequences were available on GenBank. Of the 360 species, there were 258 angiosperms (Angiospermae), 53 gymnosperms (Acrogymnospermae, including three Gnetophyta), seven monilophytes (Monilophyta), four lycophytes (Lycopodiophyta), three liverworts (Marchantiophyta), one hornwort (Anthocerotophyta), two mosses (Bryophyta), six taxa from the paraphyletic streptophytic algae, and 26 chlorophytic algae (Chlorophyta). The phylogenetic character matrices contained sequences from 78 genes and the following number of alignment positions: 58,347 bp for the matrix containing all nucleotide positions (ntAll) and the RY-coded (RY) version of the ntAll matrix; 38,898 bp in the matrix containing only the first and second codon positions (ntNo3rd), and 19,449 amino acids (AA). The number of genes present per taxon varied from 18 to 78 (mean = 70), while the number of taxa present per gene ranged from 228 to 356 (mean = 322; see Additional file 2). Taxa with few genes present, such as Helicosporidium (18 genes) and Rhizanthella (19 genes), represent highly modified complete plastid genomes of non-photosynthetic species [70,71]. The percentage of missing data (gaps and ambiguous characters) was ~15.6% for each of the four data sets. The pattern of data across each of the four matrices is decisive, meaning that it can uniquely define a single tree for all taxa [72]. The data contain 100% of all possible triplets of taxa, and are decisive for 100% of all possible trees. All alignments have been deposited in the Dryad Data Repository [73].
GC bias
GC content varied considerably both among lineages and also within single genomes, and chi-square tests rejected the null hypothesis of homogeneous base frequencies (Table 1). The average GC content in the ntAll matrix was 38.9%, and it ranged from 54.3% in Selaginella uncinata to 27.5% in Helicosporidium sp. (Figure 1, Additional file 3). Also, the average GC content varied among first, second, and third codon positions, with by far the most variation among lineages at the third codon position (Figure 1, Additional file 3). Although there was extensive heterogeneity in GC content across all species, there was relatively little variation among the seed plant taxa (Figure 2). There also was significant correlation between nucleotide composition and amino acid composition. Plastid genomes that are GC-rich had a significantly higher percentage (Figure 3; p < 0.001) of amino acids that are encoded by GC-rich codons (i.e., G, A, R, and P). Similarly, GC-rich plastid genomes had a significantly lower percentage (Figure 4; p < 0.001) of amino acids that are coded by AT-rich codons (i.e., F, Y, M, I, N, and K).
Table 1.
Chi-square tests of nucleotide composition homogeneity among lineages
| Data | χ2 | df | p |
|---|---|---|---|
| ntAll |
31350.257185 |
1077 |
< 0.0001 |
| ntNo3rd |
11968.002464 |
1077 |
< 0.0001 |
| ntAll (Position 1) |
8366.331439 |
1077 |
< 0.0001 |
| ntAll (Position 2) |
6003.338041 |
1077 |
< 0.0001 |
| ntAll (Position 3) | 46288.248785 | 1077 | < 0.0001 |
Figure 1.
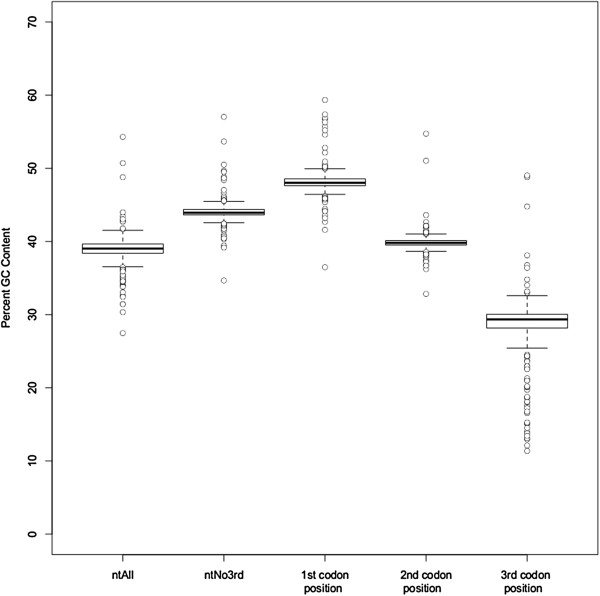
Box plots of percent GC content in the ntAll and ntNo3rd data sets as well as in the first, second, and third codon positions of the ntAll data set.
Figure 2.
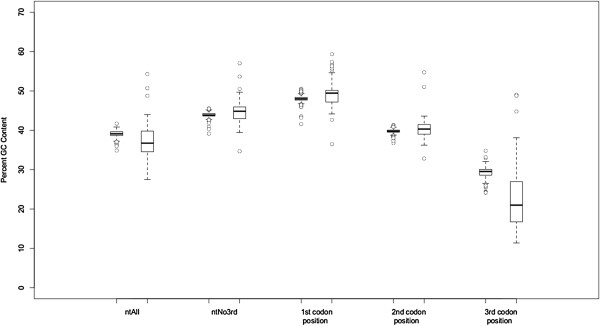
Box plots of percent GC content in seed plants (Spermatophyta; on left) and the data set as a whole (Viridiplantae; on right) in the ntAll and ntNo3rd data sets as well as the first, second, and third codon positions of the ntAll data set. For each pair of box plots, values for seed plants (Spermatophyta) are on the left, and values for all green plant taxa (Viridiplantae) are on the right.
Figure 3.
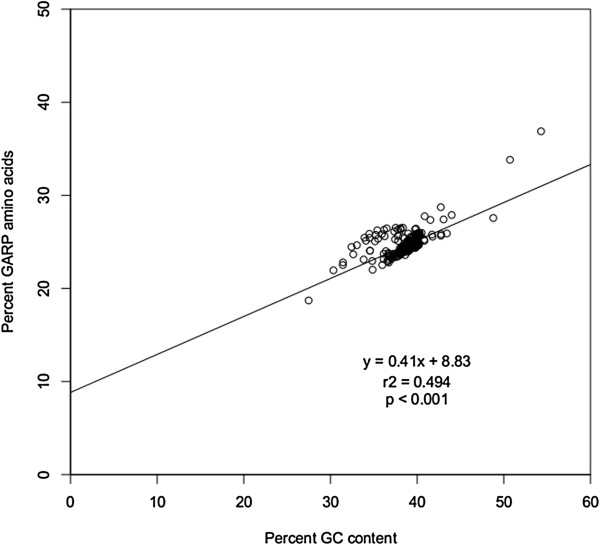
Correlation between percent GC nucleotide content in the ntAll matrix and percent of amino acids in the AA matrix that are coded for by GC-rich codons (G, A, R, and P).
Figure 4.
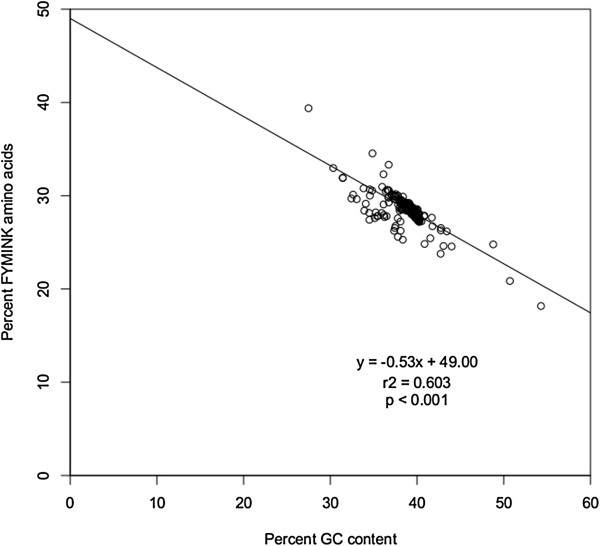
Correlation between percent GC nucleotide content in the ntAll matrix and percent of amino acids in the AA matrix that are coded for by AT-rich codons (F, Y, M, I, N, and K).
Phylogenetic analyses
In the phylogenetic analyses of all data sets and partitioning schemes, the partitioning strategy with the most partitions consistently fit the data best based on the AICc (Table 2). These best-fit models partitioned the AA matrix by gene (78 partitions) and the nucleotide (ntAll, ntNo3rd) and RY matrices by codon position and gene (234 partitions). All a posteriori bootstopping analyses indicated that convergence of support values had been reached after 100 replicates, and thus our choice of 200 replicates was more than sufficient to obtain reliable bootstrap values.
Table 2.
AICc scores for each of the phylogenetic matrix partitioning strategies
| Matrix | Number of characters | Partitioning strategy | Number of partitions | Log-likelihood | AICc | ΔAICc |
|---|---|---|---|---|---|---|
| ntAll |
58,347 |
OnePart |
1 |
−3135739.544116 |
6272952.811161 |
114533.884536 |
| CodonPart |
3 |
−3099273.099639 |
6200056.468462 |
41637.541838 |
||
| GenePart |
78 |
−3120195.077316 |
6243312.241766 |
84893.315142 |
||
|
CodonGenePart |
234 |
−3076219.426792 |
6158418.926624 |
0 |
||
| RY |
58,347 |
OnePart |
1 |
−1239354.453402 |
2480173.246480 |
21572.787069 |
| CodonPart |
3 |
−1235533.368070 |
2472537.854401 |
13937.394990 |
||
| GenePart |
78 |
−1234706.178899 |
2471197.311314 |
12596.851903 |
||
|
CodonGenePart |
234 |
−1228081.159986 |
2458600.459411 |
0 |
||
| ntNo3rd |
38,898 |
OnePart |
1 |
−1387913.034830 |
2777313.721117 |
30326.016847 |
| CodonPart |
2 |
−1385570.086154 |
2772645.570816 |
25657.866546 |
||
| GenePart |
78 |
−1376158.263023 |
2755293.787916 |
8306.083646 |
||
|
CodonGenePart |
156 |
−1371218.716450 |
2746987.704270 |
0 |
||
| AA | 19,449 | OnePart |
1 |
−1418038.152084 |
2837614.101717 |
8353.616354 |
| GenePart | 78 | −1413039.660496 | 2829260.485363 | 0 |
Partitioning strategies judged to be the best by the AICc are in bold.
We will focus on reporting the relationships of major clades of Viridiplantae shown in the 50% maximum likelihood (ML) majority-rule bootstrap consensus summary trees for each data set: ntAll (Figure 5), ntNo3rd (Figure 6), RY (Figure 7), and AA (Figure 8). These summary trees collapse some clades for ease of viewing the major relationships within Viridiplantae. A summary of important results and conflicts among these four data sets is given in Table 3. We provide full majority-rule bootstrap consensus trees for the ntAll (Figures 9, 10, 11, 12, 13, and 14), ntNo3rd (Additional file 4), RY (Additional file 5), and AA (Additional file 6) data sets. ML trees with branch lengths and BS values are also provided: ntAll (Additional file 7), ntNo3rd (Additional file 8), RY (Additional file 9), and AA (Additional file 10). Average support values among all internal nodes in the ML trees were slightly higher in the ntAll phylogeny (~94% bootstrap support [BS]; Additional file 7) compared to the other data sets (~90-91% BS; Additional files 8, 9, and 10). The ntAll phylogeny also had the most clades resolved with ≥ 70% BS (92%; 327 bipartitions resolved out of 357 possible) while the ntNo3rd, RY, and AA data sets had 87%, 87%, and 86% of the possible bipartitions resolved at ≥ 70% BS, respectively. All resulting trees have been deposited in the Dryad Data Repository [73].
Figure 5.
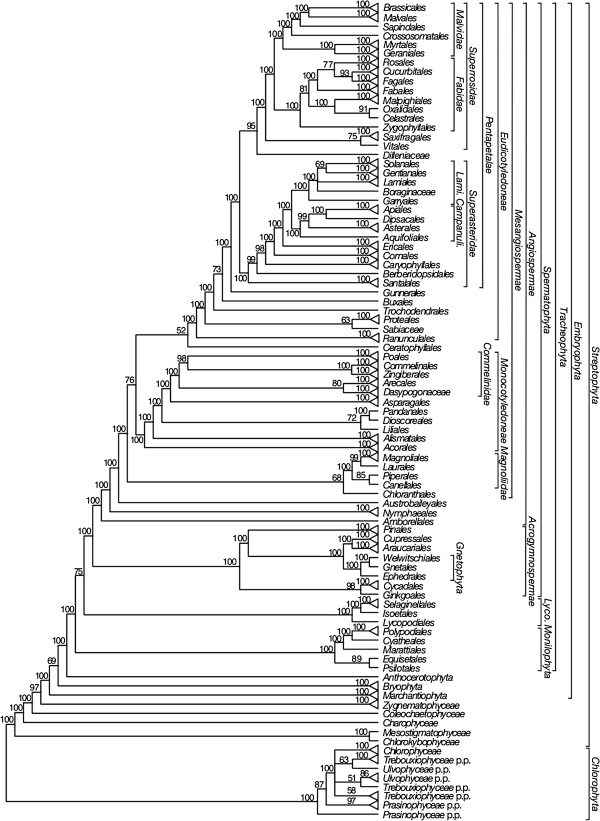
Fifty percent maximum likelihood majority-rule bootstrap consensus summary tree of Viridiplantae inferred from the all nucleotide positions (ntAll) analysis. Data set derived from 78 protein-coding genes of the plastid genome (ntax = 360; 58,347 bp; missing data ~15.6%). Bootstrap support values ≥ 50% are indicated. Terminals with a triangle represent collapsed clades with > 2 taxa. Note position of Lycopodiophyta as sister to Spermatophyta is likely caused by base composition bias (see text). See Figures 9, 10, 11, 12, 13, and 14 for the complete tree and Additional file 1 for taxonomy. Lami. = Lamiidae; Campanuli. = Campanulidae; Lyco. = Lycopodiophyta.
Figure 6.

Fifty percent maximum likelihood majority-rule bootstrap consensus summary tree of Viridiplantae inferred from the first and second codon positions (ntNo3rd) analysis. Data set derived from 78 protein-coding genes of the plastid genome (ntax = 360; 38,898 bp; missing data ~15.6%). Bootstrap support values ≥ 50% are indicated. Terminals with a triangle represent collapsed clades with > 2 taxa. See Additional file 4 for the complete tree and Additional file 1 for taxonomy. Lami. = Lamiidae; Campanuli. = Campanulidae.
