

Abstract
We are now witnessing a resurgence of theories of development and carcinogenesis in which the environment is again being accepted as a major player in phenotype determination. Perturbations in the fetal environment predispose an individual to disease that only becomes apparent in adulthood. For example, gestational exposure to diethylstilbestrol resulted in clear cell carcinoma of the vagina and breast cancer. In this review the effects of the endocrine disruptor bisphenol-A (BPA) on mammary development and tumorigenesis in rodents is used as a paradigmatic example of how altered prenatal mammary development may lead to breast cancer in humans who are also widely exposed to it through plastic goods, food and drink packaging, and thermal paper receipts. Changes in the stroma and its extracellular matrix led to altered ductal morphogenesis. Additionally, gestational and lactational exposure to BPA increased the sensitivity of rats and mice to mammotropic hormones during puberty and beyond, thus suggesting a plausible explanation for the increased incidence of breast cancer.
Keywords: xenoestrogen, progesterone receptor, beaded duct, ecological developmental biology, tissue organization field theory, neoplasia
Introduction
Perturbations in the fetal environment predispose individuals to disease that will only manifest in adulthood [1, 2]. This fact has prompted scientists to hypothesize that fetal exposure to environmental estrogens is an underlying cause of the increased incidence of uterine leiomyoma, testicular cancer and breast cancer observed in European and US populations over the last 50 years [3–5].
Estrogen levels in the fetal environment have long-term consequences regarding the risk of developing breast cancer during adult life [6–8]. Given the long latency period between exposure and effect, epidemiological studies designed to explore this hypothesis have used prenatal markers of in utero estrogen exposure because direct estrogen measurements are not available from birth records. Dizygotic twin pregnancy, which is associated with high estrogen levels, and pre-eclampsia, which is associated with low levels, were used as surrogates for high and low estrogen exposure, respectively. Dizygotic birth correlated with increased risk of breast cancer in the offspring while pre-eclampsia was associated with lowered risk [7, 9, 10]. Direct evidence of a link between prenatal estrogen exposure and breast cancer risk has been gathered from the cohort of women born to mothers treated with the potent synthetic estrogen diethylstilbestrol (DES) during pregnancy. DES was administered to women to prevent miscarriages. In 1971 the Food and Drug Administration (FDA) issued a Drug Bulletin advising physicians to stop prescribing DES to pregnant women because a rare neoplasm, clear cell adenocarcinoma of the vagina, was reported to occur in young women exposed in utero to DES [11, 12].
Breast cancer risk at 40 years of age and older is 2.5 fold higher in DES-exposed women than in unexposed women of the same age [13]. In rats, prenatal exposure to DES also resulted in increased mammary cancer incidence during adulthood when these animals were challenged with the chemical carcinogen dimethylbenzanthracene (DMBA) at puberty [14]. DES was administered to rats at pharmacological doses to mimic its medical use.
In addition to exposures to natural and pharmacological estrogens, there is the inadvertent and continuous exposure of human fetuses to endocrine disrupting chemicals (EDCs) released in the environment. EDCs like the herbicide atrazine, dioxins resulting from incineration and fuel combustion and the surfactant perfluorooctanoic acid delay mammary gland development in rodents as a result of gestational and lactational exposures [15–17]. Exposure to estrogens such as BPA and DES resulted in the long-term increase in the number of epithelial structures and the development of pre-cancerous and cancerous lesions in the mammary glands of rodents that manifested in adulthood [18, 19]. To date, BPA is the best-studied EDC and is the only one for which effects of exposure have been described at multiple time points spanning fetal development and postnatal life.
1. The complexity of endocrine disruptors
In the Statement of Principles from The Endocrine Society, an EDC is defined as “an exogenous chemical, or mixture of chemicals, that interferes with any aspect of hormone action” [20]. Several EDCs were found to have multiple hormonal activities. For example, the pesticide DDT is an estrogen agonist, while one of its metabolites is anti-androgenic [21]. BPA has estrogenic activity and is a thyroid hormone antagonist [22, 23]. Xenoestrogens are usually less potent than estradiol regarding their binding affinity to nuclear estrogen receptors (ER); effects manifesting at low doses are explained by the fact that they act additively with endogenous estrogens [24]. Xenoestrogens bind to plasma carrier proteins with significantly lower affinities than those of natural estrogens, and thus are more readily available to target organs [25]. Furthermore, some xenoestrogens like BPA produced low-dose effects by acting through ER located in the plasma membrane, including the ERα and β and the estrogen-binding protein GPR30 [26, 27].
1.1 Medical hypotheses derived from the concept of endocrine disruption
The “EDC hypothesis” was first proposed at the Wingspread Conference held in Racine, Wisconsin, in 1991 to explain the detrimental effects observed in exposed wildlife and the likelihood of harm to humans [28]. The proceedings from this conference [31] prompted medical hypotheses proposing that the increased incidence of uterine leiomyoma, testicular and breast cancer, of malformations of the male genital tract and of the decreased sperm quality observed in European populations over the previous fifty years were due to fetal exposure to EDCs [3, 5, 29].
2 Fetal origins of cancer: theoretical foundations
The hypothesis that gestational exposure to EDCs predisposes an individual to cancer, challenged the entrenched beliefs that mammalian development is the execution of a genetic program, and that only mutagenic agents can cause cancer [30, 31]. The theoretical bases of this novel perspective are briefly presented below.
2.1 Is development the unfolding of a program or an open ended process?
The adoption of animal models such as Drosophila, Xenopus, and rodents that reproduce all year long in laboratory conditions promoted a concentration on genetics with the exclusion of evolution and ecology from embryology; this approach plus the success of molecular biology led to the notion of a “developmental program” whereby genes had a privileged role on the determination of the phenotype; F. Jacob expressed this concept succinctly: “For it is during embryonic development that the instructions contained in the genetic program of an organism are expressed, that the genotype is converted into phenotype”[32]. The idea of a “program” is contested by i) gene splicing which challenges the one-to-one correlation between gene, mRNA and protein, a sine-qua-non of programmability, from gene to phenotype, and ii) stochastic gene expression, which is contrary to a Laplacian determination1 inherent in the notion of a program [33]. These arguments buttress the relevance of the environment in the expression of phenotypes, which is equally important to those of genes and other resources present in the embryo, and justifies why Medicine is now embracing the old tradition of ecological developmental biology [34].
