

Abstract
Statins are widely prescribed for lowering plasma low-density lipoprotein (LDL) concentrations and cardiovascular disease risk1, but there is considerable interindividual variation in treatment response2,3 and increasing concern regarding the potential for adverse effects, including myopathy4 and type 2 diabetes5. Despite evidence for substantial genetic influence on LDL concentrations6, pharmacogenomic trials have failed to identify genetic variations with large effects on either statin efficacy7-9 or toxicity10, and have yielded little information regarding mechanisms that modulate statin response. Here we identify a downstream target of statin treatment by screening for the effects of in vitro statin exposure on genetic associations with gene expression levels in lymphoblastoid cell lines derived from 480 participants of a clinical trial of simvastatin treatment7. This analysis identified six expression quantitative trait loci (eQTLs) that interacted with simvastatin exposure including rs9806699, a cis-eQTL for the gene GATM that encodes glycine amidinotransferase, a rate-limiting enzyme in creatine synthesis. We found this locus to be associated with incidence of statin-induced myotoxicity in two separate populations (meta-analysis odds ratio = 0.60, 95% confidence interval = 0.45-0.81, P=6.0×10-4). Furthermore, we found that GATM knockdown in hepatocyte-derived cell lines attenuated transcriptional response to sterol depletion, demonstrating that GATM may act as a functional link between statin-mediated cholesterol lowering and susceptibility to statin-induced myopathy.
Analyzing individual variation in transcriptional response to drug treatment has been successful in identifying regulatory genetic variants that interact with treatment in model organisms11 and human tissues12-15. Cellular transcriptional analysis may be particularly useful for investigating genetic influences on statin efficacy, since statin-induced plasma LDL lowering is controlled through sterol-response element binding protein (SREBP)–mediated transcriptional regulation16. Therefore, to identify novel regulatory variants that interact with statin exposure, we conducted a genome-wide eQTL analysis based on comparing simvastatin- versus control-exposure of 480 lymphoblastoid cell lines (LCLs) derived from European American participants in the Cholesterol and Pharmacogenetics (CAP) trial. LCLs have proven to be a useful model system for the study of genetic regulation of gene expression17,18. Although non-genetic sources of variation, if uncontrolled, may limit the utility of LCLs for transcriptional perturbation analyses19,20, there has been increasing use of these cells to screen for genetic variants associated with molecular response to drug intervention20. Furthermore, many features of statin-mediated regulation of cholesterol metabolism are operative in LCLs21.
Simvastatin exposure had a significant effect on gene expression levels for 5,509 of 10,195 expressed genes (54%, false discovery rate (FDR)<0.0001). The magnitude of change in expression across all responsive genes was small (0.12±0.08 mean absolute log2 change±SD, Fig. 1) with 1,952 genes exhibiting ≥10% change in expression and only 21 genes exhibiting ≥50% change in expression. Among the strongest responders were 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR), which encodes the direct target of simvastatin inhibition (0.49±0.29 mean log2 change±SD, P<0.0001, N=480), and low density lipoprotein receptor (LDLR), which encodes the receptor responsible for internalization of LDL particles (0.50±0.35 mean log2 change±SD, P<0.0001). As expected, surface expression of the LDLR protein was also increased following simvastatin exposure (1.6±0.11 mean log2 change±SD, P<0.0001, N=474). Gene set enrichment analysis showed a treatment-dependent increase in expression of genes involved in steroid biosynthesis, consistent with the mechanism responsible for the lipid-lowering response to statin, and a decrease in expression of genes involved in RNA splicing, consistent with evidence for statin regulation of alternative splicing of genes involved in cellular cholesterol homeostasis22 (Supplementary Fig. 1).
Figure 1. Simvastatin treatment alters transcript expression in LCLs.