Figure 7.

Fifty percent maximum likelihood majority-rule bootstrap consensus summary tree of Viridiplantae inferred from the RY-coded (RY) analysis. Data set derived from 78 protein-coding genes of the plastid genome (ntax = 360; 58,347 bp; missing data ~15.6%). Bootstrap support values ≥ 50% are indicated. Terminals with a triangle represent collapsed clades with > 2 taxa. See Additional file 5 for the complete tree and Additional file 1 for taxonomy. Lami. = Lamiidae; Campanuli. = Campanulidae.
Figure 8.

Fifty percent maximum likelihood majority-rule bootstrap consensus summary tree of Viridiplantae inferred from the amino acid (AA) analysis. Data set derived from 78 protein-coding genes of the plastid genome (ntax = 360; 19,449 AAs; missing data ~15.6%). Bootstrap support values ≥ 50% are indicated. Terminals with a triangle represent collapsed clades with > 2 taxa. See Additional file 6 for the complete tree and Additional file 1 for taxonomy. Lami. = Lamiidae; Campanuli. = Campanulidae.
Table 3.
Summary of selected similarities and conflicts between bootstrap consensus topologies derived from the four data sets
| Taxon | ntAll | ntNo3rd | RY | AA |
|---|---|---|---|---|
|
Amborellales |
sister to all other Angiospermae (100%/100%) |
sister to all other Angiospermae (100%/81% ) |
sister to all other Angiospermae (100%/100%) |
sister to all other Angiospermae (100%/70%) |
|
Anthocerotophyta |
sister to Tracheophyta (100%/100%) |
sister to Tracheophyta (94%/100%) |
sister to Tracheophyta (95%/100%) |
sister to Tracheophyta (53%/90%) |
|
Ceratophyllales |
sister to Eudicotyledoneae (52%/100%) |
sister to Monocotyledoneae + Eudicotyledoneae (52%/54%) |
sister to Eudicotyledoneae (75%/100%) |
sister to Piperales (67%) |
| COM clade |
within Fabidae (100%) |
within Fabidae (70%) |
within Fabidae (99%) |
sister to a clade including Cucurbitales, Rosales, Fabales, Fagales (69%/100%; Fabidae not monophyletic) |
|
Dilleniales |
sister to Superrosidae (95%/100%) |
sister to Superrosidae (77%/100%) |
sister to Superrosidae (57%/100%) |
sister to Superasteridae (70%/66%) |
|
Ginkgoales |
sister to Cycadales (98%/100%) |
sister to Cycadales (100%/100%) |
sister to Cycadales (51%/100%) |
sister to Cycadales (100%/100%) |
|
Gnetophyta |
sister to Cupressales + Araucariales (100%/100%) |
sister to Cupressales + Araucariales (100%/100%) |
sister to Cupressales + Araucariales (87%/100%) |
sister to Cupressales + Araucariales (100%/100%) |
|
Marchantiophyta |
sister to all other Embryophyta (100%/69%) |
sister to Bryophyta (78%/100%) |
sister to all other Embryophyta (100%/89%) |
sister to Bryophyta (99%/100%) |
|
Monilophyta |
sister to Lycopodiophyta + Spermatophyta (100%/75%) |
sister to Spermatophyta (93%/100%) |
sister to Spermatophyta (100%/100%) |
sister to Spermatophyta (89%/100%) |
|
Prasinophyceae |
not monophyletic; Nephroselmis sister to all other Chlorophyta (100%/87%) |
not monophyletic; Nephroselmis sister to all other Chlorophyta (100%/78%) |
not monophyletic; Nephroselmis sister to all other Chlorophyta (100%/92%) |
not monophyletic; Nephroselmis sister to all other Chlorophyta (100%/96%) |
| Zygnematophyceae | sister to Embryophyta (97%/100%) | sister to Embryophyta (99%/100%) | sister to Embryophyta (86%/100%) | sister to Embryophyta (93%/100%) |
Bootstrap support (BS) values >50% are shown as percentages. When sister groups for the taxon of interest are listed, bootstrap support (BS) values on the left are for the clade including the taxon of interest and its sister group within Viridiplantae, while BS values on the right are for the more inclusive clade excluding the taxon of interest. If only one BS value is given for a sister relationship, only two terminals are involved (see also Figures 5, 6, 7, and 8).
Figure 9.
Fifty percent maximum likelihood majority-rule bootstrap consensus tree of Viridiplantae inferred from the all nucleotide positions (ntAll) analysis. Portion of tree showing Chlorophyta, Chlorokybophyceae, Mesostigmatophyceae, Charophyceae, Coleochaetophyceae, Zygnematophyceae, Marchantiophyta, Bryophyta, and Anthocerotophyta. Data set derived from 78 protein-coding genes of the plastid genome (ntax = 360; 58,347 bp; missing data ~15.6%). Bootstrap support values ≥ 50% are indicated. See also Figure 5 for a summary tree of major Viridiplantae clades and Additional file 1 for taxonomy. Tree continued in Figure 10.
Figure 10.

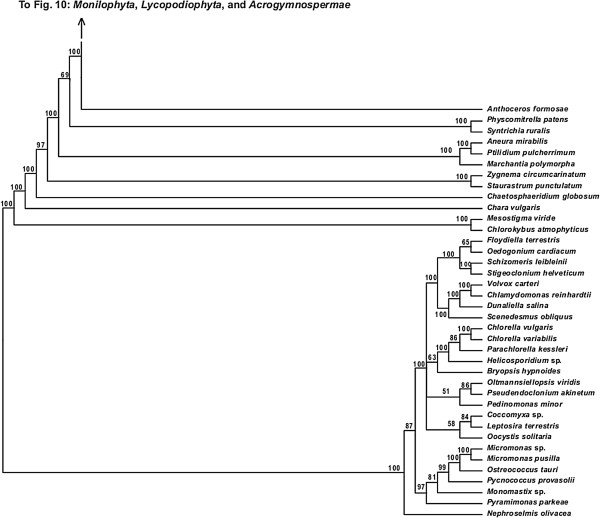
Fifty percent maximum likelihood majority-rule bootstrap consensus tree of Viridiplantae inferred from the all nucleotide positions (ntAll) analysis. Portion of tree showing Monilophyta, Lycopodiophyta, and Acrogymnospermae. Data set derived from 78 protein-coding genes of the plastid genome (ntax = 360; 58,347 bp; missing data ~15.6%). Bootstrap support values ≥ 50% are indicated. See also Figure 5 for a summary tree of major Viridiplantae clades and Additional file 1 for taxonomy. Note position of Lycopodiophyta as sister to Spermatophyta is likely caused by base composition bias (see text). Tree continued in Figures 9 and 11.
Figure 11.

Fifty percent maximum likelihood majority-rule bootstrap consensus tree of Viridiplantae inferred from the all nucleotide positions (ntAll) analysis. Portion of tree showing Amborellales, Nymphaeales, Austrobaileyales, Chloranthales, and Magnoliidae. Data set derived from 78 protein-coding genes of the plastid genome (ntax = 360; 58,347 bp; missing data ~15.6%). Bootstrap support values ≥ 50% are indicated. See also Figure 5 for a summary tree of major Viridiplantae clades and Additional file 1 for taxonomy. Tree continued in Figures 10 and 12.
Figure 12.

Fifty percent maximum likelihood majority-rule bootstrap consensus tree of Viridiplantae inferred from the all nucleotide positions (ntAll) analysis. Portion of tree showing Monocotyledoneae. Data set derived from 78 protein-coding genes of the plastid genome (ntax = 360; 58,347 bp; missing data ~15.6%). Bootstrap support values ≥ 50% are indicated. See also Figure 5 for a summary tree of major Viridiplantae clades and Additional file 1 for taxonomy. Tree continued in Figures 11 and 13.
Figure 13.
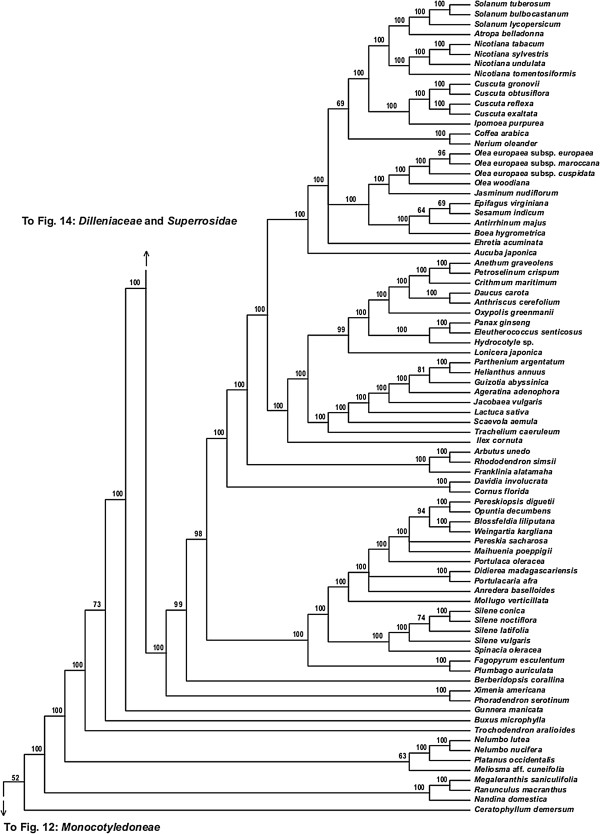
Fifty percent maximum likelihood majority-rule bootstrap consensus tree of Viridiplantae inferred from the all nucleotide positions (ntAll) analysis. Portion of tree showing Ceratophyllales, Ranunculales, Sabiaceae, Proteales, Trochodendrales, Buxales, Gunnerales, and Superasteridae. Data set derived from 78 protein-coding genes of the plastid genome (ntax = 360; 58,347 bp; missing data ~15.6%). Bootstrap support values ≥ 50% are indicated. See also Figure 5 for a summary tree of major Viridiplantae clades and Additional file 1 for taxonomy. Tree continued in Figures 12 and 14.
Figure 14.
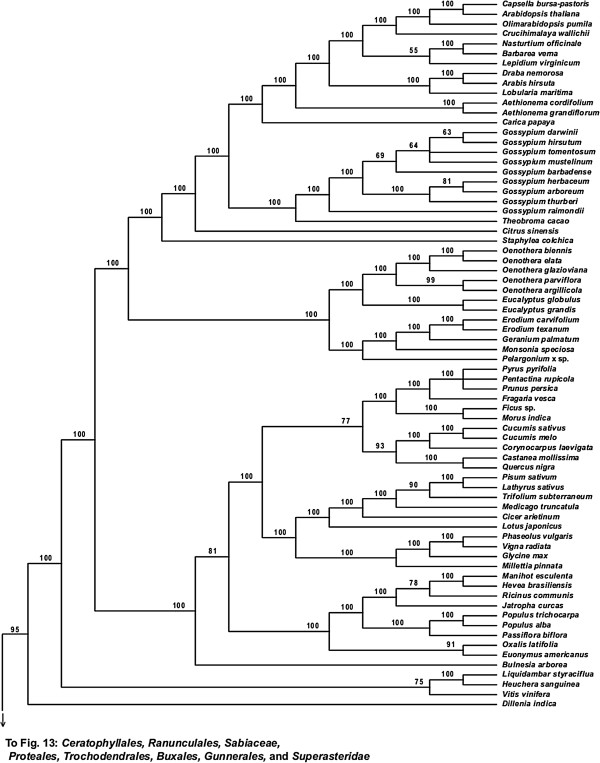
Fifty percent maximum likelihood majority-rule bootstrap consensus tree of Viridiplantae inferred from the all nucleotide positions (ntAll) analysis. Portion of tree showing Dilleniaceae and Superrosidae. Data set derived from 78 protein-coding genes of the plastid genome (ntax = 360; 58,347 bp; missing data ~15.6%). Bootstrap support values ≥ 50% are indicated. See also Figure 5 for a summary tree of major Viridiplantae clades and Additional file 1 for taxonomy. Tree continued in Figure 13.
The monophyly of Chlorophyta receives 100% BS in all analyses. Prasinophyceae are consistently not monophyletic. Instead, the prasinophyte Nephroselmis is sister to all other Chlorophyta (Figure 9; Additional files 4, 5, and 6), while remaining Prasinophyceae form a clade that is variously supported (ntAll 97% BS, ntNo3rd 78% BS, RY 93% BS, and AA 68% BS) and is sister to a clade of the remaining Chlorophyta. Chlorophyceae are monophyletic (100% BS in all analyses), but Trebouxiophyceae and Ulvophyceae are not monophyletic, and the relationship of Chlorophyceae to these lineages is unresolved.
We consistently recovered a single set of relationships among the streptophytic algae subtending the land plant clade. Zygnematophyceae are sister to land plants, Coleochaetophyceae are sister to Zygnematophyceae + Embryophyta, Charophyceae are sister to Coleochaetophyceae + (Zygnematophyceae + Embryophyta), and a clade of Mesostigmatophyceae + Chlorokybophyceae is sister to all other Streptophyta. Each of these relationships has ≥86% BS support (Figures 5, 6, 7, and 8).
The branching order of the non-vascular land plant lineages differs among analyses. In analyses of the ntAll and RY data sets, Marchantiophyta (liverworts), followed by Bryophyta (mosses), and then Anthocerotophyta (hornworts) are the earliest-branching land plant lineages, with Anthocerotophyta the immediate sister to the vascular plants (Tracheophyta; Figures 5 and 7). In the ntAll and RY analyses, these relationships had ≥89% BS support except for the Bryophyta + (Anthocerophyta + Tracheophyta) relationship in the ntAll analysis, which received only 69% BS (Figure 5). In contrast, in the ntNo3rd and AA analyses, Bryophyta and Marchantiophyta formed a clade (78% BS [Figure 6] and 99% BS [Figure 8], respectively), followed by Anthocerophyta as sister to Tracheophyta (94% [Figure 6] and 53% BS [Figure 8], respectively).