2.2 Carcinogenesis: development gone awry?
For about a century, it has been assumed that carcinogenesis is a cell-based process caused by DNA mutations in a single founder cell and that those mutations cause uncontrolled cell proliferation [35]. These are the tenets of the somatic mutation theory (SMT) [36]. When it became evident that non-mutagenic agents also cause cancer, a “course correction” was proposed whereby changes in the epigenome play a central role through dysregulated cell proliferation [30, 37]. An alternative theory of carcinogenesis, i.e., the tissue organization field theory (TOFT), instead, places carcinogenesis at the tissue level of organization, a concept spanning embryology and pathology [38]. The TOFT posits that the persistence of morphogenetic fields throughout adult life orchestrate histogenesis and organogenesis before birth and tissue remodeling and regeneration during postnatal life [39]. From this perspective, carcinogens cause neoplastic development by altering the reciprocal interactions between the mesenchyme/stroma and the parenchyma in a tissue or organ.
Experiments were conducted to resolve the controversy of whether the target of the carcinogen was a cell in the rat mammary gland epithelium or the stroma of the gland [40]. Recombination of stroma exposed to a carcinogen with normal, unexposed epithelial cells resulted in neoplasms in the epithelium [40]. The reverse combination did not. This observation suggests that the stroma, rather than individual cells in the epithelium, is the target of carcinogens. Moreover, the cancer phenotype was reversed when mammary cancer epithelial cells were placed into a normal, unexposed mammary stroma [41]. Equally significant, normal mammary epithelial cells can also contribute to the normalization of cancerous epithelial cells [42]. These results point to the contextuality of the neoplastic phenotype and the centrality of stroma-epithelium interaction in carcinogenesis.
3. A model for fetal origin of cancer: The xenoestrogen BPA
BPA was described as a “synthetic estrogenic agent” in 1936 [43]; however, it was never used as an estrogen due to its low potency when compared to DES which synthesis and characterization was published in 1938 [44]. Twenty years later, polycarbonate plastics, which are made from BPA monomers, were introduced. Worldwide, approximately 3 million metric tons of BPA are produced per year [45]. Besides food and beverage polycarbonate containers, BPA is also used in some cash register receipts, medical devices, contact lenses and other consumer products. As a result, humans are routinely exposed, most likely throughout life [46].
BPA is present in most human tissues and fluids including blood, breast milk, and amniotic fluid, typically at levels of approximately 1ng/ml [47]. In this review we focus on the effects observed at doses that result in plasma levels below or equal to those found in humans; these doses are referred to as “low dose” [48]. The pharmacokinetics of BPA appear to be similar across mice, rats, non-human primates and humans [48].
3.1 Exposure to BPA alters fetal mammary gland morphogenesis
The mammary gland develops through complex reciprocal interactions between mesenchyme and epithelium [49, 50] (see also Propper et al in this volume). Based on our published experimental data showing morphological alterations in both compartments [51, 52] and that of others showing that estrogen receptors are only expressed in the stroma during fetal development [53, 54], we hypothesized that BPA, acting as an estrogen, would interfere with those reciprocal interactions. BPA was administered to mouse dams from gestational day (GD) 8 to GD18 through subcutaneous osmotic pumps delivering 0.25 μg/kg maternal bw/day (from here on μg/kg bw/day); this BPA dose is 1000-fold lower than that needed to produce levels comparable to those found in human plasma. Mammary glands of embryos at day 18 (E18) were examined. In the primary periductal mesenchyme, where nuclear ER α and β [54] and trans-membrane ER GPR30 are expressed [52], BPA altered extracellular matrix (ECM) organization [55]. Collagen fiber density increased in the stroma abutting the epithelium and decreased in the loose connective tissue further away. There was a marked decrease of tenascin C (TnC) in the periductal stroma (Figure 1) [52]. The number of cells containing lipid droplets in the periductal stroma increased, and accelerated adipocyte differentiation was also observed in the presumptive fat pad. Additionally, the cell number/area was significantly decreased in the presumptive fat pad and the proportion of Bax-positive cells was increased, suggesting increased apoptotic activity. Transcriptome analysis of the primary periductal stroma confirmed an increased expression of adipogenesis regulatory genes [PPARγ, low density lipoprotein receptor (Ldlr), G protein-coupled receptor 81 (GPR81), and Fabp4] while a decreased expression of ECM components, such as TnC, was observed [52].
Figure 1. Effects of exposure to 250ng BPA/kg/day from E8 to E18 on mice.

Trichrome stained 5 μm section of control (A, E) and BPA-exposed (B, F) E18 mammary glands (EP denotes epithelium). Collagen stains deep blue; note the reduction of collagen deposition in the mesenchyme distal to the epithelium. Immuno-histochemical localization of TnC in controls (C) and BPA-exposed (D) mammary glands. Red alkaline phosphatase staining of TnC is observed in the stroma surrounding the epithelial ducts. The density of collagen in the entire stromal compartment is significantly decreased in BPA-exposed females compared with controls, p=0.010 (G). However, the density of collagen within 10μm of the epithelial ducts is significantly increased in BPA females, p=0.042 (H). All scale bars represent 100 μm. (I) Quantification showed that the number of adipocytes was significantly increased within 1 mm from the developing epithelium in BPA-exposed females (squares), compared with controls (circles). **,P <0.02; ***, P < 0.005. Copyright 2007, The Endocrine Society, (A–F) and (I) reprinted with permission [55]
Within the epithelium, BPA increased the area subtended by the ductal tree, the ductal extension (distance from the nipple to the furthest point of growth) and delayed lumen formation [51]. At the transcriptome level, there was an increased expression of anti-apoptotic genes [52]. Because mammary gland development is dependent on reciprocal interactions between these compartments, the changes in the primary periductal stroma involving ECM and adipogenesis, as well as the advanced fat pad maturation, may be responsible for the altered growth and the delayed lumen formation recorded in the ducts (Figure 2).