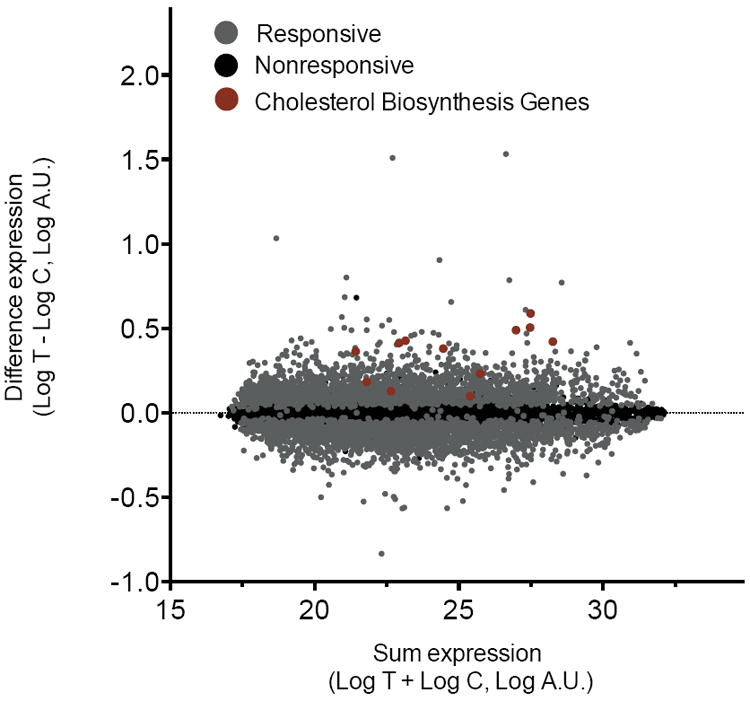
Log change in expression following simvastatin- and control-exposed lymphoblastoid cell lines (n=480) displayed as a function of log sum of expression traits. Grey: genes for which expression was significantly changed in response to simvastatin exposure (N=5509/10105, 0.12±0.08 mean absolute log2 change±SD, q<0.0001); Black: genes for which expression was not significantly changed (N=4686). Red: genes in the cholesterol biosynthesis pathway, all of which exhibited significant changes in expression.
We first identified eQTLs without considering whether they interact with simvastatin exposure. We computed Bayes factors (BFs)23 to quantify evidence for association between every single nucleotide polymorphism (SNP) and the expression level of each gene, and we used permutations to estimate FDRs (see Methods). This analysis identified 4590 genes with cis-eQTLs, defined as eQTLs within 1Mb of the gene’s transcription start or end site (FDR=1%, log10BF≥3.24, Supplementary Table 1). Statistical power to detect eQTLs was substantially increased by controlling for known covariates and unknown confounders (represented by principal components of the gene expression data24,25) and by testing for association with expression traits averaged across paired simvastatin- and control-exposed samples to reduce measurement error (Supplementary Table 2 and Supplementary Fig. 2). Our analysis also identified 98 trans-eQTLs at the same stringent FDR (FDR=1%, log10BF≥7.20, Supplementary Table 3).
To identify eQTLs that interact with simvastatin exposure (i.e., eQTLs with different effects in control- versus simvastatin-exposed samples, or differential eQTLs; deQTLs), we used two approaches14: i) univariate association mapping of log fold expression change between paired control- and simvastatin-exposed samples; ii) bivariate association mapping of paired control- and simvastatin-exposed samples. This bivariate approach aims to improve power and interpretability by explicitly distinguishing among different modes of interaction (see Methods), which the univariate approach does not distinguish. The univariate approach identified cis-deQTLs for four genes: GATM, RSRC1, VPS37D, and OR11L1 (FDR=20%, log10BF≥4.9, Supplementary Table 4 and 5). No trans-deQTLs were identified at an FDR of 20%, so trans analyses were not further pursued (see Supplementary Table 6 for top trans-deQTLs). The bivariate approach identified cis-deQTLs for six genes (FDR=20%, log10BF≥5.1; Supplementary Tables 4 and 7, Supplementary Fig. 3 and Supplementary Data), including two genes not identified in the univariate analysis: ATP5SL and ITFG2. Both GATM and VPS37D had significantly stronger eQTL associations under simvastatin-exposed conditions in comparison to control, whereas the other four genes had significantly stronger eQTL associations under control-exposed conditions (Fig. 2a, Supplementary Table 4 and Supplementary Fig. 3). As in similar studies12-14,17, we found many fewer deQTLs than stable eQTLs, or SNPs with similar effects across both conditions. The finding of relatively few gene by exposure interactions, and of relatively modest effect sizes of those interactions, appears remarkably consistent across studies regardless of method (including family-based comparisons), exposure, sample size, sample source, or number of stable eQTLs detected. We focus further analysis on our most significant differential association from the bivariate model, the GATM locus, for which we observed stronger evidence for eQTL association following statin exposure and for which there was evidence for biological relevance to pathways involved in lipoprotein metabolism and myopathy (see Supplementary data).