Within Tracheophyta, the ntNo3rd, RY, and AA data sets all place Lycopodiophyta sister to a Euphyllophyta clade (Monilophyta + Spermatophyta; ≥89% BS, Figures 6, 7, and 8). However, the analysis of the ntAll data set places Monilophyta sister to a clade of Lycopodiophyta + Spermatophyta (75% BS, Figures 5, 6, 7, 8, 9, and 10).
Our analyses of Monilophyta generally reveal strong support for a clade of Equisetales + Psilotales as sister to Marattiales + leptosporangiate ferns (represented by Cyatheales and Polypodiales). The lowest support obtained was for Equisetales + Psilotales in the ntNo3rd analysis (84% BS; Figure 6) and ntAll (89% BS; Figure 5); all other nodes in all analyses received > 90% BS, with Marattiales + leptosporangiate ferns receiving ≥ 99% BS.
Within Spermatophyta, all analyses place the extant gymnosperms (Acrogymnospermae) sister to Angiospermae with 100% BS. Within extant gymnosperms, Cycadales and Ginkgoales form a clade (≥ 98% BS in ntAll, ntNo3rd, and AA; 51% BS in RY) that is sister to a clade in which Gnetophyta (100% BS in all analyses) are nested within the paraphyletic conifers. There is generally high support (100% BS in ntAll [Figure 5], ntNo3rd [Figure 6], and AA [Figure 7]; 87% BS [Figure 8] in RY) placing Gnetophyta as sister to a clade of Araucariales + Cupressales. This “Gnecup” clade [sensu 16, 30, 41] is then sister to Pinales, which has 100% BS in all analyses.
In all analyses, Angiospermae receive 100% BS, and Amborella (Amborellales) is sister to all other angiosperms, followed by Nymphaeales, and then Austrobaileyales. These relationships are mostly supported by 100% BS. However, Nymphaeales + (Austrobaileyales + Mesangiospermae) receives 81% BS (Figure 6) in the ntNo3rd analyses and 70% BS (Figure 8) in the AA analyses. The remaining angiosperms (Mesangiospermae) receive 100% BS in all analyses. Within Mesangiospermae, the relationships among Monocotyledoneae, Magnoliidae, Eudicotyledoneae, and Ceratophyllum (Ceratophyllales) are not well supported and vary depending on the analysis. The strongest support for the placement of Ceratophyllales is 75% BS as sister to Eudicotyledoneae in the RY analysis (Figure 7).
Chloranthales receive 61-69% BS as sister to the well-supported (100% BS in ntAll, RY; 83% BS in ntNo3rd) Magnoliidae. However, Magnoliidae are not monophyletic in the AA analyses, where Piperales are sister to Ceratophyllales (67% BS; Figure 8).
Within the monocot clade (Monocotyledoneae), Acorales, followed by Alismatales, have 100% BS in all analyses as subsequent sisters to the remaining monocots. In three of our analyses (ntAll, ntNo3rd, and AA), a variously supported clade (72%, 69%, and 80% BS, respectively) of Liliales + (Pandanales + Dioscoreales) is sister to a clade (>95% BS in these three analyses) of the remaining monocots (Asparagales + Commelinidae). However, in the RY-coded analysis, Pandanales + Dioscoreales (100% BS) is sister to a clade of Liliales + (Asparagales + Commelinidae), which receives 69% BS (Figure 7). Here Asparagales + Commelinidae is supported by 80% BS.
Within the eudicots (Eudicotyledoneae), which receive 100% BS in all analyses, Ranunculales are sister to the remaining taxa. In the ntAll, ntNo3rd, RY, and AA analyses, the clade of these remaining taxa receives 100%, 85%, 100%, and 62% BS, respectively. Relationships vary among Sabiaceae, Proteales, and a clade of the remaining taxa, depending on the analysis. In the ntAll and ntNo3rd analyses, Proteales + Sabiaceae are supported as a clade, although with only 63% and 60% BS, respectively. However, in the RY analysis, Proteales are sister to a clade containing Sabiaceae plus the remaining taxa, which has 79% BS. In the AA analysis, relationships among these three clades are unresolved.
Among the remaining eudicots, we consistently recovered Trochodendrales as sister to Buxales + Pentapetalae and Gunnerales as sister to the remaining lineages of Pentapetalae: Dilleniaceae, Superrosidae, and Superasteridae. The placement of Dilleniaceae remains uncertain. The family is sister to Superrosidae in the ntAll (95% BS), ntNo3rd (77% BS), and RY (57% BS) analyses, but appears as sister to Superasteridae (70% BS) in the AA analysis.
Within Superrosidae, a clade of Vitales + Saxifragales is supported in the ntAll (75% BS), ntNo3rd (70% BS), and AA (78% BS) analyses. In the RY analysis, the relationship among Saxifragales, Vitales, and remaining Rosidae (Fabidae + Malvidae) is unresolved. Fabidae and Malvidae are both recovered with ≥ 99% BS in the ntAll and RY analyses. However, each clade receives only 70% BS in the ntNo3rd analysis. In the AA analysis neither clade is monophyletic; Zygophyllales are embedded (68% BS) within a clade of Malvidae taxa. The COM clade (Celastrales, Oxalidales, Malpighiales) is sister to a clade of Fagales, Cucurbitales, Rosales, and Fabales in Fabidae in the AA (69% BS; Figure 8), RY (82% BS; Figure 7), and ntAll (81% BS; Figure 5) trees and forms a trichotomy with Zygophyllales and the clade of Fagales, Cucurbitales, Rosales, and Fabales in the ntNo3rd tree (70% BS; Figure 6). Zygophyllales are sister to Geraniales (69% BS; Figure 8) in the AA tree and sister to all other Fabidae in the ntAll and RY trees (with 100% [Figure 5] and 99% BS [Figure 7], respectively).
Superasteridae (Santalales, Berberidopsidales, Caryophyllales, and Asteridae) are recovered in all analyses. This clade receives 100% BS in the ntAll and RY analyses, 95% BS in the ntNo3rd analysis, and 66% BS in the AA analysis. Santalales and Berberidopsidales are strongly supported as subsequent sisters to Caryophyllales + Asteridae. Within Asteridae, Cornales, followed by Ericales, are subsequent sisters to a strongly supported clade that comprises strongly supported Campanulidae and Lamiidae clades. Within Lamiidae, the placement of Boraginaceae is weak among the various analyses. Boraginaceae are sister to Gentianales (59% BS; Figure 8) in the AA tree, part of a trichotomy (100% BS; Figure 5) with Lamiales and Solanales + Gentianales in the ntAll tree, and sister to a weakly supported clade including Gentianales, Lamiales, and Solanales in the ntNo3rd (Figure 6) and RY (Figure 7) trees.
Analysis of only the third codon positions (nt3rdOnly, Additional file 11) resulted in several very strong conflicts along the backbone of Viridiplantae when compared to the topology from the ntNo3rd analyses. These conflicts include the backbone relationships within Chlorophyta, the placements of Cycadales and Lycopodiophyta, the relationships of the three major bryophyte lineages, and backbone relationships within Poales. Removal of four taxa (Epifagus, Helicosporidium, Neottia, and Rhizanthella) with elevated rates of molecular evolution and few genes present in the data sets did not significantly affect the resulting topologies.
Discussion
While the enormous phylogenetic data sets that result from new genome or transcriptome sequencing efforts can ameliorate the effects of random or stochastic error, they also may exacerbate the effects of systematic error, or error resulting from problems in the analysis, such as model inaccuracy. The high amount of agreement among our various analyses and strong support for results generally consistent with previous studies (many of which also used plastid genes) suggest that plastid genome sequence data hold much promise for resolving relationships throughout the green plants. However, several areas of conflict between analyses using different character-coding strategies demonstrate that plastid genome phylogenetics is also susceptible to systematic error. Here we evaluate the phylogenetic results, emphasizing areas of agreement and concern, and then address some of the methodological issues raised by our results.
Evaluation of phylogenetic relationships
Historically, Chlorophyta have been divided into Prasinophyceae, Trebouxiophyceae, Chlorophyceae, and Ulvophyceae based on the ultrastructure of the flagellar apparatus and features related to cytokinesis [74,75]. The current status of green algae phylogenetics (Chlorophyta and streptophytic algae) has been reviewed recently [26,76,77]. The most comparable study to ours in terms of data and taxon sampling is by Lang and Nedelcu [26], who constructed a phylogeny of green algae with plastid genome sequence data. However, they analyzed only an amino acid data set using Bayesian inference and the CAT model [78,79]. We found a paraphyletic Prasinophyceae (not including Pedinomnas; Figures 5, 6, 7 and 8), which agrees with previous molecular analyses [26,76,77]. However, Lang and Nedelcu [26] recovered a monophyletic Prasinophyceae, albeit with little support. Chlorophyceae are monophyletic (100% BS in all of our analyses), which agrees with the results of Lang and Nedelcu [26]. We also find that Trebouxiophyceae and Ulvophyceae are not monophyletic, and that the relationship of Chlorophyceae to these lineages is unresolved. The branching order of the various Trebouxiophyceae, Ulvophyceae, and Chlorophyceae lineages within Chlorophyta, unresolved in our analyses, was also uncertain in earlier analyses (reviewed in [26,76,77]). Similarly in Lang and Nedelcu [26], Trebouxiophyceae and Ulvophyceae were not supported as monophyletic, although unlike our results, almost all nodes in their phylogeny were maximally supported.
Our analyses provide consistent, strong support for the relationships of streptophytic algae to land plants, and all analyses support Zygnematophyceae as the sister to land plants (Figures 5, 6, 7, and 8). Relationships among these lineages and the closest relatives of land plants have varied in previous studies depending on taxon sampling and gene choice. Some studies agree with our results placing Zygnematophyceae as sister to land plants [25,27,80-82], while other phylogenetic analyses indicate that Charophyceae[23,83,84] or Coleochaetophyceae[26,40,85,86] occupy this position. Depending on the analysis, Zhong et al. [87] found either Zygnematophyceae alone or a clade of Zygnematophyceae + Coleochaetophyceae as sister to land plants. In particular, the results of Lang and Nedelcu [26] conflict with our results regarding the sister group to Embryophyta. While we find a clade of Coleochaetophyceae + (Zygnematophyceae + Embryophyta), their results strongly support Zygnematophyceae + (Coleochaetophyceae + Embryophyta).
Phylogenetic relationships among bryophytes (mosses, hornworts, and liverworts) are also contentious, and nearly every possible relationship among these lineages has been reported, often with strong support. Most studies have shown the bryophytes as paraphyletic with respect to Tracheophyta rather than as a clade [30-33]. As recovered in our ntAll and RY analyses (Figures 5 and 7), liverworts (Marchantiophyta) often are placed sister to all other land plants, followed by mosses (Bryophyta), and with hornworts (Anthocerotophyta) sister to Tracheophyta[29,34,47,50,88,89]. A sister relationship between mosses and liverworts, found in our ntNo3rd and AA analyses (Figures 6 and 8), was proposed previously based on morphological [90-93] and molecular data [27,30,94,95] and has been recovered with numerous nuclear genes (Wickett et al., in review). This relationship was also recovered in analyses of complete plastid genome data by Karol et al. [34] when divergent taxa (i.e., Selaginella spp.) were excluded from phylogenetic analyses and also by Wolf and Karol [35] when third positions were excluded.
Our results placing Lycopodiophyta sister to Euphyllophyta in all but the ntAll analysis agree with most molecular phylogenetic analyses [29,96,97]. This split is also supported by analyses of morphological characters in fossil [15] and extant taxa [98]. Monilophyta and Spermatophyta also possess a 30-kb inversion in the large single-copy region of the plastid genome not found in Lycopodiophyta and the three bryophyte clades [99]. In the ntAll analysis, Euphyllophyta are not monophyletic (Figure 5); Lycopodiophyta, rather than Monilophyta, are sister to Spermatophyta. This relationship has been reported previously [34]; however, it likely is a phylogenetic artifact, perhaps related to base composition bias (see below). The plastid genome of the lycophyte Selaginella has an especially high GC content [21], with Selaginella unicata having the highest GC content in our ntAll data set (54.3%; Figure 1).
In some previous studies, relationships among lineages of Monilophyta have not been well resolved or supported (e.g., [29,89,96-98]). As a result, the relationships among Equisetales, Psilotales, Marattiales, and leptosporangiate ferns are often represented as a polytomy (e.g., [35]). In contrast, most of our analyses recovered strong support for a clade of Equisetales + Psilotales as sister to Marattiales + leptosporangiate ferns (represented here by Cyatheales and Polypodiales). These relationships agree with recent studies of monilophyte relationships based on plastid genome sequence data [34,35], although support is stronger here. Unfortunately, Ophioglossales, which often appear as sister to Psilotales, lacked a sequenced plastome at the time of our analyses. However, plastid genome data for Ophioglossales have subsequently been published and analyzed in a phylogenetic context [100], with strong support for Ophioglossales as sister to Psilotales and weak support for this clade as sister to Equisetales. Results from that study with regard to Marattiales and leptosporangiate ferns agree with the relationships presented here.
Relationships among the lineages of extant seed plants, and especially the placement of Gnetophyta, have long been debated [38,39,43,51,89,101]. Gnecup trees, found in all of our analyses, were initially recovered by Nickrent et al. [30], and then more recently by Zhong et al. [41]. However, Zhong et al. [41] suggested that the support for Gnecup may be the result of long-branch attraction; by removing highly variable proteins, support for Gnecup decreased. Furthermore, by removing what they considered parallel substitutions between lineages leading to Gnetophyta and to Cryptomeria (the sole Cupressales in their analyses), a Gnepine topology was recovered. Although several different placements for Gnetophyta have been recovered and strongly supported, many studies involving multiple genes have placed Gnetophyta sister to Pinales (Gnepine; [38,39,43,89], Wickett et al., in review). Using both coalescent and concatenation analyses, Xi et al. [102] found that the phylogenetic placement of Gnetophyta differs between the nuclear and plastid genomes. In their analyses using nuclear data, the Gnepine hypothesis is supported, while their analyses of plastid data support the Gnecup hypothesis. In contrast, Lee et al. [46] found strong support for Gnetophyta sister to the remaining gymnosperms [(Cycadales + Ginkgoales) + conifers)] in an ML analysis of 22,833 sets of nuclear gene orthologs from 101 land plant genera.