Figure 2. Effects of exposure to 250ng BPA/kg/day on mice exposed from E8 to E18.
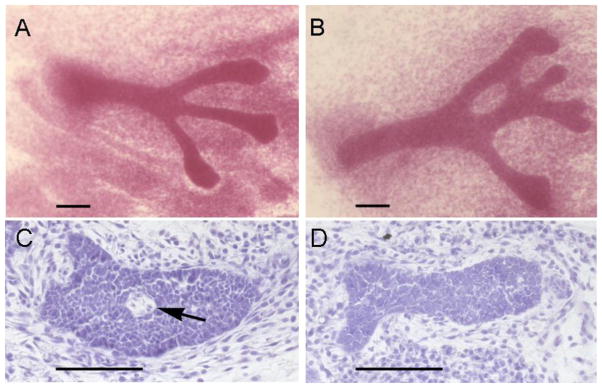
Carmine –stained whole mounts of E18 4th mammary gland of controls (A), and BPA exposed embryos (B). Note the increased size of the epithelial cords, compared with controls. Lumen formation in E18 mouse mammary glands is inhibited by BPA treatment. Lumen formation [arrow] was observed in 38% of control animals (C) but in none of the histological sections of animals exposed in utero to 250ng BPA/kg BW/day from E9(D). Scale bars represent 100μm. Copyright 2007, The Endocrine Society, reprinted with permission [55]
When the mammary glands were grouped by the fetal position in the uterus, significant differences became apparent between females positioned between two males (2M) and females placed between 2 females (0M), suggesting hormone mediation of this differential effect. In the control group, the epithelium of the 2M glands had more branches and larger area than the 0M. Treatment with BPA obliterated this difference; that is, the epithelium of the 0M glands of BPA exposed females subtended larger areas and branched more than the non-exposed 0M [55].
3.2 Does BPA act as an estrogen in the fetal mammary gland?
BPA displays estrogenic activity at low concentrations (1–100 nM) [26, 27] and interferes with thyroid hormone action at higher concentrations (100 nM-10 μM) [56]. In order to examine whether the effects of BPA were due to its estrogenic effect, the transcriptomes of the periductal stroma and the epithelium of mouse embryos exposed in utero from E8 to E19 to a reference estrogen, i.e., 10 ng ethinylestradiol/kg bw/day, were compared to those of embryos exposed to 0.25 μg BPA/kg bw/day [52]. The similarity of the E19 transcriptomes of these two compounds strongly suggests that BPA acts mainly as an estrogen [52]. Of note, the similarity of the transcription profiles of the two hormones is higher in the epithelium than the periductal stroma [52]. These observations suggest that the mesenchyme, which expresses both nuclear ERs [54] and GPR30 [52], responds to different estrogenic agents in distinct manners but “integrates” estrogenic effects of diverse substances into a common set of “instructions” for the epithelium.
3.3 Effects of perinatal BPA exposure manifest in adult life
The post-natal mammary glands of CD1 female mice exposed pre-natally (0.025 or 0.25 μg BPA/kg bw/day through osmotic pumps) had a significantly enhanced response to estrogens when administered after ovariectomy [57]. At post-natal day (PND) 30 the mammary glands of intact animals exposed pre-natally to BPA exhibited an increased number of terminal end buds (TEBs) relative to the ductal area [57], decreased apoptosis in the TEBs and an increased number of epithelial cells expressed progesterone receptor (PR) [57]. A study using C57BL/6 mice exposed through drinking water from conception to weaning found a dose dependent increase in TEBs and the mRNA expression levels of estrogen-regulated genes such as amphiregulin [58] and secretory leukoprotease inhibitor (unpublished observation, Brisken). At 3 months of age, a significant increase of the mammary cell number was observed in both the epithelial and the stromal compartments. There was an increase in the number of PR-positive cells in mammary epithelium at 6 and 12 months of age (Figure 3). The response to progesterone by mammary epithelial organoids obtained from 3 month-old animals exposed from conception to weaning to BPA was also increased when compared to unexposed controls [58].
Figure 3. BPA exposure increases the number of progesterone receptor positive cells as seen in sections stained with an antibody against PR.

Sections from an inguinal mammary gland of a mouse exposed from E8 to birth showing a cluster of PR positive cells at one month of age indicating a presumptive branching point (arrow) (A), Copyright 2005, The Endocrine Society reprinted with permission [57]; a control (B), and a mouse exposed to BPA from conception to weaning show a differential staining pattern of PR expression (C) Copyright 2011, The Endocrine Society reprinted with permission [58]. Note that the ducts of the mammary glands of BPA-exposed mice contain more PR-positive luminal epithelial cells than the unexposed controls
Progesterone increases lateral branching [59]. The increase in the proportion of epithelial cells expressing PR and their organization in clusters at 1 month of age was followed by a significant increase of lateral branching observed at 4 months of age in mice exposed gestationally to BPA [57] (Figure 4). At 6 months of age, there was an overall increase in epithelial structures including terminal ends and a premature appearance of alveolar buds which are normally associated with pregnancy in mice [60]. These findings are consistent with the persistence of an increased number of epithelial cells expressing PR at 6 and 12 months of age [58]. When gestational exposure was extended through the lactation period, mammary glands of the offspring of BPA-treated mothers had an increased volume fraction of alveolar buds at 3 and 9 months of age, and an increased volume fraction of ducts at 9 months of age; these changes occurred at doses of 0.25 μg BPA/kg bw/day. Animals exposed to 2.5 μg/kg bw/day also showed an increase in alveolar buds at 9 months of age [61]. Perinatal exposure to estrogens also induced intraductal hyperplasias, which in whole mounts is recognized by the appearance of “beaded ducts”. Animals exposed pre- and post-natally (up to day 16) to low doses of BPA ranging from 0.25 to 25 μg BPA/kg bw/day developed beaded ducts [61] (Figure 5).
Figure 4. Prenatal exposure to BPA results in increased branching in adulthood.