Figure 2. Treatment-specific QTL associated with GATM expression.
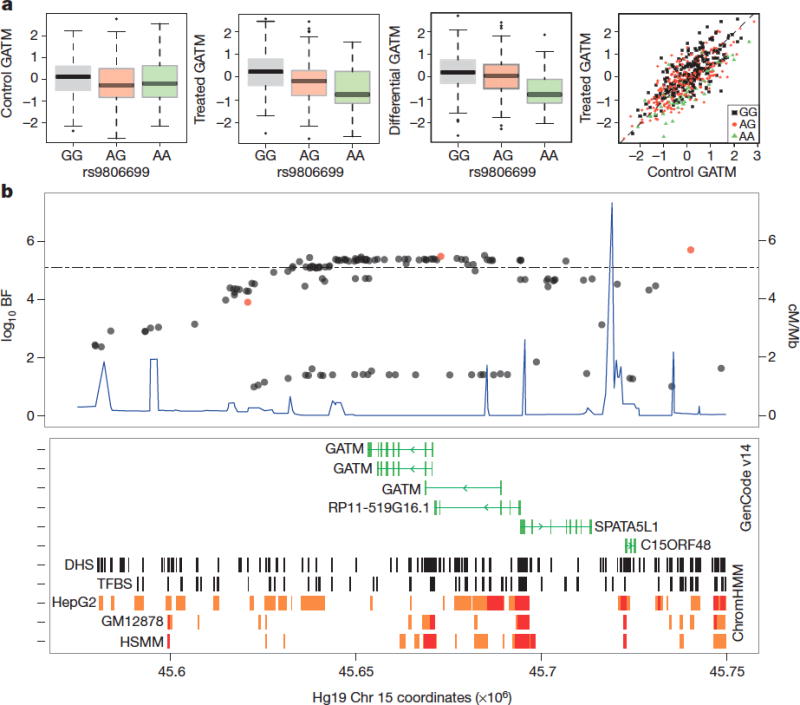
(a) Association of rs9806699 with quantile normalized GATM expression levels following (i) control exposure (not significant); (ii) simvastatin exposure (log10BF=5.1, effect size = -0.43). (iii), fold change (log10BF=5.7, effect size = -0.40); (iv), control versus simvastatin-exposed GATM expression (black: GG, N=225; red: GA, N=207; green: AA, N=48). Box height and whiskers described in Supplemental Methods. (b) SNPs associated with GATM expression (log10BF, left y-axis); SNPs associated with statin-induced myopathy (red), significance threshold (dotted line) recombination rates (blue, right y-axis); Bottom panel: transcribed genes (green), DNAse I hypersensitive (DHS) sites and transcription factor binding sites (TFBS; black), predicted chromosomal enhancers (orange) and promoters (red) as identified in hepatocyte (HepG2), lymphoblastoid (GM12878) and myocyte (HSMM) cell lines.
GATM encodes glycine amidinotransferase, an enzyme required for synthesis of creatine. We observed evidence for deQTL association with GATM (log10BF>5.1) across a group of 51 SNPs within the GATM locus that are in linkage disequilibrium (chr15: 45627979-45740392, hg19, r2= 0.85 – 0.99, N=587). The most significant deQTL association was observed with SNP rs9806699 (MAF=0.32), for which we observed stronger evidence for an association with GATM expression following simvastatin exposure (log10BF = 5.1, effect size= -0.43) than following control exposure (log10BF=0.52, effect size = -0.17, Fig. 2a). SNPs at this locus also had a stable association with expression of a neighboring gene, SPATA5L1 (deQTL rs9806699 log10BF = -0.33, stable eQTL rs9806699 log10BF=21.75, Supplementary Fig. 4). This locus has been shown previously to be associated with reduced glomerular filtration rate (GFR)26 with a small effect size (<1%). This association was specific to GFR as estimated from plasma creatinine but not from a second biomarker of renal function (e.g., cystatin C), suggesting that the association was related to variation in creatinine production rather than renal elimination. We found evidence for SNP differential association with GATM that spans the GATM coding region and includes multiple SNPs located within DNAse I hypersensitive sites, active promoters as well as several alternative GATM transcription start sites (Fig. 2b).