The backbone relationships among angiosperm (Angio-spermae) lineages generally agree with results from recent analyses, including a 17-gene analysis of 632 angiosperms [103] and previous analyses of plastid genome data sets [63,104-106]. The position of Ceratophyllum (Ceratophyllales), and thus the relationships among Monocotyledoneae, Eudicotyledoneae, and Magnoliidae, varies among our analyses, although without strong support. This contrasts with several other large, multi-gene analyses in which Monocotyledoneae are sister to Ceratophyllales + Eudicotyledoneae[63,103,106]. Interestingly, the strongest support for the placement of Ceratophyllales sister to Eudicotyledoneae is in the RY analysis (75% BS; Figure 7). However, in that analysis, the relationships among Ceratophyllales + Eudicotyledoneae, Monocotyledoneae, and Magnoliidae are unresolved.
Within the angiosperms, some relationships that have been uncertain, particularly at deep levels (reviewed in [103,107]), receive moderate to strong support in at least some of our analyses. For example, the placement of Myrtales and Geraniales in the Malvidae is supported with 70% BS (Figure 6) in the ntNo3rd tree and ≥ 99% BS in the RY (Figure 7) and ntAll (Figure 5) trees. Myrtales and Geraniales are also placed in a clade with the Malvidae taxa in the AA analysis (68% BS; Figure 8); however, Zygophyllales are also included within this clade, making Malvidae non-monophyletic. Likewise, Chloranthales are sister to Magnoliidae in all trees, but with weaker support (61% BS for RY and ntNo3rd, 68% BS for ntAll, and 69% BS for AA, but with Piperales removed from Magnoliidae in the latter). In two cases, all analyses but RY resolve relationships (although often with only moderate support), with RY producing a polytomy that does not conflict with the resolutions found in the other analyses. These two cases are as follows: (1) Vitales + Saxifragales supported by ≥ 70% BS in all analyses but RY, with Saxifragales, Vitales, and remaining Rosidae forming a polytomy in the RY tree (Figure 7); (2) Dasypogonaceae + Arecales in all but RY (52%, 78%, and 80% BS in the ntNo3rd, AA, and ntAll trees, respectively) and a trichotomy of Dasypogonaceae, Arecales, and Poales + (Zingiberales + Commelinales) in the RY tree (Figure 7). In two additional cases when RY is compared to the other three analyses, the RY analysis produced either stronger support for the placement of a taxon or a different placement altogether. First, in the ntAll, ntNo3rd, and AA analyses, the position of Sabiaceace among the early-diverging lineages of Eudicotyledoneae is weakly supported. However, in the RY analysis, Sabiaceae receive moderate support (79% BS; Figure 7) as sister to a strongly supported (100% BS; Figure 7) clade of Trochodendrales + (Buxales (Gunnerales + Pentapetalae)). This contrasts with previous studies that often place Sabiaceae as sister to Proteales[103]. An example of a different placement of a taxon in the RY analysis when compared to the other analyses involves Liliales. The ntAll, ntNo3rd, and the AA analyses support Liliales as sister to a clade of Dioscoreales + Pandanales with 72%, 69%, and 80% BS, respectively. This placement of Liliales was also recovered in Barrett et al. [108]. In contrast, in the RY analysis, Liliales are placed in a clade with Asparagales + Commelinidae with moderate support (69% BS; Figure 7). This latter placement of Liliales was strongly supported in an analysis with much better taxon sampling [103].
Some taxa that have been problematic in previous studies (e.g., Boraginaceae, Ceratophyllales, the COM clade, Dilleniaceae, and Zygophyllaceae) continue to defy definitive placement. Their positions vary among our analyses, although they are generally not well supported in some, or all, of the trees. Despite its general placement of the COM clade in Fabidae in these and other plastid analyses, this clade is more closely related to Malvidae in some analyses, particularly those using mitochondrial gene sequences (reviewed in [103]). Recent analyses of plastid, mitochondrial, and nuclear data suggest that the COM clade may represent ancient reticulation involving Fabidae and Malvidae during the rapid radiation of Rosidae (Sun et al., in prep.).
Methodological issues of plastid phylogenomic analyses
To address potential systematic error in large-scale phylogenetic analyses, scientists often either try to improve the fit of models to the data or change or remove problematic data. With increasing sequence length and number of genes, it is more likely that a sequence alignment will contain regions with heterogeneous processes of molecular evolution. We see evidence of this high heterogeneity with our model-fitting experiments, which always favor the most parameter-rich models (Table 2). Thus, defining partitioning schemes and models that can accurately reflect the true processes of molecular evolution while not over-parameterizing the analysis remains critically important for phylogenetic analyses of large plastid data sets. Although we assessed models that account for heterogeneity in patterns of molecular evolution among genes and in some cases codon positions, our model selection tests only evaluated a small selection of possible models and partitioning schemes. It is possible that other partitioning schemes could enable simpler models.
Most conventional phylogenetic models, like those used in our analyses, also assume homogeneous processes of evolution throughout the tree. Yet when the branches of the phylogeny encompass over one billion years of evolutionary history, as likely do those in the green plants, the patterns of evolution almost certainly differ among lineages and through time. This is apparent from the often good fit of covarion models (which may better describe rate shifts through time) to plastid genes [109,110] and the presence of nucleotide compositional heterogeneity, which can confound conventional phylogenetic analyses (e.g., [111,112]). Also, our models do not account for shifts in selective pressure or instances of positive selection that will affect nucleotide and amino acid substitution patterns (e.g., [113,114]).
Nucleotide compositional heterogeneity remains a concern for green plant plastid genome analyses. This variation is most evident in non-seed plant taxa (Figure 2), and thus it has not been a focus of many previous phylogenetic analyses of plastid genome sequences. A GC bias in itself is not necessarily problematic for phylogenetic analyses, but nearly all commonly used models for likelihood-based phylogenetic analyses assume single equilibrium nucleotide frequencies. Given that GC content appears to vary by codon position in plants (Figures 1 and 2) [115-117], a partitioning scheme that estimates separate nucleotide frequencies for each codon position may account for some of the spatial heterogeneity in GC content in the plastid genome, but it does not address the differences in GC frequency among lineages.
A commonly used strategy to reduce the effects of GC heterogeneity across lineages is RY-coding, in which the purines (A and G) are coded as Rs and the pyrimidines (C and T) are coded as Ys [118]. RY-coding can reduce the compositional variability among lineages, improve the fit of models, and increase the signal for internal branches [118-121]. An obvious disadvantage to RY-coding is that by coding the sequences with two character states instead of four, it reduces the amount of information in the sequences. In general, we see little overall reduction, and even some gains, in bootstrap support when using RY-coding compared to the use of all nucleotide data (ntAll), suggesting that the benefits of RY-coding make up for any potential costs of information loss. Perhaps the biggest topological difference in the RY phylogeny (Figure 7) compared to ntAll (Figure 5) is the placement of Monilophyta rather than Lycopodiophyta as sister to seed plants. The unexpected placement of Lycopodiophyta as the sister to seed plants in the ntAll analysis (Figure 5) is almost certainly an artifact of systematic error; several other lines of evidence support Monilophyta as the sister group of seed plants (see above).
Approaches to reducing systematic errors by excluding problematic data, which often include fast-evolving or saturated sites, also have been suggested for plastid genome analyses [20,41,80,110,122]. With the proper model of molecular evolution and adequate taxon sampling, fast sites are not necessarily problematic; they are only problematic insofar as they are difficult to model. Yet with heterogeneous processes of molecular evolution throughout the tree, the fast-evolving or saturated sites can produce a significant non-phylogenetic signal (e.g., [123]). Indeed, the third codon positions appear to have especially high levels of compositional heterogeneity, potentially causing systematic error (Figures 1 and 2), and an analysis of just the third codon positions (nt3rdOnly) conflicts with the analyses of other data sets in several critical parts of the tree (Additional file 11). However, third codon positions also represent a large proportion of the variable sites in the alignment, and removing them may exclude much of the phylogenetic information in some parts of the tree. With regard to backbone relationships in our phylogeny, excluding the third position sites (ntNo3rd) produces several interesting changes in contrast to ntAll: 1) it supports the sister relationship of mosses and liverworts, 2) monilophytes, not lycophytes, are placed sister to seed plants as expected, and 3) support for some of the backbone angiosperm relationships is reduced. Thus, the effects of removing the third codon position sites appear to vary in different parts of the tree.
Another strategy for overcoming potential error associated with fast-evolving sites is to code the sequences as amino acids rather than nucleotides. This does not necessarily eliminate problems of compositional heterogeneity, as the GC bias also may bias amino acid composition (Figures 3 and 4) [124]. Regarding backbone green plant relationships, the AA analysis provided similar results to analyses of only first and second codon positions. AA analysis also produced some weakly supported, questionable relationships among angiosperm lineages (i.e., Piperales + Ceratophyllales; Figure 8). In previous deep-level plant analyses, analyses of amino acid data have resulted in arguably more problematic or questionable relationships than analyses of nucleotide data [29,80]. However, these results are likely due to inappropriate models of amino acid evolution [125], and with better models, optimized for plastid evolution, amino acid data may be a valuable source of phylogenetic information.
Taxon sampling is also important for plastid phylogenomic studies, especially when the model of evolution is inadequate [56,58,126-131], and genome-scale analyses often have limited taxon sampling. New methods for rapid and inexpensive plastid genome sequencing (e.g., [132]) may ameliorate the effects of insufficient sampling of extant taxa; however, many major lineages of green plants are now extinct, precluding their inclusion in analyses of molecular data (but see [133-136]). In addition, ancient, rapid radiations abound within portions of the green plant tree of life, creating extremely difficult phylogenetic problems no matter the taxon sampling [63,69,107,137].
Furthermore, even in the absence of systematic error, it is possible that a tree built from plastid genome data will not reflect species relationships. The plastid genome represents a single locus of linked genes (i.e., a single coalescent history). For phylogenetic analyses, this can be beneficial because combining genes with different evolutionary histories into a single character matrix can lead to phylogenetic error [138-140]. However, incomplete lineage sorting or ancient reticulation could lead to conflict between the plastid gene tree and the species phylogeny [141]. For this reason, it will be interesting to compare phylogenetic hypotheses from the plastid genome with independent phylogenetic estimates from numerous nuclear and mitochondrial loci.
Finally, while full plastid genome sequence data provide much power for resolving difficult phylogenetic relationships, it is not clear that they can resolve all plant relationships. Theoretical work suggests that extremely large data sets may be necessary to resolve some relationships when the internal nodes are separated by very short branches [142], and recent analyses indicate that full plastid genomes are not sufficient to reject alternative topologies among monocots [108]. Indeed, the unresolved or conflicting parts of the green plant phylogeny in our analyses are generally associated with short internal branch lengths (see Additional files 7, 8, 9, 10, and 11). Thus, even if the model of evolution accurately reflects the true process of molecular evolution, and there is no systematic error, plastid genome data alone may not be sufficient to resolve all parts of the green plant tree of life. That is, the topology may not be identifiable with the plastid data alone. A recent analysis using a new diagnostic test for phylogenetic identifiability based on data cloning suggested that a backbone topology of angiosperms was identifiable from plastid sequence data using the GTR + Γ model [143], but the tree in this paper is much larger and the models more complex. In any case, it will be necessary to include perspectives from the nuclear genome and phenotypic data before we are confident about all deep-level relationships among green plants.
Conclusions
Our diverse analyses provide a first approach to addressing some of the difficult issues associated with plastid phylogenetic analyses at this evolutionary depth and level of taxon sampling. The results of the analyses using different models, character-coding strategies, and character subsets suggest that much of the tree is robust to many different phylogenetic approaches, and they highlight regions of the tree that need more scrutiny (i.e., those relationships not consistent across analyses). More sophisticated modelling approaches may more accurately characterize the heterogeneous processes of molecular evolution, but it is also crucial that the parameters of these complex models can be estimated by the data at hand [143]. While it may be impossible for any model to reflect perfectly the complexities of molecular evolution, as we better characterize these processes it will be possible to examine through simulations their possible effects on phylogenetic analyses and to recognize phylogenetic error caused by model misspecification.
Methods
Taxon and sequence sampling
Protein-coding data, including nucleotides and their corresponding amino acid sequences, for all Viridiplantae taxa that had complete or nearly complete plastid genome sequences were downloaded from GenBank on February 28, 2012. If there were multiple genome sequences from the same taxon, we included the sequence with the most data. Our sampling included most major lineages of Viridiplantae. A complete list of taxa and GenBank accession numbers is available in Additional file 1.
Taxonomic names (Additional file 1) follow various references. Four classes of chlorophytic algae (Chlorophyta) are recognized following a traditional classification [26,76]. Classes of streptophytic algae and orders for both chlorophytic and streptophytic algae follow Leliaert et al. [76]. Names for the three main bryophyte clades follow recent classifications: mosses (Bryophyta[144]), hornworts (Anthocerotophyta[145]), and liverworts (Marchantiophyta[146]). Major clades of tracheophytes follow Cantino et al. [147] and Soltis et al. [103]. Familial and ordinal names within major clades of land plants follow these references: Bryophyta[144]; Anthocerotophyta[145]; Marchantiophyta[146]; lycophytes (Lycopodiophyta) and ferns (Monilophyta) [148]; gymnosperms (Acrogymnospermae[149]); and angiosperms (Angiospermae[150]). All scientific names are italicized to distinguish common names from scientific names [147,151].
Building the phylogenetic character matrix
To build the phylogenetic matrix, first we used a clustering approach to identify homologous gene sequences. Amino acid sequences from all downloaded genomes were compared to each other using BLASTP v.2.2.26 [152]. Significant BLAST hits were defined as those having a maximum e-value of 1.0e-5 and having the hit region cover at least 40% of the target and query sequences. Based on the BLAST hits, we formed clusters of putative homologs using single-linkage clustering. This approach identified groups of sequences that had a significant BLAST hit with at least one other sequence in the cluster and were connected to each other by a path of significant BLAST hits. The resulting clusters were modified in two ways. First, clusters that contained two or more different genes from a single taxon were re-clustered at a more stringent e-value to separate the genes. Second, when it appeared that a single gene was split into multiple clusters, we combined them. Some clusters contained multiple sequences from the same species when the gene was present in the inverted repeat region in the plastid genome. If the sequences were identical, only one was retained for analysis. In cases where the two sequences differed slightly, we removed both sequences. Only clusters containing sequences from at least 50% of the 360 taxa were retained for the phylogenetic analyses.