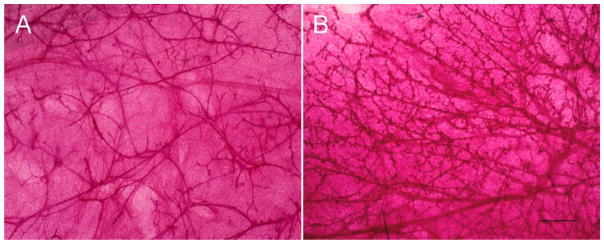
Photomicrographs of mammary gland whole mounts of 4 month old control mice (A) and mice exposed to 250ng BPA/kg bw/d from E8 to birth (B). Scale bar represents 1 mm.
Figure 5. Beaded ducts in whole-mounted mouse mammary glands from BPA-exposed offspring.

Mammary glands from control mice do not develop beaded ducts (A); glands from exposed females do. Panel B shows a mammary gland from a 9 month-old female exposed to BPA during perinatal development (E8 to PND21) containing beaded ducts. Beads are marked by arrowheads. Inset illustrates a higher magnification of a beaded duct. In comparison to a normal duct (C), confocal images of beaded ducts in BPA-exposed females (D) demonstrate the presence of cells obliterating the ductal lumen. Areas with cells inside the ductal lumen are marked by yellow arrows. All photographed mammary glands were stained with carmine alum. Magnification: A and B = 32x; C and D = 200x. Adapted from from Reproductive Toxicology [61] with permission from Elsevier. (E) Histology section of ductal contents stained with H&E.
In rats, as in mice, pre-natal exposure (from E10 to birth) to 250 μg BPA/kg bw/day via gavage resulted in an increase in the number of TEBs at PND21, an increased number of terminal ducts at PND 21 and 100, and an increased number of lobules type 1 at PND 35. Although morphological changes were only observed with the 250 μg/kg bw/day exposure, changes in gene expression were observed after exposure to both doses [62], albeit in a dose-dependent and age-dependent manner. The proteomic profile of the mammary glands at PND 21 identified 11 proteins differentially expressed in the BPA group when compared to controls [63]. At PND 50, 10 proteins were differentially expressed in the BPA animals compared to control; they included vimentin, adiponectin, desmin and the matricellular protein SPARC (secreted protein acidic and rich in cysteine) which affects ECM composition and collagen assembly [64]. In sum, BPA induces alterations in the mammary gland that are manifested regardless of route of exposure (oral vs. subcutaneous), the type of exposure (bolus vs. continuous), the timing of exposure (in utero vs. postnatal) and the species (mouse vs. rat). The outcomes can be either obvious, such as altered tissue architecture, or more subtle, such as differential gene and/or protein expression.
4. Exposure to BPA during organogenesis predisposes to mammary gland neoplasia
The main risk factor for breast cancer is lifetime exposure to ovarian hormones. Mammary glands of gestationally BPA-exposed mice showed an enhanced sensitivity to estradiol and to progesterone [57, 58]. BPA-exposed mice had an increased number of TEBs, TEB area, TEB density and ductal extension [65]. Intraductal hyperplasias, a pre-cancerous lesion that gives rise to mammary adenocarcinomas after transplantation into syngeneic mice were observed in female mice exposed from E8 through weaning to 0.25, 2.5 and 25 μg BPA/kg BW/day. These pre-cancerous lesions appeared in 3 month–old mice exposed to the lowest dose [61] and were characterized by the presence of epithelial cells growing inside the ductal lumen or even spanning the entire luminal diameter. The proliferation index of these cells was 5 times higher than that of cells in normal ducts. Interestingly, epithelial cells from alveolar buds adjacent to the hyperplastic ducts also had a high proliferation index. The stroma associated with intraductal hyperplasias showed macrophages and mast cells and it was high in fibrous collagen [61].
To explore the hypothesis that developmental exposure to low doses of BPA induces mammary neoplasias long after the exposure ended, we used a rat model because their mammary glands respond to carcinogens by developing tumors that better mimic the human breast disease regarding estrogen dependence and histopathology [66, 67]. BPA was administered to fetuses at doses ranging from 2.5 to 1000 μg/kg bw/day; this exposure resulted in the development of carcinomas in situ in the mammary glands of 33% of the rats exposed to 250 and 1000 μg BPA/kg bw/day2 while none of the unexposed animals developed carcinomas in situ [68]. Neoplasias were observed in young adult rats (PND 50 and 95). Fetal exposure to BPA also increased by 3–4 fold the number of intraductal hyperplasias [69]. Lesions in BPA-exposed animals were highly proliferative and contained numerous ERα positive cells [68, 70], suggesting that the proliferative activity in these lesions may be estrogen-mediated. As mentioned previously, rat mammary carcinomas as well as those in humans are predominantly estrogen-dependent, a feature that strengthens the relevance of these findings.
5. Is BPA a carcinogen?
According to the Environmental Protection Agency (EPA), a carcinogen is a chemical or physical agent capable of causing cancer [http://www.epa.gov/airtoxics/nata/gloss1.html], a definition that does not specify the mechanism(s) by which the cancer is induced or the time lapse between exposure and diagnosis; it only identifies the consequence of an insult. Thus, by this definition BPA is a carcinogen.
6. Linking existing data into an explanation of BPA-driven mammary carcinogenesis
In the embryo, BPA binds to ER present in the primary mesenchyme and induces changes in gene expression that lead to accelerated maturation of periductal adipocytes which in turn, accelerates ductal growth and branching [31, 51]. Simultaneous and profound changes in the primary periductal matrix composition and organization lead to alterations of its biomechanical properties, which in turn affect the growth and branching of the epithelium [71, 72]. Because ERs are not expressed in the embryonic mammary epithelium, the induction of anti-apoptotic genes (which mediate delayed lumen formation) suggests mediation by biochemical factors (morphogens) secreted by the carcinogen-damaged periductal stroma and/or by biomechanical factors [73] (Figure 6). Altered tissue development and remodeling of the mammary gland during each ovarian cycle may also contribute to neoplastic development. This remodeling activity is likely to be augmented due to the increased sensitivity to estradiol and progesterone observed in adult animals who were exposed to BPA from E8 to PND16 [57, 58]. In addition, morphological changes observed during adulthood such as increased abundance of alveolar buds and lobulo-alveolar structures suggest the presence of increased prolactin plasma levels and/or increased sensitivity to prolactin. BPA alters the architecture of hypothalamic nuclei [74], which may generate altered circulating hormone levels that would facilitate the development of intra-ductal hyperplasias and carcinomas in situ. The involvement of both the mammary gland and the hypothalamus is supported by the fact that the mammary gland phenotype is more severe in mice exposed pre- and postnatally to BPA (from E8 to PND 16) which spans a critical window for differentiation of cyclical vs. tonic gonadotropin release. Such exposed animals showed changes in a hypothalamic region essential for cyclic gonadotropin release [74] and altered estrous cycles [75, 76], which in turn, may abnormally affect the development and the remodeling of the mammary gland, thus exacerbating the observed neoplastic phenotypes of these animals.