Phosphorylation of creatine, the primary downstream product of GATM activity, is a major mechanism for energy storage in muscle and is mediated by creatine kinase, the primary plasma biomarker of statin-induced myopathy. To test the relationship of this locus with statin-induced myotoxicity, we examined the association of the GATM deQTL locus with statin-induced myopathy in a population-based cohort comprised of 72 cases of myopathy and 220 matched controls (Marshfield cohort)27. In this cohort, we observed that the minor allele at the GATM deQTL locus was associated with reduced incidence of statin-induced myopathy (odds ratio=0.61, 95% Confidence Interval (CI)=0.39-0.95, P=0.03; Table 1). This association replicated in a second cohort consisting of 100 cases of myopathy identified within the Study of Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH)10 (odds ratio for rs1719247 = 0.61, CI=0.42-0.88, P=0.01; r2=0.70 to rs9806699; Table 1). Meta-analysis of these two cohorts showed an overall odds ratio of 0.60 (CI=0.45-0.81, P=6.0×10-4, log10BF=1.5, Table 1). Because myopathy is defined in part through elevation in plasma creatine kinase concentrations, we also tested for a direct association of this locus with this enzyme in statin-treated populations in which myopathy was not observed. Within CAP (40mg/d simvastatin exposure for 6 weeks), no association of rs9806699 was observed with plasma creatine kinase either before simvastatin exposure (N=575, P=0.83) or following exposure (N=574, P=0.48). This lack of association was confirmed in a second statin study (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin, or JUPITER, trial, 20mg/d rosuvastatin, median follow-up=1.9 years, NCT00239681) both prior to rosuvastatin exposure (N=8504, P=0.54) and following treatment (N=3052, P=0.83)3. These findings suggest that the observed association of the GATM locus with risk for statin-induced myopathy is independent of an association with plasma creatine kinase. While the present studies do not address the mechanism for the link between reduced GATM expression and protection from statin-induced myopathy, it is thought that diminished capacity for phosphocreatine storage modifies cellular energy storage and adenosine monophosphate-activated protein kinase (AMPK) signaling28,29 in a manner that is protective against cellular stress as induced by glucose deprivation29 or, potentially, by cholesterol depletion. Given that myocellular creatine stores are predominantly derived from renal and hepatic creatine biosynthesis, these results raise the possibility that statins may predispose to muscle toxicity in part through metabolic effects in the liver, the major site of statin’s pharmacologic actions (Supplementary Fig. 5). On the other hand, the finding of severe myopathy in two cases of extreme genetic GATM deficiency30 suggests that this protective effect may be overcome if creatine synthesis is insufficient to support myocellular energy needs.
Table 1.
Associations of SNPs at GATM locus with statin-induced myopathy.
| N, Cases | N, Controls | SNP | Position | LD (r2) | MAF, Cases | MAF, Controls | Effect size | P value | |
|---|---|---|---|---|---|---|---|---|---|
| Marshfield | 72 | 220 | rs9806699 | Chr15: 43,527,684 | 1.0 | 0.21 | 0.30 | 0.61 (0.39-0.95) | 3.2×10-2 |
| 72 | 220 | rs1719247 | Chr15: 43,408,027 | 0.76 | 0.19 | 0.29 | 0.59 (0.36-0.93) | 2.4×10-2 | |
| 72 | 220 | rs1346268 | Chr15: 43,460,321 | 0.80 | 0.21 | 0.29 | 0.66 (0.41-1.02) | 6.4×10-2 | |
| SEARCH | 100 | 4021 | rs1719247 | Chr15: 43,408,027 | 0.70 | 0.17 | 0.25 | 0.61 (0.42-0.88) | 1.0×10-2 |
| 100 | 4029 | rs1346268 | Chr15: 43,460,321 | 0.74 | 0.18 | 0.26 | 0.62 (0.43-0.90) | 1.0×10-2 | |
| Meta-analysis | rs1719247 | Chr15: 43,408,027 | 0.18 | 0.25 | 0.60 (0.45-0.81) | 7.0×10-4 | |||
| rs1346268 | Chr15: 43,460,321 | 0.19 | 0.26 | 0.63 (0.48-0.84) | 1.8×10-3 |
LD, linkage disequilibrium with respect to top deQTL SNP, rs9806699 based on Pearson correlation (r2). deQTL associations with GATM expression in CAP were: log10BF=6.22 (rs9806699), log10BF=4.35 (rs1719247), and log10BF=5.96 (rs1346268). All SNPs were in Hardy-Weinberg equilibrium in these populations. Effect size reported as odds ratio with 95% confidence interval.