Each remaining amino acid cluster (78 total) was aligned with MAFFT v. 6.859 [153] using the L-INS-i algorithm, and subsequently, poorly aligned regions were removed using trimAl v.1.2rev59 [154]. After using trimAl, we also visually inspected the trimmed alignments and removed poorly aligned regions. The nucleotide sequences for each cluster were aligned with PAL2NAL v.14 [155] to correspond to the trimmed amino acid alignment and ensure that the correct reading frame was maintained. We checked for anomalous sequences by building ML trees from each of the aligned clusters with RAxML [156,157] following the search strategies outlined below. These topologies were visually examined, and sequences in obviously spurious locations in the tree were removed. If any sequences were removed from a cluster alignment, we realigned and edited the cluster’s untrimmed data as described above. Alignments for each gene were concatenated using FASconCAT v.1.0 [158].
From this data set, we generated an amino acid (AA) alignment, two nucleotide alignments, and a binary character alignment. The first nucleotide alignment contained all nucleotide positions (ntAll), while the second contained only the first and second codon positions (ntNo3rd). The binary character alignment was an RY-coded version (RY) of the ntAll data set. RY-coding [159] involves recoding the nucleotides as binary characters, either purines (A or G = R) or pyrimidines (C or T = Y). RY-coding has been used to ameliorate biases caused by saturation, rate heterogeneity, and base composition [119,160,161]. To determine if the data sets were decisive using our selected partitioning schemes (see below), we followed the approach used in Sanderson et al. [72].
We assessed base composition bias in the nucleotide data set (ntAll) by conducting a chi-square test using PAUP* v.4.0b10 [162] to determine if the base frequencies across taxa were homogeneous. To determine if base composition of the nucleotide sequences in the ntAll matrix could affect the composition of amino acid sequences in the AA matrix, we conducted linear regressions in R [163]. We examined the relationship of percent GC content to the percent of amino acids that are coded for by GC-rich codons (i.e., G, A, R, and P) as well as the relationship of percent GC content to the percent of amino acids that are coded for by AT-rich codons (i.e., F, Y, M, I, N, and K).
Phylogenetic analyses
All ML phylogenetic analyses were implemented with RAxML v. 7.3.0 [156,157]. The optimal partitioning scheme for each alignment was chosen from among several commonly used partitioning strategies using the corrected Akaike information criterion (AICc) [164,165]. This penalizes models for additional parameters and should account for the trade-off between increased model fit and over-parameterization when choosing the best model. For the nucleotide (ntAll and ntNo3rd) and RY-coded data, we examined four possible partitioning strategies: 1) no partitioning, 2) partitioning by each codon position (three partitions), 3) partitioning by gene (78 partitions), and 4) partitioning by each codon position within each gene (234 partitions). For the AA data, we tested two partitioning strategies: 1) no partitioning, and 2) partitioning by gene (78 partitions). A novel approach for determining partitions of phylogenomic data sets a posteriori using a Bayesian mixture model has recently been proposed [69]. Additionally, the program PartitionFinder [166] allows for the statistical comparison of multiple a priori partitioning schemes. We explored both of these methods, but we were unable to complete the analyses due to computational limitations resulting from the large size of our data set.
To determine which partitioning scheme was optimal for each data set, we first obtained the optimal ML tree for each data set under each partitioning scheme as follows. For the nucleotide (ntAll, ntNo3rd) and RY-coded data, we ran 10 ML searches from different starting trees. We used the GTR+Γ model of evolution for each partition in the nucleotide data set and the binary model of evolution (BINGAMMA) for the RY data set. For the AA data, we ran 3 ML searches from different starting trees. To select the best amino acid substitution model for each partition of the AA data set, we used the Perl script (ProteinModelSelection.pl) included in the RAxML distribution package. For each ML search, we estimated a separate substitution rate matrix for each partition but a single set of branch length parameters for all partitions. We then optimized the model and branch lengths on each resulting ML tree using RAxML (-f e). AICc values for each partitioning scheme were then calculated by using the log-likelihood, number of estimable parameters, and sample size given by RAxML. The optimal partitioning strategy for each data set was then used in subsequent ML bootstrap analyses. Bootstrap searches (200 replicates for each matrix) were executed separately from the search for the best ML tree using the standard bootstrap option in RAxML. To determine if 200 replicates were adequate for estimating bootstrap values, we conducted a posteriori bootstopping analyses (-I autoMRE) as implemented in RAxML and described in Pattengale et al. [167]. All trees were rooted at the branch between Chlorophyta and Streptophyta[23,24].
To further explore our data, we conducted the following phylogenetic analyses using the methods described above unless otherwise noted. To determine if there is conflict between the phylogenetic signal in the ntNo3rd data set and the data set containing only third positions (nt3rdOnly), we analyzed the nt3rdOnly data partitioned by gene region. We also conducted phylogenetic analyses on each of the four main data sets (ntAll, ntNo3rd, RY, and AA) with four taxa removed: Neottia nidus-avis and Rhizanthella gardneri (mycoheterotrophic orchids), Epifagus virginiana (a parasitic flowering plant), and Helicosporidium sp. (a parasitic green alga). These taxa have elevated rates of molecular evolution and relatively few genes present in the data sets (see Additional file 2). We removed them to ensure that their inclusion did not cause any phylogenetic artifacts.
Availability of supporting data
The data sets supporting the results of this article are available in the Dryad Digital Repository: http://doi.org/10.5061/dryad.k1t1f.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
BRR conceived the study. BRR, PSS, DES, and JGB participated in the design of the study. BRR, MAG, and JGB analyzed the data. BRR, PSS, DES, and JGB wrote the paper. All authors read and approved the final manuscript.
Supplementary Material
Taxon sampling. Taxa included in this study, their GenBank accession numbers, original publications, and their higher taxonomy.
Genes sampled and missing data for each taxon. Information on taxa sampled for each gene included, and the percent of missing data for each taxon in each data set. Number of genes present per taxon and number of taxa present per gene are also given.
GC content for each taxon in the ntAll and ntNo3rd data sets as well as in the first, second, and third codon positions of the ntAll data set.
Fifty percent maximum likelihood majority-rule bootstrap consensus summary tree of Viridiplantae inferred from the first and second codon positions (ntNo3rd) analysis. See also Figure 6 for a summary tree of major Viridiplantae clades and Additional file 1 for taxonomy. Data set derived from 78 protein-coding genes of the plastid genome (ntax = 360, 38,898 bp, missing data ~15.6%,). Bootstrap support values ≥ 50% are indicated.
Fifty percent maximum likelihood majority-rule bootstrap consensus tree of Viridiplantae inferred from the RY-coded (RY) analysis. See also Figure 7 for a summary tree of major Viridiplantae clades and Additional file 1 for taxonomy. Data set derived from 78 protein-coding genes of the plastid genome (ntax = 360, 58,347 bp, missing data ~15.6%,). Bootstrap support values ≥ 50% are indicated.
Fifty percent maximum likelihood majority-rule bootstrap consensus tree of Viridiplantae inferred from the amino acid (AA) analysis. See also Figure 8 for a summary tree of major Viridiplantae clades and Additional file 1 for taxonomy. Data set derived from 78 protein-coding genes of the plastid genome (ntax = 360, 19,449 AAs, missing data ~15.6%,). Bootstrap support values ≥ 50% are indicated.
Maximum likelihood tree of Viridiplantae inferred from the all nucleotide positions (ntAll) analysis. Cladogram of the maximum likelihood bipartition tree is shown on the left with bootstrap values indicated above the branches. The phylogram of same tree is shown on the right. Data set derived from 78 protein-coding genes of the plastid genome (ntax = 360; 58,347 bp; missing data ~15.6%). Bootstrap support values ≥ 50% are indicated.
Maximum likelihood tree of Viridiplantae inferred from the first and second codon positions (ntNo3rd) analysis. Cladogram of the maximum likelihood bipartition tree is shown on the left with bootstrap values indicated above the branches. The phylogram of same tree is shown on the right. Data set derived from 78 protein-coding genes of the plastid genome (ntax = 360, 38,898 bp, missing data ~15.6%,). Bootstrap support values ≥ 50% are indicated.
Maximum likelihood tree of Viridiplantae inferred from the RY-coded (RY) analysis. Cladogram of the maximum likelihood bipartition tree is shown on the left with bootstrap values indicated above the branches. The phylogram of same tree is shown on the right. Data set derived from 78 protein-coding genes of the plastid genome (ntax = 360, 58,347 bp, missing data ~15.6%,). Bootstrap support values ≥ 50% are indicated.
Maximum likelihood tree of Viridiplantae inferred from the amino acid (AA) analysis. Cladogram of the maximum likelihood bipartition tree is shown on the left with bootstrap values indicated above the branches. The phylogram of same tree is shown on the right. Data set derived from 78 protein-coding genes of the plastid genome (ntax = 360, 19,449 AAs, missing data ~15.6%,). Bootstrap support values ≥ 50% are indicated.
Maximum likelihood tree of Viridiplantae inferred from the third codon position (nt3rdOnly) analysis. Cladogram of the maximum likelihood bipartition tree is shown on the left with bootstrap values indicated above the branches. The phylogram of same tree is shown on the right. Data set derived from 78 protein-coding genes of the plastid genome (ntax = 360, 19,449 bp, missing data ~15.6%,). Bootstrap support values ≥ 50% are indicated.
Contributor Information
Brad R Ruhfel, Email: brad.ruhfel@eku.edu.
Matthew A Gitzendanner, Email: magitz@ufl.edu.
Pamela S Soltis, Email: psoltis@flmnh.ufl.edu.
Douglas E Soltis, Email: dsoltis@ufl.edu.
J Gordon Burleigh, Email: gburleigh@ufl.edu.
Acknowledgements
This research was supported in part by the iPlant Tree of Life Project (iPlant Collaborative, funded by NSF grant DBI-0735191) and the Open Tree of Life Project (NSF grant #DEB-12008809). We would also like to thank E. L. Braun and Z. Xi for their input regarding aspects of our data analyses and E. V. Mavrodiev for help with the figures.