Figure 6.
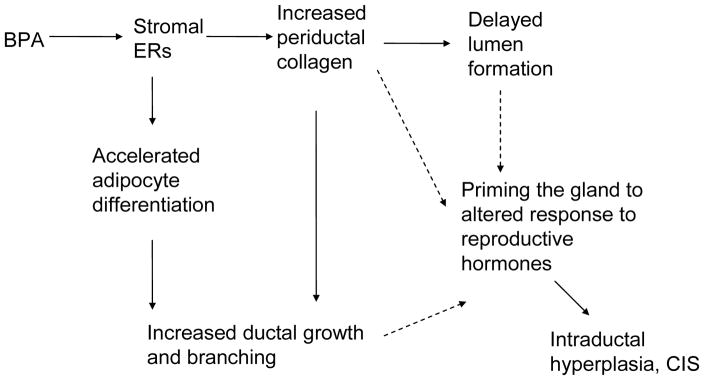
Proposed causal links tying BPA exposure, mammary gland development and carcinogenesis: BPA binds to the ERs present in the primary mesenchyme which alters the peri-ductal stroma, increasing peri-ductal collagen deposition and thus tissue rigidity. Increased rigidity is known to block or delay lumen formation. BPA also induces adipocyte differentiation in the primary periductal stroma and fat pad, which in turn causes increased duct elongation and branching. These changes lead to an increased sensitivity to mammotropic hormones such as estrogens and progesterone and likely to prolactin. The solid arrows link observations at E18 with postulated causal links. Dashed arrows link the observed effects at E18 with effects observed during puberty and adulthood. Not represented here are the effects of BPA on the hypothalamus, where it alters the control of ovarian cyclicity and likely the control of prolactin production.
CONCLUSIONS
The causal link between fetal exposure to estrogens and the development of breast cancer that was first suggested by epidemiologists has now been confirmed by the increased risk to develop breast cancer during adulthood of women exposed to DES during their fetal life. Fetal and neonatal exposures to EDCs cause persistent alterations in the mammary glands of rodents, including pre- and neoplastic lesions, long after the exposure ended. In the case of BPA, mammary neoplasias may have their origin in the altered mammary morphogenesis that occurs during fetal and neonatal exposure. The data obtained from laboratory animals support the extrapolation that exposure to BPA and other xenoestrogens during organogenesis in humans contributes to the increase in the incidence of breast cancer observed over recent decades.
Acknowledgments
We thank Tessie Paulose and Nicole Acevedo for their editorial assistance. This research was supported by The Avon Foundation grant #02-2009-093, and 02-2011-095 as well as by the National Institute of Environmental Health Sciences, Award Numbers R01ES08314, RC2ES018822 and U01ES020888. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Environmental Health Sciences or the National Institutes of Health.
Abbreviations
- EDC
Endocrine disrupting chemical
- BPA
Bisphenol-A
- DES
diethylstilbestrol
- FDA
United States Food and Drug Administration
- DMBA
dimethylbenzanthracene
- DDT
dichlorodiphenyltrichloroethane
- PCB
polychlorinated biphenyl
- SMT
somatic mutation theory
- TOFT
tissue organization field theory
- bw
bodyweight
- CDC
United States Centers for Disease Control and Prevention
- GD
Gestational day
- E
embryonic day
- ERs
Estrogen receptors
- PR
progesterone receptor
- PND
postnatal day
- ECM
extracellular matrix
- TEB
terminal end bud
- SPARC
secreted protein acidic and rich in cysteine
- EPA
United States Environmental Protection Agency
Footnotes
Laplacian determination is the one operating in computers where complete knowledge of the state at one moment completely determines its state at all future moments.
A dose of 250 μg/kg/day resulted in unconjugated BPA serum levels in the range of those found in humans (unpublished data, AMS and CS)
References
- 1.Sallout B, Walker M. The fetal origin of adult diseases. Journal of Obstetrics and Gynaecology. 2003;23:555–60. doi: 10.1080/0144361031000156483. [DOI] [PubMed] [Google Scholar]
- 2.Barker DJP, Hanson MA. Altered regional blood flow in the fetus: the origins of cardiovascular disease? Acta Paediatricia. 2004;93:1559–60. [PubMed] [Google Scholar]
- 3.Sharpe RM, Skakkebaek NE. Are oestrogens involved in falling sperm count and disorders of the male reproductive tract? Lancet. 1993;341:1392–95. doi: 10.1016/0140-6736(93)90953-e. [DOI] [PubMed] [Google Scholar]
- 4.Skakkebaek NE, Meyts ER, Jorgensen N, et al. Germ cell cancer and disorders of spermatogenesis: an environmental connection? APMIS. 1998;106:3–12. doi: 10.1111/j.1699-0463.1998.tb01314.x. [DOI] [PubMed] [Google Scholar]
- 5.Markey CM, Rubin BS, Soto AM, Sonnenschein C. Endocrine disruptors from Wingspread to environmental developmental biology. J Steroid Biochem Molec Biol. 2003;83:235–44. doi: 10.1016/s0960-0760(02)00272-8. [DOI] [PubMed] [Google Scholar]
- 6.Trichopoulos D. Is breast cancer initiated in utero? Epidemiology. 1990;1:95–96. [PubMed] [Google Scholar]
- 7.Braun MM, Ahlbom A, Floderus B, Brinton LA, Hoover RN. Effect of twinship on incidence of cancer of the testis, breast, and other sites (Sweden) CCC. 1995;6:519–24. doi: 10.1007/BF00054160. [DOI] [PubMed] [Google Scholar]
- 8.Ekbom A, Trichopoulos D, Adami HO, Hsieh CC, Lan SJ. Evidence of prenatal influences on breast cancer risk. Lancet. 1992;340:1015–18. doi: 10.1016/0140-6736(92)93019-j. [DOI] [PubMed] [Google Scholar]
- 9.Potischman N, Troisi R. In-utero and early life exposures in relation to risk of breast cancer. CCC. 1999;10:561–73. doi: 10.1023/a:1008955110868. [DOI] [PubMed] [Google Scholar]
- 10.Tamimi R, Lagiou P, Vatten LJ, et al. Pregnancy hormones, pre-eclampsia, and implications for breast cancer risk in the offspring. Cancer Epidemiol Biomarkers Prev. 2003;12:647–50. [PubMed] [Google Scholar]
- 11.Herbst AL, Ulfelder H, Poskanzer DC. Adenocarcinoma of the vagina: association of maternal stilbestrol therapy with tumor appearance in young women. New Engl J Med. 1971;284:878–81. doi: 10.1056/NEJM197104222841604. [DOI] [PubMed] [Google Scholar]
- 12.Mittendorf R. Teratogen update: carcinogenesis and teratogenesis associated with exposure to diethylstilbestrol (DES) in utero. Teratology. 1995;51:435–45. doi: 10.1002/tera.1420510609. [DOI] [PubMed] [Google Scholar]
- 13.Palmer JR, Hatch EE, Rosenberg CL, et al. Risk of breast cancer in women exposed to diethylstilbestrol in utero: preliminary results (United States) CCC. 2002;13:753–58. doi: 10.1023/a:1020254711222. [DOI] [PubMed] [Google Scholar]
- 14.Boylan ES, Calhoon RE. Transplacental action of diethylstilbestrol on mammary carcinogenesis in female rats given one or two doses of 7,12-dimethylbenz(a)anthracene. Cancer Res. 1983;43:4879–84. [PubMed] [Google Scholar]
- 15.Rayner JL, Enoch RR, Fenton SE. Adverse effects of prenatal exposure to atrazine during a critical period of mammary gland growth. Toxicol Sci. 2005;87:255–66. doi: 10.1093/toxsci/kfi213. [DOI] [PubMed] [Google Scholar]
- 16.White SS, Calafat AM, Kuklenyik Z, et al. Gestational PFOA exposure of mice is associated with altered mammary gland development in dams and female offspring. Toxicol Sci. 2007;96:133–44. doi: 10.1093/toxsci/kfl177. [DOI] [PubMed] [Google Scholar]
- 17.Fenton SE, Hamm JT, Birnbaum L, Youngblood GL. Persistent abnormalities in the rat mammary gland following gestational and lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) Toxicol Sci. 2002;67:63–74. doi: 10.1093/toxsci/67.1.63. [DOI] [PubMed] [Google Scholar]
- 18.Tomooka Y, Bern HA. Growth of mouse mammary glands after neonatal sex hormone treatment. J Nat Cancer Inst. 1982;69:1347–52. [PubMed] [Google Scholar]
- 19.Vandenberg LN, Maffini MV, Sonnenschein C, Rubin BS, Soto AM. Bisphenol-A and the great divide: A review of controversies in the field of endocrine disruption. Endocr Rev. 2009;30:75–95. doi: 10.1210/er.2008-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zoeller RT, Brown TR, Doan L, et al. Endocrine-dsrupting chemicals and public health protection: A statement of principles from The Endocrine Society. Endocrinology. 2012;153:4097–110. doi: 10.1210/en.2012-1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kelce WR, Stone CR, Laws SC, Gray LE, Kemppainen JA, Wilson EM. Persistent DDT metabolite p,p′-DDE is a potent androgen receptor antagonist. Nature. 1995;375:581–85. doi: 10.1038/375581a0. [DOI] [PubMed] [Google Scholar]
- 22.Moriyama K, Tagami T, Akamizu T, et al. Thyroid hormone action is disrupted by Bisphenol A as an antagonist. J Clin Endocrinol Metab. 2002;87:5185–90. doi: 10.1210/jc.2002-020209. [DOI] [PubMed] [Google Scholar]
- 23.Zoeller RT, Bansal R, Parris C. Bisphenol-A, an environmental contaminant that acts as a thyroid hormone receptor antagonist in vitro, increases serum thyroxine, and alters RC3/neurogranin expression in the developing rat brain. Endocrinology. 2005;146:607–12. doi: 10.1210/en.2004-1018. [DOI] [PubMed] [Google Scholar]
- 24.Silva E, Rajapakse N, Kortenkamp A. Something from “nothing” -- eight weak estrogenic chemicals combined at concentrations below NOECs produce significant mixture effects. Environ Sci Technol. 2002;36:1751–56. doi: 10.1021/es0101227. [DOI] [PubMed] [Google Scholar]