References
- Govaerts R. How many species of seed plants are there? - a response. Taxon. 2003;52(3):583–584. doi: 10.2307/3647457. [DOI] [Google Scholar]
- Govaerts R. How many species of seed plants are there? Taxon. 2001;50(4):1085–1090. doi: 10.2307/1224723. [DOI] [Google Scholar]
- Judd WS, Campbell CS, Kellogg EA, Stevens PF, Donoghue MJ. Plant systematics : a phylogenetic approach. 3. Sunderland, MA: Sinauer Associates; 2008. [Google Scholar]
- Charophycean green algae. [ http://www.life.umd.edu/labs/delwiche/Charophyte.html]
- AlgaeBase. [ http://www.algaebase.org]
- Guiry MD. How many species of algae are there? J Phycol. 2012;48(5):1057–1063. doi: 10.1111/j.1529-8817.2012.01222.x. [DOI] [PubMed] [Google Scholar]
- Courties C, Vaquer A, Troussellier M, Lautier J, Chretiennot-Dinet MJ, Neveux J, Machado C, Claustre H. Smallest eukaryotic organism. Nature. 1994;370(6487):255. [Google Scholar]
- Butterfield NJ. Modes of pre-Ediacaran multicellularity. Precambrian Res. 2009;173(1–4):201–211. [Google Scholar]
- Butterfield NJ, Knoll AH, Swett K. Paleobiology of the Neoproterozoic Svanbergfjellet Formation, Spitsbergen. Fossils Strata. 1994;34:1–84. [Google Scholar]
- Halverson GP, Maloof AC, Schrag DP, Dudas FO, Hurtgen M. Stratigraphy and geochemistry of a ca 800 Ma negative carbon isotope interval in northeastern Svalbard. Chem Geol. 2007;237(1–2):5–27. [Google Scholar]
- Yoon HS, Hackett JD, Ciniglia C, Pinto G, Bhattacharya D. A molecular timeline for the origin of photosynthetic eukaryotes. Mol Biol Evol. 2004;21(5):809–818. doi: 10.1093/molbev/msh075. [DOI] [PubMed] [Google Scholar]
- Hedges SB, Blair JE, Venturi ML, Shoe JL. A molecular timescale of eukaryote evolution and the rise of complex multicellular life. BMC Evol Biol. 2004;4:2. doi: 10.1186/1471-2148-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herron MD, Hackett JD, Aylward FO, Michod RE. Triassic origin and early radiation of multicellular volvocine algae. Proc Natl Acad Sci USA. 2009;106(9):3254–3258. doi: 10.1073/pnas.0811205106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parfrey LW, Lahr DJG, Knoll AH, Katz LA. Estimating the timing of early eukaryotic diversification with multigene molecular clocks. Proc Natl Acad Sci USA. 2011;108(33):13624–13629. doi: 10.1073/pnas.1110633108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenrick P, Crane PR. The origin and early evolution of plants on land. Nature. 1997;389:33–39. doi: 10.1038/37918. [DOI] [Google Scholar]
- Doyle JA. Seed ferns and the origin of angiosperms. J Torrey Bot Soc. 2006;133(1):169–209. doi: 10.3159/1095-5674(2006)133[169:SFATOO]2.0.CO;2. [DOI] [Google Scholar]
- Hilton J, Bateman RM. Pteridosperms are the backbone of seed-plant phylogeny. J Torrey Bot Soc. 2006;133(1):119–168. doi: 10.3159/1095-5674(2006)133[119:PATBOS]2.0.CO;2. [DOI] [Google Scholar]
- Rothfels CJ, Larsson A, Kuo LY, Korall P, Chiou WL, Pryer KM. Overcoming deep roots, fast rates, and short internodes to resolve the ancient rapid radiation of eupolypod II ferns. Syst Biol. 2012;61(3):490–509. doi: 10.1093/sysbio/sys001. [DOI] [PubMed] [Google Scholar]
- Soltis PS, Soltis DE, Savolainen V, Crane PR, Barraclough TG. Rate heterogeneity among lineages of tracheophytes: integration of molecular and fossil data and evidence for molecular living fossils. Proc Natl Acad Sci USA. 2002;99(7):4430–4435. doi: 10.1073/pnas.032087199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong B, Deusch O, Goremykin VV, Penny D, Biggs PJ, Atherton RA, Nikiforova SV, Lockhart PJ. Systematic error in seed plant phylogenomics. Genome Biol Evol. 2011;3:1340–1348. doi: 10.1093/gbe/evr105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DR. Unparalleled GC content in the plastid DNA of Selaginella. Plant Mol Biol. 2009;71(6):627–639. doi: 10.1007/s11103-009-9545-3. [DOI] [PubMed] [Google Scholar]
- Smith SA, Donoghue MJ. Rates of molecular evolution are linked to life history in flowering plants. Science. 2008;322(5898):86–89. doi: 10.1126/science.1163197. [DOI] [PubMed] [Google Scholar]
- Karol KG, McCourt RM, Cimino MT, Delwiche CF. The closest living relatives of land plants. Science. 2001;294:2351–2353. doi: 10.1126/science.1065156. [DOI] [PubMed] [Google Scholar]
- Lemieux C, Otis C, Turmel M. Ancestral chloroplast genome in Mesostigma viride reveals an early branch of green plant evolution. Nature. 2000;403(6770):649–652. doi: 10.1038/35001059. [DOI] [PubMed] [Google Scholar]
- Wodniok S, Brinkmann H, Glockner G, Heidel AJ, Philippe H, Melkonian M, Becker B. Origin of land plants: do conjugating green algae hold the key? BMC Evol Biol. 2011;11:104. doi: 10.1186/1471-2148-11-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang BF, Nedelcu AM. In: Genomics of Chloroplasts and Mitochondria, Volume 35. Bock R, Knoop V, editor. Netherlands: Springer; 2012. Plastid genomes of algae; pp. 59–87. [Google Scholar]
- Turmel M, Otis C, Lemieux C. The chloroplast genome sequence of Chara vulgaris sheds new light into the closest green algal relatives of land plants. Mol Biol Evol. 2006;23(6):1324–1338. doi: 10.1093/molbev/msk018. [DOI] [PubMed] [Google Scholar]
- Turmel M, Pombert J, Charlebois P, Otis C, Lemieux C. The green algal ancestry of land plants as revealed by the chloroplast genome. Int J Pl Sci. 2007;168(5):679–689. doi: 10.1086/513470. [DOI] [Google Scholar]
- Qiu YL, Li L, Wang B, Chen Z, Knoop V, Groth-Malonek M, Dombrovska O, Lee J, Kent L, Rest J. et al. The deepest divergences in land plants inferred from phylogenomic evidence. Proc Natl Acad Sci USA. 2006;103(42):15511–15516. doi: 10.1073/pnas.0603335103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickrent DL, Parkinson CL, Palmer JD, Duff RJ. Multigene phylogeny of land plants with special reference to bryophytes and the earliest land plants. Mol Biol Evol. 2000;17:1885–1895. doi: 10.1093/oxfordjournals.molbev.a026290. [DOI] [PubMed] [Google Scholar]
- Renzaglia KS, Schuette S, Duff RJ, Ligrone R, Shaw AJ, Mishler BD, Duckett JG. Bryophyte phylogeny: advancing the molecular and morphological frontiers. Bryologist. 2007;110(2):179–213. doi: 10.1639/0007-2745(2007)110[179:BPATMA]2.0.CO;2. [DOI] [Google Scholar]
- Mishler BD, Churchill SP. A cladistic approach to the phylogeny of the “bryophytes”. Brittonia. 1984;36:406–424. doi: 10.2307/2806602. [DOI] [Google Scholar]
- Shaw J, Renzaglia K. Phylogeny and diversification of bryophytes. Amer J Bot. 2004;91(10):1557–1581. doi: 10.3732/ajb.91.10.1557. [DOI] [PubMed] [Google Scholar]
- Karol KG, Arumuganathan K, Boore JL, Duffy AM, Everett KDE, Hall JD, Hansen SK, Kuehl JV, Mandoli DF, Mishler BD. et al. Complete plastome sequences of Equisetum arvense and Isoetes flaccida: implications for phylogeny and plastid genome evolution of early land plant lineages. BMC Evol Biol. 2010;10:321. doi: 10.1186/1471-2148-10-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf PG, Karol KG. In: Genomics of Chloroplasts and Mitochondria, Volume 35. Bock R, Knoop V, editor. Netherlands: Springer; 2012. Plastomes of bryophytes, lycophytes and ferns; pp. 89–102. [Google Scholar]
- Crane PR. Phylogenetic analysis of seed plants and the origin of angiosperms. Ann Missouri Bot Gard. 1985;72:716–793. doi: 10.2307/2399221. [DOI] [Google Scholar]
- Chaw SM, Zharkikh A, Sung HM, Lau TC, Li WH. Molecular phylogeny of extant gymnosperms and seed plant evolution: analysis of nuclear 18S rRNA sequences. Mol Biol Evol. 1997;14(1):56–68. doi: 10.1093/oxfordjournals.molbev.a025702. [DOI] [PubMed] [Google Scholar]
- Bowe LM, Coat G, dePamphilis CW. Phylogeny of seed plants based on all three genomic compartments: extant gymnosperms are monophyletic and Gnetales’ closest relatives are conifers. Proc Natl Acad Sci USA. 2000;97(8):4092–4097. doi: 10.1073/pnas.97.8.4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaw SM, Parkinson CL, Cheng YC, Vincent TM, Palmer JD. Seed plant phylogeny inferred from all three plant genomes: monophyly of extant gymnosperms and origin of Gnetales from conifers. Proc Natl Acad Sci USA. 2000;97(8):4086–4091. doi: 10.1073/pnas.97.8.4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finet C, Timme RE, Delwiche CF, Marleta F. Multigene phylogeny of the green lineage reveals the origin and diversification of land plants. Curr Biol. 2010;20(24):2217–2222. doi: 10.1016/j.cub.2010.11.035. [DOI] [PubMed] [Google Scholar]
- Zhong B, Yonezawa T, Zhong Y, Hasegawa M. The position of Gnetales among seed plants: overcoming pitfalls of chloroplast phylogenomics. Mol Biol Evol. 2010;27(12):2855–2863. doi: 10.1093/molbev/msq170. [DOI] [PubMed] [Google Scholar]
- Mathews S. Phylogenetic relationships among seed plants: persistent questions and the limits of molecular data. Amer J Bot. 2009;96(1):228–236. doi: 10.3732/ajb.0800178. [DOI] [PubMed] [Google Scholar]
- Burleigh JG, Mathews S. Phylogenetic signal in nucleotide data from seed plants: implications for resolving the seed plant tree of life. Amer J Bot. 2004;91(10):1599–1613. doi: 10.3732/ajb.91.10.1599. [DOI] [PubMed] [Google Scholar]
- Bhattacharya D, Medlin L. Algal phylogeny and the origin of land plants. Plant Physiol. 1998;116(1):9–15. doi: 10.1104/pp.116.1.9. [DOI] [Google Scholar]
- Soltis PS, Soltis DE, Wolf PG, Nickrent DL, Chaw S-M, Chapman RL. The phylogeny of land plants inferred from 18S rDNA sequences: pushing the limits of rDNA signal? Mol Biol Evol. 1999;16:1774–1784. doi: 10.1093/oxfordjournals.molbev.a026089. [DOI] [PubMed] [Google Scholar]
- Lee EK, Cibrian-Jaramillo A, Kolokotronis S-O, Katari MS, Stamatakis A, Ott M, Chiu JC, Little DP, Stevenson DW, McCombie WR. et al. A functional phylogenomic view of the seed plants. PLoS Genet. 2011;7(12):e1002411. doi: 10.1371/journal.pgen.1002411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu YL, Cho Y, Cox JC, Palmer JD. The gain of three mitochondrial introns identifies liverworts as the earliest land plants. Nature. 1998;394:671–674. doi: 10.1038/29286. [DOI] [PubMed] [Google Scholar]
- Duff RJ, Nickrent DL. Phylogenetic relationships of land plants using mitochondrial small-subunit rDNA sequences. Amer J Bot. 1999;86:372–386. doi: 10.2307/2656759. [DOI] [PubMed] [Google Scholar]
- Qiu YL, Palmer JD. Phylogeny of early land plants: insights from genes and genomes. Trends Plant Sci. 1999;4(1):26–30. doi: 10.1016/S1360-1385(98)01361-2. [DOI] [PubMed] [Google Scholar]
- Qiu YL. Phylogeny and evolution of charophytic algae and land plants. J Syst Evol. 2008;46(3):287–306. [Google Scholar]
- Magallon S, Sanderson MJ. Relationships among seed plants inferred from highly conserved genes: Sorting conflicting phylogenetic signals among ancient lineages. Amer J Bot. 2002;89(12):1991–2006. doi: 10.3732/ajb.89.12.1991. [DOI] [PubMed] [Google Scholar]
- Smith S, Beaulieu J, Donoghue M. Mega-phylogeny approach for comparative biology: an alternative to supertree and supermatrix approaches. BMC Evol Biol. 2009;9(1):37. doi: 10.1186/1471-2148-9-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicke S, Schneeweiss G, dePamphilis C, Müller K, Quandt D. The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol Biol. 2011;76(3):273–297. doi: 10.1007/s11103-011-9762-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer JD, Nugent JM, Herbon LA. Unusual structure of geranium chloroplast dna - a triple-sized inverted repeat, extensive gene duplications, multiple inversions, and 2 repeat families. Proc Natl Acad Sci USA. 1987;84(3):769–773. doi: 10.1073/pnas.84.3.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegemann S, Keuthe M, Greiner S, Bock R. Horizontal transfer of chloroplast genomes between plant species. Proc Natl Acad Sci USA. 2012;109(7):2434–2438. doi: 10.1073/pnas.1114076109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltis DE, Soltis PM. In: Molecular Systematics of Plants II. Soltis DE, Soltis PM, Doyle J, editor. Boston: Kluwer; 1998. Choosing an approach and an appropriate gene for phylogenetic analysis; pp. 1–42. [Google Scholar]
- Olmstead RG, Palmer JD. Chloroplast DNA systematics - a review of methods and data-analysis. Amer J Bot. 1994;81(9):1205–1224. doi: 10.2307/2445483. [DOI] [Google Scholar]
- Chase MW, Soltis DE, Olmstead RG, Morgan D, Les DH, Mishler BD, Duvall MR, Price RA, Hills HG, Qiu Y-L. et al. Phylogenetics of seed plants: An analysis of nucleotide sequences from the plastid gene rbcL. Ann Missouri Bot Gard. 1993;80:528–580. doi: 10.2307/2399846. [DOI] [Google Scholar]
- Savolainen V, Chase MW. A decade of progress in plant molecular phylogenetics. Trends Gen. 2003;19(12):717–724. doi: 10.1016/j.tig.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Shinozaki K. et al. The complete nucleotide sequence of tobacco chloroplast genome: its gene organization and expression. EMBO J. 1986;5:2043–2049. doi: 10.1002/j.1460-2075.1986.tb04464.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohyama K, Fukuzawa H, Kohchi T, Shirai H, Sano T, Sano S, Umesono K, Shiki Y, Takeuchi M, Chang Z. et al. Chloroplast gene organization deduced from complete sequence of liverwort Marchantia polymorpha chloroplast DNA. Nature. 1986;322(6079):572–574. doi: 10.1038/322572a0. [DOI] [Google Scholar]
- Moore MJ, Dhingra A, Soltis PS, Shaw R, Farmerie WG, Folta KM, Soltis DE. Rapid and accurate pyrosequencing of angiosperm plastid genomes. BMC Plant Biol. 2006;6:17. doi: 10.1186/1471-2229-6-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Soltis PS, Bell CD, Burleigh JG, Soltis DE. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc Natl Acad Sci USA. 2010;107(10):4623–4628. doi: 10.1073/pnas.0907801107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronn R, Knaus BJ, Liston A, Maughan PJ, Parks M, Syring JV, Udall J. Targeted enrichment strategies for next-generation plant biology. Amer J Bot. 2012;99(2):291–311. doi: 10.3732/ajb.1100356. [DOI] [PubMed] [Google Scholar]
- Cronn R, Liston A, Parks M, Gernandt DS, Shen R, Mockler T. Multiplex sequencing of plant chloroplast genomes using Solexa sequencing-by-synthesis technology. Nucleic Acids Res. 2008;36(19):1–11. doi: 10.1093/nar/gkn502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks M, Cronn R, Liston A. Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 2009;7:84. doi: 10.1186/1741-7007-7-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub SCK, Parks M, Weitemier K, Fishbein M, Cronn RC, Liston A. Navigating the tip of the genomic iceberg: next-generation sequencing for plant systematics. Amer J Bot. 2012;99(2):349–364. doi: 10.3732/ajb.1100335. [DOI] [PubMed] [Google Scholar]
- Jansen RK, Ruhlman TA. In: Genomics of Chloroplasts and Mitochondria, Volume 35. Bock R, Knoop V, editor. Netherlands: Springer; 2012. Plastid genomes of seed plants. [Google Scholar]
- Xi Z, Ruhfel BR, Schaefer H, Amorim AM, Sugumaran M, Wurdack KJ, Endress PK, Matthews ML, Stevens PF, Mathews S. et al. Phylogenomics and a posteriori data partitioning resolve the Cretaceous angiosperm radiation Malpighiales. Proc Natl Acad Sci USA. 2012;109(43):17519–17524. doi: 10.1073/pnas.1205818109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Koning AP, Keeling PJ. The complete plastid genome sequence of the parasitic green alga Helicosporidium sp. is highly reduced and structured. BMC Biol. 2006;4:10. doi: 10.1186/1741-7007-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delannoy E, Fujii S, Colas des Francs-Small C, Brundrett M, Small I. Rampant gene loss in the underground orchid Rhizanthella gardneri highlights evolutionary constraints on plastid genomes. Mol Biol Evol. 2011;28(7):2077–2086. doi: 10.1093/molbev/msr028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson MJ, McMahon MM, Steel M. Phylogenomics with incomplete taxon coverage: the limits to inference. BMC Evol Biol. 2010;10:13. doi: 10.1186/1471-2148-10-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhfel BR, Gitzendanner MA, Soltis PS, Soltis DE, Burleigh JG. Data from: from algae to angiosperms:–inferring the phylogeny of green plants (Viridiplantae) from 360 plastid genomes. Dryad Data Repository. 2014. doi:10.5061/dryad.k1t1f. [DOI] [PMC free article] [PubMed]
- Lewis LA, McCourt RM. Green algae and the origin of land plants. Amer J Bot. 2004;91(10):1535–1556. doi: 10.3732/ajb.91.10.1535. [DOI] [PubMed] [Google Scholar]
- Mattox KR, Stewart KD. In: The Systematics of Green Algae. Irvin DEG, John DM, editor. London, UK: Academic Press; 1984. Classification of the green algae: a concept based on comparative cytology; pp. 29–72. [Google Scholar]
- Leliaert F, Smith DR, Moreau H, Herron MD, Verbruggen H, Delwiche CF, De Clerck O. Phylogeny and molecular evolution of the green algae. CRC Crit Rev Plant Sci. 2012;31(1):1–46. doi: 10.1080/07352689.2011.615705. [DOI] [Google Scholar]
- Leliaert F, Verbruggen H, Zechman FW. Into the deep: New discoveries at the base of the green plant phylogeny. Bioessays. 2011;33(9):683–692. doi: 10.1002/bies.201100035. [DOI] [PubMed] [Google Scholar]
- Lartillot N, Brinkmann H, Philippe H. Suppression of long-branch attraction artefacts in the animal phylogeny using a site-heterogeneous model. BMC Evol Biol. 2007;7:14. doi: 10.1186/1471-2148-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lartillot N, Philippe H. A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol Biol Evol. 2004;21:1095. doi: 10.1093/molbev/msh112. [DOI] [PubMed] [Google Scholar]
- Lemieux C, Otis C, Turmel M. A clade uniting the green algae Mesostigma viride and Chlorokybus atmophyticus represents the deepest branch of the Streptophyta in chloroplast genome-based phylogenies. BMC Biol. 2007;5:2. doi: 10.1186/1741-7007-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Ezpeleta N, Philippe H, Brinkmann H, Becker B, Melkonian M. Phylogenetic analyses of nuclear, mitochondrial, and plastid multigene data sets support the placement of Mesostigma in the Streptophyta. Mol Biol Evol. 2007;24(3):723–731. doi: 10.1093/molbev/msl200. [DOI] [PubMed] [Google Scholar]
- Timme RE, Bachvaroff TR, Delwiche CF. Broad phylogenomic sampling and the sister lineage of land plants. PLoS ONE. 2012;7(1):e29696. doi: 10.1371/journal.pone.0029696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turmel M, Otis C, Lemieux C. An unexpectedly large and loosely packed mitochondrial genome in the charophycean green alga Chlorokybus atmophyticus. BMC Genomics. 2007;8:12. doi: 10.1186/1471-2164-8-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocquyt E, Verbruggen H, Leliaert F, De Clerck O. Evolution and cytological diversification of the green seaweeds (Ulvophyceae) Mol Biol Evol. 2010;27(9):2052–2061. doi: 10.1093/molbev/msq091. [DOI] [PubMed] [Google Scholar]
- Turmel M, Gagnon M-C, O’Kelly CJ, Otis C, Lemieux C. The chloroplast genomes of the green algae Pyramimonas, Monomastix, and Pycnococcus shed new light on the evolutionary history of prasinophytes and the origin of the secondary chloroplasts of euglenids. Mol Biol Evol. 2009;26(3):631–648. doi: 10.1093/molbev/msn285. [DOI] [PubMed] [Google Scholar]
- Turmel M, Otis C, Lemieux C. The chloroplast genomes of the green algae Pedinomonas minor, Parachlorella kessleri, and Oocystis solitatia reveal a shared ancestry between the Pedinomonadales and Chlorellales. Mol Biol Evol. 2009;26(10):2317–2331. doi: 10.1093/molbev/msp138. [DOI] [PubMed] [Google Scholar]
- Zhong B, Xi Z, Goremykin VV, Fong R, Mclenachan PA, Novis PM, Davis CC, Penny D. Streptophyte algae and the origin of land plants revisited using heterogeneous models with three new algal chloroplast genomes. Mol Biol Evol. 2014;31(1):177–183. doi: 10.1093/molbev/mst200. [DOI] [PubMed] [Google Scholar]
- Groth-Malonek M, Pruchner D, Grewe F, Knoop V. Ancestors of trans-splicing mitochondrial introns support serial sister group relationships of hornworts and mosses with vascular plants. Mol Biol Evol. 2005;22(1):117–125. doi: 10.1093/molbev/msh259. [DOI] [PubMed] [Google Scholar]
- Qiu YL, Li L, Wang B, Chen Z, Dombrovska O, Lee JH, Kent L, Li RQ, Jobson RW, Hendry TA. et al. A nonflowering land plant phylogeny inferred from nucleotide sequences of seven chloroplast, mitochondrial, and nuclear genes. Int J Pl Sci. 2007;168(5):691–708. doi: 10.1086/513474. [DOI] [Google Scholar]
- Renzaglia KS, Duff RJ, Nickrent DL, Garbary DJ. Vegetative and reproductive innovations of early land plants: implications for a unified phylogeny. Philos Transs R Soc Lon B. 2000;355:769–793. doi: 10.1098/rstb.2000.0615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renzaglia KS, Garbary DJ. Motile gametes of land plants: diversity, development, and evolution. CRC Crit Rev Plant Sci. 2001;20(2):107–213. doi: 10.1080/20013591099209. [DOI] [Google Scholar]
- Garbary DJ, Renzaglia KS, Duckett JG. The phylogeny of land plants-a cladistic analysis based on male gametogenesis. Pl Syst Evol. 1993;188:237–269. [Google Scholar]
- Garbary DJ, Renzaglia KS. In: Bryology for the twenty-first century. Bates JW, Ashton NW, Duckett JG, editor. Leeds, U.K: Maney Publishing and British Bryological Society; 1998. Bryophyte phylogeny and the evolution of land plants: evidence from development and ultrastructure; pp. 45–63. [Google Scholar]
- Nishiyama T, Wolf PG, Kugita M, Sinclair RB, Sugita M, Sugiura C, Wakasugi T, Yamada K, Yoshinaga K, Yamaguchi K. et al. Chloroplast phylogeny indicates that bryophytes are monophyletic. Mol Biol Evol. 2004;21(10):1813–1819. doi: 10.1093/molbev/msh203. [DOI] [PubMed] [Google Scholar]
- Goremykin VV, Hellwig FH. Evidence for the most basal split in land plants dividing bryophyte and tracheophyte lineages. Pl Syst Evol. 2005;254(1–2):93–103. [Google Scholar]
- Pryer KM, Schneider H, Smith AR, Cranfill R, Wolf PG, Hunt JS, Sipes SD. Horsetails and ferns are a monophyletic group and the closest living relatives to seed plants. Nature. 2001;409:618–622. doi: 10.1038/35054555. [DOI] [PubMed] [Google Scholar]
- Pryer KM, Schneider H, Magallón S. In: Assembling the Tree of Life. Cracraft J, Donoghue MJ, editor. New York: University Press; 2004. The radiation of vascular plants; pp. 138–153. [Google Scholar]
- Kranz HD, Huss VAR. Molecular evolution of pteridophytes and their relationship to seed plants: evidence from complete 18S rRNA gene sequences. Pl Syst Evol. 1996;202(1–2):1–11. [Google Scholar]
- Raubeson LA, Jansen RK. Chloroplast DNA evidence on the ancient evolutionary split in vascular land plants. Science. 1992;255(5052):1697–1699. doi: 10.1126/science.255.5052.1697. [DOI] [PubMed] [Google Scholar]
- Grewe F, Guo W, Gubbels E, Hansen AK, Mower J. Complete plastid genomes from Ophioglossum californicum, Psilotum nudum, and Equisetum hyemale reveal an ancestral land plant genome structure and resolve the position of Equisetales among monilophytes. BMC Evol Biol. 2013;13(1):8. doi: 10.1186/1471-2148-13-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltis DE, Soltis PS, Zanis MJ. Phylogeny of seed plants based on evidence from eight genes. Amer J Bot. 2002;89(10):1670–1681. doi: 10.3732/ajb.89.10.1670. [DOI] [PubMed] [Google Scholar]
- Xi Z, Rest J, Davis CC. Phylogenomics and coalescent analyses resolve extant seed plant relationships. PLoS ONE. 2013;8(11):e80870. doi: 10.1371/journal.pone.0080870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltis DE, Smith SA, Cellinese N, Wurdack KJ, Tank DC, Brockington SF, Refulio-Rodriguez NF, Walker JB, Moore MJ, Carlsward BS. et al. Angiosperm phylogeny: 17 genes, 640 taxa. Am J Bot. 2011;98(4):704–730. doi: 10.3732/ajb.1000404. [DOI] [PubMed] [Google Scholar]
- Jansen RK, Saski C, Lee SB, Hansen AK, Daniell H. Complete plastid genome sequences of three rosids (Castanea, Prunus, Theobroma): evidence for at least two independent transfers of rpl22 to the nucleus. Mol Biol Evol. 2011;28(1):835–847. doi: 10.1093/molbev/msq261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen RK, Cai Z, Raubeson LA, Daniell H, Depamphilis CW, Leebens-Mack J, Muller KF, Guisinger-Bellian M, Haberle RC, Hansen AK. et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc Natl Acad Sci USA. 2007;104:19369–19374. doi: 10.1073/pnas.0709121104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Bell CD, Soltis PS, Soltis DE. Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc Natl Acad Sci USA. 2007;104(49):19363–19368. doi: 10.1073/pnas.0708072104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltis DE, Soltis PS, Endress PK, Chase MW. Phylogeny and evolution of angiosperms. Sunderland, Mass: Sinauer Associates; 2005. [Google Scholar]
- Barrett CF, Davis JI, Leebens-Mack J, Conran JG, Stevenson DW. Plastid genomes and deep relationships among the commelinid monocot angiosperms. Cladistics. 2013;29(1):65–87. doi: 10.1111/j.1096-0031.2012.00418.x. [DOI] [PubMed] [Google Scholar]
- Ane C, Burleigh JG, McMahon MM, Sanderson MJ. Covarion structure in plastid genome evolution: a new statistical test. Mol Biol Evol. 2005;22(4):914–924. doi: 10.1093/molbev/msi076. [DOI] [PubMed] [Google Scholar]
- Goremykin VV, Nikiforova SV, Biggs PJ, Zhong BJ, Delange P, Martin W, Woetzel S, Atherton RA, McLenachan PA, Lockhart PJ. The evolutionary root of flowering plants. Syst Biol. 2013;62(1):50–61. doi: 10.1093/sysbio/sys070. [DOI] [PubMed] [Google Scholar]
- Foster PG. Modeling compositional heterogeneity. Syst Biol. 2004;53(3):485–495. doi: 10.1080/10635150490445779. [DOI] [PubMed] [Google Scholar]
- Jermiin LS, Ho SYW, Ababneh F, Robinson J, Larkum AWD. The biasing effect of compositional heterogeneity on phylogenetic estimates may be underestimated. Syst Biol. 2004;53(4):638–643. doi: 10.1080/10635150490468648. [DOI] [PubMed] [Google Scholar]
- Erixon P, Oxelman B. Whole-gene positive selection, elevated synonymous substitution rates, duplication, and indel evolution of the chloroplast clpP1 gene. PLoS ONE. 2008;3(1):10. doi: 10.1371/journal.pone.0001386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guisinger MM, Kuehl JV, Boore JL, Jansen RK. Genome-wide analyses of Geraniaceae plastid DNA reveal unprecedented patterns of increased nucleotide substitutions. Proc Natl Acad Sci USA. 2008;105(47):18424–18429. doi: 10.1073/pnas.0806759105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai ZQ, Penaflor C, Kuehl JV, Leebens-Mack J, Carlson JE, dePamphilis CW, Boore JL, Jansen RK. Complete plastid genome sequences of Drimys, Liriodendron, and Piper: implications for the phylogenetic relationships of magnoliids. BMC Evol Biol. 2006;6:20. doi: 10.1186/1471-2148-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raubeson LA, Peery R, Chumley TW, Dziubek C, Fourcade HM, Boore JL, Jansen RK. Comparative chloroplast genomics: analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genomics. 2007;8:27. doi: 10.1186/1471-2164-8-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guisinger MM, Kuehl JV, Boore JL, Jansen RK. Extreme reconfiguration of plastid genomes in the angiosperm family Geraniaceae: rearrangements, repeats, and codon usage. Mol Biol Evol. 2011;28(1):583–600. doi: 10.1093/molbev/msq229. [DOI] [PubMed] [Google Scholar]
- Phillips MJ, Penny D. The root of the mammalian tree inferred from whole mitochondrial genomes. Mol Phylogenet and Evol. 2003;28(2):171–185. doi: 10.1016/S1055-7903(03)00057-5. [DOI] [PubMed] [Google Scholar]
- Phillips MJ, Delsuc F, Penny D. Genome-scale phylogeny and the detection of systematic biases. Mol Biol Evol. 2004;21:1455. doi: 10.1093/molbev/msh137. [DOI] [PubMed] [Google Scholar]