- 25.Nagel SC, vom Saal FS, Thayer KA, Dhar MG, Boechler M, Welshons WV. Relative binding affinity-serum modified access (RBA-SMA) assay predicts the relative in vivo bioactivity of the xenoestrogens bisphenol A and octylphenol. Environ Health Perspect. 1997;105:70–76. doi: 10.1289/ehp.9710570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watson CS, Bulayeva NN, Wozniak AL, Alyea RA. Xenoestrogens are potent activators of nongenomic estrogenic responses. Steroids. 2007;72:124–34. doi: 10.1016/j.steroids.2006.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soriano S, Alonso-Magdalena P, Garcia-Arevalo M, et al. Rapid insulinotropic action of low doses of Bisphenol-A on mouse and human Islets of Langerhans: Role of estrogen receptor beta. PLoS ONE. 2012;7:e31109. doi: 10.1371/journal.pone.0031109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chemically induced alterations in sexual and functional development: the wildlife/human connection. Princeton: Princeton Scientific Publishing; [Google Scholar]
- 29.Davis DL, Bradlow HL, Wolff M, Woodruff T, Hoel DG, Anton-Culver H. Medical hypothesis: xenoestrogens as preventable causes of breast cancer. Environ Health Perspect. 1993;101:372–77. doi: 10.1289/ehp.93101372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keri RA, Ho S-M, Hunt PA, Knudsen KE, Soto AM, Prins GS. An evaluation of evidence for the carcinogenic activity of bisphenol A. Reproductive Toxicology. 2007;24:240–252. doi: 10.1016/j.reprotox.2007.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Soto AM, Sonnenschein C. Environmental causes of cancer: endocrine disruptors as carcinogens. Nat Rev Endocrinol. 2010;6:363–70. doi: 10.1038/nrendo.2010.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jacob F. The Possible and the Actual. Seattle, WA: University of Washington Press; [Google Scholar]
- 33.Longo G, Miquel P-A, Sonnenschein C, Soto AM. Is information a proper observable for biological organization? Prog Biophys Mol Biol. 2012;109:108–14. doi: 10.1016/j.pbiomolbio.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gilbert SF, Epel D. Ecological Developmental Biology: Integrating Epigenetics, Medicine, and Evolution. Sunderland MA: Sinauer Associates; [Google Scholar]
- 35.Weinberg RA. One renegade cell: how cancer begins. New York: Basic Books; [Google Scholar]
- 36.Vaux DL. In defense of the somatic mutation theory of cancer. BioEssays. 2011;33:341–43. doi: 10.1002/bies.201100022. [DOI] [PubMed] [Google Scholar]
- 37.Ho S-M, Tang WY, Belmonte de Frausto J, Prins GS. Developmental exposure to estradiol and bisphenol a increases susceptibility to prostate carcinogenesis and epigenetically regulates phosphodiesterase type 4 variant 4. Cancer Res. 2006;66:5624–32. doi: 10.1158/0008-5472.CAN-06-0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soto AM, Sonnenschein C. The somatic mutation theory of cancer: growing problems with the paradigm? BioEssays. 2004;26:1097–107. doi: 10.1002/bies.20087. [DOI] [PubMed] [Google Scholar]
- 39.Sonnenschein C, Soto AM. The Society of Cells: Cancer and Control of Cell Proliferation. New York: Springer Verlag; [Google Scholar]
- 40.Maffini MV, Soto AM, Calabro JM, Ucci AA, Sonnenschein C. The stroma as a crucial target in rat mammary gland carcinogenesis. J Cell Sci. 2004;117:1495–502. doi: 10.1242/jcs.01000. [DOI] [PubMed] [Google Scholar]
- 41.Maffini MV, Calabro JM, Soto AM, Sonnenschein C. Stromal regulation of neoplastic development: Age-dependent normalization of neoplastic mammary cells by mammary stroma. Am J Pathol. 2005;167:1405–10. doi: 10.1016/S0002-9440(10)61227-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Booth BW, Boulanger CA, Anderson LH, Smith GH. The normal mammary microenvironment suppresses the tumorigenic phenotype of mouse mammary tumor virus-neu-transformed mammary tumor cells. Oncogene. 2011;30:679–89. doi: 10.1038/onc.2010.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dodds EC, Lawson W. Synthetic estrogenic agents without the phenanthrene nucleus. Nature. 1936;137:996. [Google Scholar]
- 44.Dodds EC, Goldberg L, Lawson W, Robinson R. Estrogenic activity of certain synthetic compounds. Nature. 1938;141:247–48. [Google Scholar]
- 45.Burridge E. Chemical profile: bisphenol A. ICIS Chemical Business. 2008;274:48. [Google Scholar]
- 46.Vandenberg LN, Hauser R, Marcus M, Olea N, Welshons WV. Human exposure to bisphenol A (BPA) Reproductive Toxicology. 2007;24:139–77. doi: 10.1016/j.reprotox.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 47.Vandenberg LN, Chauhoud I, Heindel JJ, Padmanabhan V, Paumgartten FJ, Schoenfelder G. Urinary, circulating and tissue biomonitoring studies indicate widespread exposure to Bisphenol A. Environ Health Perspect. 2010;118:1055–70. doi: 10.1289/ehp.0901716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Taylor JA, vom Saal FS, Welshons WV, et al. Similarity of bisphenol A pharmacokinetics in rhesus monkeys and mice: relevance for human exposure. Environ Health Perspect. 2011;119:422–30. doi: 10.1289/ehp.1002514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Veltmaat JM, Mailleux AA, Thiery JP, Bellusci S. Mouse embryonic mammogenesis as a model for the molecular regulation of pattern formation. Differentiation. 2003;71:1–17. doi: 10.1046/j.1432-0436.2003.700601.x. [DOI] [PubMed] [Google Scholar]
- 50.Cowin P, Wysolmerski J. Molecular mechanisms guiding embryonic mammary gland development. Cold Spring Harb Perspect Biol. 2010;2:a003251. doi: 10.1101/cshperspect.a003251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vandenberg LN, Maffini MV, Wadia PR, Sonnenschein C, Rubin BS, Soto AM. Bisphenol A exposure alters fetal mammary gland development. Endocrinology Society Meeting P1–177. 2006:205. [Google Scholar]
- 52.Wadia PR, Cabaton NJ, Borrero MD, Rubin BS, Sonnenschein C, Shioda T, Soto AM. Low-dose BPA exposure alters the mesenchymal and epithelial transcriptomes of the mouse fetal mammary gland. PLoS One. 2013 doi: 10.1371/journal.pone.0063902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Narbaitz R, Stumpf WE, Sar M. Estrogen receptors in the mammary gland primordia of fetal mouse. Anat Embryol. 1980;158:161–66. doi: 10.1007/BF00315903. [DOI] [PubMed] [Google Scholar]