- Ishikawa SA, Inagaki Y, Hashimoto T. RY-coding and non-homogeneous models can ameliorate the maximum-likelihood inferences from nucleotide sequence data with parallel compositional heterogeneity. Evol Bioinform. 2012;8:357–371. doi: 10.6026/97320630008357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delsuc F, Phillips MJ, Penny D. Comment on “Hexapod origins: monophyletic or paraphyletic?”. Science. 2003;301(5639):1482. doi: 10.1126/science.1086558. [DOI] [PubMed] [Google Scholar]
- Parks M, Cronn R, Liston A. Separating the wheat from the chaff: mitigating the effects of noise in a plastome phylogenomic data set from Pinus L. (Pinaceae) BMC Evol Biol. 2012;12(1):100. doi: 10.1186/1471-2148-12-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffroy O, Brinkmann H, Delsuc F, Philippe H. Phylogenomics: the beginning of incongruence? Trends Genet. 2006;22(4):225–231. doi: 10.1016/j.tig.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Foster PG, Hickey DA. Compositional bias may affect both DNA-based and protein-based phylogenetic reconstructions. J Mol Evol. 1999;48(3):284–290. doi: 10.1007/PL00006471. [DOI] [PubMed] [Google Scholar]
- Mathews S, Clements MD, Beilstein MA. A duplicate gene rooting of seed plants and the phylogenetic position of flowering plants. Philos Trans R Soc B-Biol Sci. 2010;365(1539):383–395. doi: 10.1098/rstb.2009.0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltis DE, Albert VA, Savolainen V, Hilu K, Qiu YL, Chase MW, Farris JS, Stefanovic S, Rice DW, Palmer JD. et al. Genome-scale data, angiosperm relationships, and “ending incongruence”: a cautionary tale in phylogenetics. Trends Plant Sci. 2004;9(10):477–483. doi: 10.1016/j.tplants.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Graybeal A. Is it better to add taxa or characters to a difficult phylogenetic problem? Syst Biol. 1998;47:9–17. doi: 10.1080/106351598260996. [DOI] [PubMed] [Google Scholar]
- Hillis DM. Taxonomic sampling, phylogenetic accuracy, and investigator bias. Syst Biol. 1998;47(1):3–8. doi: 10.1080/106351598260987. [DOI] [PubMed] [Google Scholar]
- Zwickl DJ, Hillis DM. Increased taxon sampling greatly reduces phylogenetic error. Syst Biol. 2002;51:588–598. doi: 10.1080/10635150290102339. [DOI] [PubMed] [Google Scholar]
- Hillis DM, Pollock DD, McGuire JA, Zwickl DJ. Is sparse taxon sampling a problem for phylogenetic inference? Syst Biol. 2003;52:124–126. doi: 10.1080/10635150390132911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leebens-Mack J, Raubeson LA, Cui L, Kuehl JV, Fourcade MH, Chumley TW, Boore JL, Jansen RK, dePamphilis CW. Identifying the basal angiosperm node in chloroplast genome phylogenies: sampling one’s way out of the Felsenstein Zone. Mol Biol Evol. 2005;22(10):1948–1963. doi: 10.1093/molbev/msi191. [DOI] [PubMed] [Google Scholar]
- Stull GW, Moore MJ, Mandala VS, Douglas NA, Kates H-R, Qi X, Brockington SF, Soltis PS, Soltis DE, Gitzendanner MA. A targeted enrichment strategy for massively parallel sequencing of angiosperm plastid genomes. Appl Plant Sci. 2013;1(2):1200497. doi: 10.3732/apps.1200497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiens JJ. Missing data, incomplete taxa, and phylogenetic accuracy. Syst Biol. 2003;52(4):528–538. doi: 10.1080/10635150390218330. [DOI] [PubMed] [Google Scholar]
- Wiens JJ, Moen DS. Missing data and the accuracy of Bayesian phylogenetics. J Syst Evol. 2008;46(3):307–314. [Google Scholar]
- Ruhfel BR, Stevens PF, Davis CC. Combined morphological and molecular phylogeny of the clusioid clade (Malpighiales) and the placement of the ancient rosid macrofossil Paleoclusia. Int J Pl Sci. 2013;174(6):910–936. doi: 10.1086/670668. [DOI] [Google Scholar]
- Wiens JJ. Paleontology, genomics, and combined-data phylogenetics: can molecular data improve phylogeny estimation for fossil taxa? Syst Biol. 2009;58(1):87–99. doi: 10.1093/sysbio/syp012. [DOI] [PubMed] [Google Scholar]
- Wang H, Moore MJ, Soltis PS, Bell CD, Brockington SF, Alexandre R, Davis CC, Latvis M, Manchester SR, Soltis DE. Rosid radiation and the rapid rise of angiosperm-dominated forests. Proc Natl Acad Sci USA. 2009;106(10):3853–3858. doi: 10.1073/pnas.0813376106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubatko LS, Degnan JH. Inconsistency of phylogenetic estimates from concatenated data under coalescence. Syst Biol. 2007;56(1):17–24. doi: 10.1080/10635150601146041. [DOI] [PubMed] [Google Scholar]
- Matsen FA, Steel M. Phylogenetic mixtures on a single tree can mimic a tree of another topology. Syst Biol. 2007;56(5):767–775. doi: 10.1080/10635150701627304. [DOI] [PubMed] [Google Scholar]
- Penny D, White WT, Hendy MD, Phillips MJ. A bias in ML estimates of branch lengths in the presence of multiple signals. Mol Biol Evol. 2008;25(2):239–242. doi: 10.1093/molbev/msm263. [DOI] [PubMed] [Google Scholar]
- Maddison WP. Gene trees in species trees. Syst Biol. 1997;46(3):523–536. doi: 10.1093/sysbio/46.3.523. [DOI] [Google Scholar]
- Mossel E, Steel M. In: Mathematics of Evolution and Phylogeny. Gascuel O, Steel M, editor. Oxford: Oxford University Press; 2005. How much can evolved characters tell us about the tree that generated them? pp. 384–412. [Google Scholar]
- Ponciano JM, Burleigh JG, Braun EL, Taper ML. Assessing parameter identifiability in phylogenetic models using data cloning. Syst Biol. 2012;61(6):955–972. doi: 10.1093/sysbio/sys055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffinet B, Buck WR, Shaw AJ. In: Bryophyte Biology. 2. Goffinet B, Shaw AJ, editor. Cambridge, UK: Cambridge University Press; 2008. Morphology and classification of the Bryophyta; pp. 55–138. [Google Scholar]
- Stotler RE, Crandall-Stotler B. A revised classification of the Anthocerotophyta and a checklist of the hornworts of North America, north of Mexico. Bryologist. 2005;108(1):16–26. doi: 10.1639/0007-2745(2005)108[16:ARCOTA]2.0.CO;2. [DOI] [Google Scholar]
- Crandall-Stotler B, Stotler RE, Long DG. Phylogeny and classification of the Marchantiophyta. Edinb J Bot. 2009;66(1):155–198. doi: 10.1017/S0960428609005393. [DOI] [Google Scholar]
- Cantino PD, Doyle JA, Graham SW, Judd WS, Olmstead RG, Soltis DE, Soltis PS, Donoghue MJ. Towards a phylogenetic nomenclature of Tracheophyta. Taxon. 2007;56(3):1E–44E. [Google Scholar]
- Christenhusz MJM, Zhang X-C, Schneider H. A linear sequence of extant families and genera of lycophytes and ferns. Phytotaxa. 2011;19:7–54. [Google Scholar]
- Christenhusz MJM, Reveal JL, Farjon A, Gardner MF, Mill RR, Chase MW. A new classification and linear sequence of extant gymnosperms. Phytotaxa. 2011;19:55–70. [Google Scholar]
- III A. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG III. Bot J Linn Soc. 2009;161(2):105–121. [Google Scholar]
- McNeill J, Barrie FR, Buck WR, Demoulin V, Greuter W, Hawkworth DL, Herendeen PS, Knapp S, Marhold K, Prado J, International code of nomenclature for algae, fungi, and plants (Melbourne code); adopted by the Eighteenth International Botanical Congress, Melbourne, Australia, July 2011. Königstein, Germany: Koeltz Scientific Books; 2012. [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang JH, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma KÄ, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30(14):3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25(15):1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suyama M, Torrents D, Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006;34(suppl 2):W609–W612. doi: 10.1093/nar/gkl315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22(21):2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- Ott M, Zola J, Aluru S, Stamatakis A. Proceedings of IEEE/ACM Supercomputing (SC2007) conference: 2007. Reno, Nevada, USA: ACM; 2007. Large-scale maximum likelihood-based phylogenetic analysis on the IBM BlueGene/L. [Google Scholar]
- Kuck P, Meusemann K. FASconCAT: Convenient handling of data matrices. Mol Phylogenet Evol. 2010;56(3):1115–1118. doi: 10.1016/j.ympev.2010.04.024. [DOI] [PubMed] [Google Scholar]
- Woese CR, Achenbach L, Rouviere P, Mandelco L. Archaeal phylogeny: reexamination of the phylogenetic position of Archaeoglobus fulgidus in light of certain composition-induced artifacts. Syst Appl Microbiol. 1991;14:364. doi: 10.1016/S0723-2020(11)80311-5. [DOI] [PubMed] [Google Scholar]
- Delsuc F, Brinkmann H, Philippe H. Phylogenomics and the reconstruction of the tree of life. Nat Rev Genet. 2005;6(5):361–375. doi: 10.1038/nrg1603. [DOI] [PubMed] [Google Scholar]
- Philippe H, Delsuc F, Brinkmann H, Lartillot N. Phylogenomics. Annu Rev Ecol Evol Syst. 2005;36(1):541–562. doi: 10.1146/annurev.ecolsys.35.112202.130205. [DOI] [Google Scholar]
- Swofford DL. PAUP*: Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4b10. Sunderland, MA: Sinauer Associates; 2003. [Google Scholar]
- Team RC. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2012. [Google Scholar]
- Hurvich CM, Tsai CL. Regression and time-series model selection in small samples. Biometrika. 1989;76(2):297–307. doi: 10.1093/biomet/76.2.297. [DOI] [Google Scholar]
- Posada D, Buckley TR. Model selection and model averaging in phylogenetics: advantages of Akaike Information Criterion and Bayesian approaches over likelihood ratio tests. Syst Biol. 2004;53(5):793–808. doi: 10.1080/10635150490522304. [DOI] [PubMed] [Google Scholar]
- Lanfear R, Calcott B, Ho SYW, Guindon S. PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol. 2012;29(6):1695–1701. doi: 10.1093/molbev/mss020. [DOI] [PubMed] [Google Scholar]
- Pattengale ND, Alipour M, Bininda-Emonds ORP, Moret BME, Stamatakis A. How many bootstrap replicates are necessary? J Comput Biol. 2010;17(3):337–354. doi: 10.1089/cmb.2009.0179. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Taxon sampling. Taxa included in this study, their GenBank accession numbers, original publications, and their higher taxonomy.
Genes sampled and missing data for each taxon. Information on taxa sampled for each gene included, and the percent of missing data for each taxon in each data set. Number of genes present per taxon and number of taxa present per gene are also given.
GC content for each taxon in the ntAll and ntNo3rd data sets as well as in the first, second, and third codon positions of the ntAll data set.
Fifty percent maximum likelihood majority-rule bootstrap consensus summary tree of Viridiplantae inferred from the first and second codon positions (ntNo3rd) analysis. See also Figure 6 for a summary tree of major Viridiplantae clades and Additional file 1 for taxonomy. Data set derived from 78 protein-coding genes of the plastid genome (ntax = 360, 38,898 bp, missing data ~15.6%,). Bootstrap support values ≥ 50% are indicated.
Fifty percent maximum likelihood majority-rule bootstrap consensus tree of Viridiplantae inferred from the RY-coded (RY) analysis. See also Figure 7 for a summary tree of major Viridiplantae clades and Additional file 1 for taxonomy. Data set derived from 78 protein-coding genes of the plastid genome (ntax = 360, 58,347 bp, missing data ~15.6%,). Bootstrap support values ≥ 50% are indicated.
Fifty percent maximum likelihood majority-rule bootstrap consensus tree of Viridiplantae inferred from the amino acid (AA) analysis. See also Figure 8 for a summary tree of major Viridiplantae clades and Additional file 1 for taxonomy. Data set derived from 78 protein-coding genes of the plastid genome (ntax = 360, 19,449 AAs, missing data ~15.6%,). Bootstrap support values ≥ 50% are indicated.
Maximum likelihood tree of Viridiplantae inferred from the all nucleotide positions (ntAll) analysis. Cladogram of the maximum likelihood bipartition tree is shown on the left with bootstrap values indicated above the branches. The phylogram of same tree is shown on the right. Data set derived from 78 protein-coding genes of the plastid genome (ntax = 360; 58,347 bp; missing data ~15.6%). Bootstrap support values ≥ 50% are indicated.
Maximum likelihood tree of Viridiplantae inferred from the first and second codon positions (ntNo3rd) analysis. Cladogram of the maximum likelihood bipartition tree is shown on the left with bootstrap values indicated above the branches. The phylogram of same tree is shown on the right. Data set derived from 78 protein-coding genes of the plastid genome (ntax = 360, 38,898 bp, missing data ~15.6%,). Bootstrap support values ≥ 50% are indicated.
Maximum likelihood tree of Viridiplantae inferred from the RY-coded (RY) analysis. Cladogram of the maximum likelihood bipartition tree is shown on the left with bootstrap values indicated above the branches. The phylogram of same tree is shown on the right. Data set derived from 78 protein-coding genes of the plastid genome (ntax = 360, 58,347 bp, missing data ~15.6%,). Bootstrap support values ≥ 50% are indicated.
Maximum likelihood tree of Viridiplantae inferred from the amino acid (AA) analysis. Cladogram of the maximum likelihood bipartition tree is shown on the left with bootstrap values indicated above the branches. The phylogram of same tree is shown on the right. Data set derived from 78 protein-coding genes of the plastid genome (ntax = 360, 19,449 AAs, missing data ~15.6%,). Bootstrap support values ≥ 50% are indicated.
Maximum likelihood tree of Viridiplantae inferred from the third codon position (nt3rdOnly) analysis. Cladogram of the maximum likelihood bipartition tree is shown on the left with bootstrap values indicated above the branches. The phylogram of same tree is shown on the right. Data set derived from 78 protein-coding genes of the plastid genome (ntax = 360, 19,449 bp, missing data ~15.6%,). Bootstrap support values ≥ 50% are indicated.