- 54.Lemmen JG, Broekhof JLM, Kuiper GGJM, Gustafsson JA, Van Der Saag PT, van der Burg B. Expression of estrogen receptor alpha and beta during mouse embryogenesis. Mech Dev. 1999;81:163–67. doi: 10.1016/s0925-4773(98)00223-8. [DOI] [PubMed] [Google Scholar]
- 55.Vandenberg LN, Maffini MV, Wadia PR, Sonnenschein C, Rubin BS, Soto AM. Exposure to environmentally relevant doses of the xenoestrogen bisphenol-A alters development of the fetal mouse mammary gland. Endocrinology. 2007;148:116–27. doi: 10.1210/en.2006-0561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zoeller RT. Environmental chemicals impacting the thyroid: targets and consequences. Thyroid. 2007;17:811–17. doi: 10.1089/thy.2007.0107. [DOI] [PubMed] [Google Scholar]
- 57.Munoz de Toro MM, Markey CM, Wadia PR, et al. Perinatal exposure to Bisphenol A alters peripubertal mammary gland development in mice. Endocrinology. 2005;146:4138–47. doi: 10.1210/en.2005-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ayyanan A, Laribi O, Schuepbach-Mallepell S, et al. Perinatal exposure to bisphenol a increases adult mammary gland progesterone response and cell number. Mol Endocrinol. 2011;25:1915–23. doi: 10.1210/me.2011-1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brisken C, Park S, Vass T, Lydon JP, O’Malley BW, Weinberg RA. A paracrine role for the epithelial progesterone receptor in mammary gland development. Proc Nat Acad Sci USA. 1998;95:5076–81. doi: 10.1073/pnas.95.9.5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Markey CM, Luque EH, Munoz de Toro MM, Sonnenschein C, Soto AM. In utero exposure to bisphenol A alters the development and tissue organization of the mouse mammary gland. Biol Reprod. 2001;65:1215–23. doi: 10.1093/biolreprod/65.4.1215. [DOI] [PubMed] [Google Scholar]
- 61.Vandenberg LN, Maffini MV, Schaeberle CM, et al. Perinatal exposure to the xenoestrogen bisphenol-A induces mammary intraductal hyperplasias in adult CD-1 mice. Reproductive Toxicology. 2008;26:210–219. doi: 10.1016/j.reprotox.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moral R, Wang R, Russo IH, Lamartiniere CA, Pereira J, Russo J. Effect of prenatal exposure to the endocrine disruptor bisphenol A on mammary gland morphology and gene expression signature. J Endocrinol. 2008;196:101–12. doi: 10.1677/JOE-07-0056. [DOI] [PubMed] [Google Scholar]
- 63.Betancourt AM, Mobley JA, Russo J, Lamartiniere CA. Proteomic analysis in mammary glands of rat offspring exposed in utero to bisphenol A. J Proteomics. 2010;73:1241–53. doi: 10.1016/j.jprot.2010.02.020. [DOI] [PubMed] [Google Scholar]
- 64.Bradshaw AD. The role of SPARC in extracellular matrix assembly. J Cell Commun Signal. 2009;3:239–46. doi: 10.1007/s12079-009-0062-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wadia PR, Vandenberg LN, Schaeberle CM, Rubin BS, Sonnenschein C, Soto AM. Perinatal Bisphenol-A exposure increases estrogen sensitivity of the mammary gland in diverse mouse strains. Environ Health Perspect. 2007;115:592–98. doi: 10.1289/ehp.9640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Singh M, McGinley JN, Thompson HJ. A comparison of the histopathology of premalignant and malignant mammary gland lesions induced in sexually immature rats with those occurring in the human. Lab Invest. 2000;80:221–31. doi: 10.1038/labinvest.3780025. [DOI] [PubMed] [Google Scholar]
- 67.Nandi S, Guzman R, Yang J. Hormones and mammary carcinogenesis in mice, rats, and humans: a unifying hypothesis. Proc Nat Acad Sci USA. 1995;92:3650–3657. doi: 10.1073/pnas.92.9.3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Murray TJ, Maffini MV, Ucci AA, Sonnenschein C, Soto AM. Induction of mammary gland ductal hyperplasias and carcinoma in situ following fetal Bisphenol A exposure. Reproductive Toxicology. 2007;23:383–90. doi: 10.1016/j.reprotox.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Medina D. The preneoplastic phenotype in murine mammary tumorigenesis. J Mammary Gland Biol Neoplasia. 2000;5:393–407. doi: 10.1023/a:1009529928422. [DOI] [PubMed] [Google Scholar]
- 70.Durando M, Kass L, Piva J, et al. Prenatal bisphenol A exposure induces preneoplastic lesions in the mammary gland in Wistar rats. Environ Health Perspect. 2007;115:80–86. doi: 10.1289/ehp.9282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dhimolea E, Maffini MV, Soto AM, Sonnenschein C. The role of collagen reorganization on mammary epithelial morphogenesis in a 3D culture model. Biomaterials. 2010;31:3622–30. doi: 10.1016/j.biomaterials.2010.01.077. [DOI] [PubMed] [Google Scholar]
- 72.Krause S, Jondeau-Cabaton A, Dhimolea E, Soto AM, Sonnenschein C, Maffini MV. Dual regulation of breast tubulogenesis using extracellular matrix composition and stromal cells. Tissue Eng Part A. 2012;18:520–532. doi: 10.1089/ten.tea.2011.0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Paszek MJ, Weaver VM. The tension mounts: mechanics meets morphogenesis and malignancy. J Mammary Gland Biol Neoplasia. 2004;9:325–42. doi: 10.1007/s10911-004-1404-x. [DOI] [PubMed] [Google Scholar]
- 74.Rubin BS, Lenkowski JR, Schaeberle CM, Vandenberg LN, Ronsheim PM, Soto AM. Evidence of altered brain sexual differentiation in mice exposed perinatally to low environmentally relevant levels of bisphenol A. Endocrinology. 2006;147:3681–91. doi: 10.1210/en.2006-0189. [DOI] [PubMed] [Google Scholar]
- 75.Rubin BS, Murray MK, Damassa DA, King JC, Soto AM. Perinatal exposure to low doses of bisphenol-A affects body weight, patterns of estrous cyclicity and plasma LH levels. Environ Health Perspect. 2001;109:675–80. doi: 10.1289/ehp.01109675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Markey CM, Coombs MA, Sonnenschein C, Soto AM. Mammalian development in a changing environment: exposure to endocrine disruptors reveals the developmental plasticity of steroid-hormone target organs. Evolution and Development. 2003;5:1–9. doi: 10.1046/j.1525-142x.2003.03011.x. [DOI] [PubMed] [Google Scholar]