Given the influence of statin exposure on regulation of GATM expression, we next tested whether GATM may modulate sterol-mediated changes in cholesterol homeostasis. Knockdown of GATM in hepatocyte-derived cell lines (HepG2 and Huh7) resulted in reduced upregulation of SREBP-responsive genes (HMGCR, LDLR, and SREBP2) by sterol depletion (Fig. 3a). Moreover, GATM knockdown decreased media accumulation of apoB, the major structural protein of LDL, in both cell lines (p<0.05; Fig. 3b), but did not alter levels of apoAI, the major structural protein in high density lipoproteins (HDL, Fig. 3b). An effect of GATM deficiency on cholesterol and lipoprotein metabolism is further supported by a recent study describing reduced plasma cholesterol concentrations in GATM knockout mice28.
Figure 3. GATM knockdown attenuated sterol-mediated induction in expression of SREBP-responsive genes.
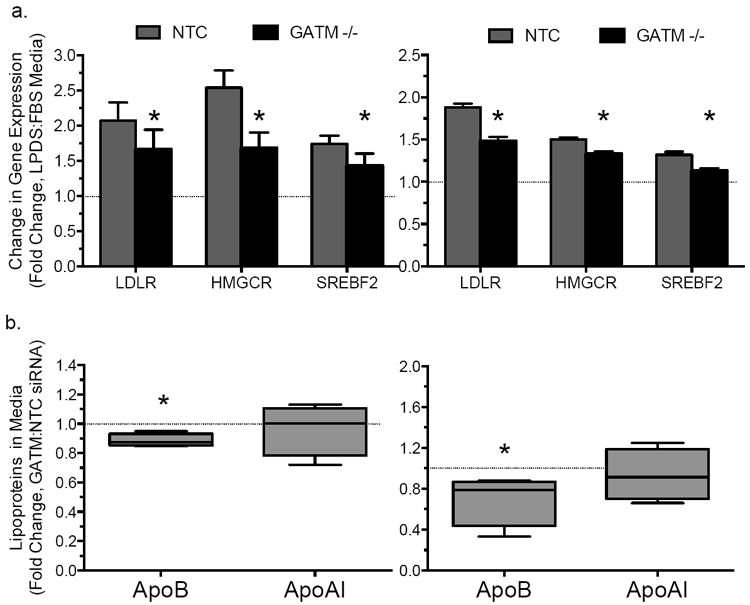
(a) Changes in transcript concentrations following sterol depletion via 24-hr exposure to lipoprotein deficient serum (LPDS)-containing media vs. standard FBS-containing media in hepatocyte-derived HepG2 (left, N=12) and Huh7 (right, N=12) cell lines. Asterisk indicates P<0.05 for the comparison of GATM versus NTC siRNA treated cells. (b) Fold changes in media accumulation of apolipoprotein B (ApoB) and apolipoprotein AI (ApoAI) following gene knockdown with GATM versus (NTC) siRNA in HepG2 cells (left, N=6-10) or Huh7 (right, N=4-6) cells under standard culture conditions. Error bars represent SEM.
In summary, this study has provided evidence that functionally significant genetic effects can be discovered using a novel cell-based screen for gene-by-treatment effects on transcriptional expression. This approach has led to the identification of GATM as a genetic locus associated with statin-induced myopathy, and as a potential link between cellular cholesterol homeostasis and energy metabolism.
Online-only Methods
In vitro simvastatin exposure of lymphoblastoid cell lines
Lymphoblastoid cell lines (LCLs), immortalized by Epstein-Barr virus transformation of lymphocytes isolated from whole blood31, were derived from European-American participants in the CAP trial, a six-week 40mg/day simvastatin trial (Supplementary Table 8)2. Simvastatin was provided by Merck Inc. (Whitehouse Station, NJ), converted to active form (beta-hydroxy simvastatin acid, SVA) and quantified by liquid chromatography-tandem mass spectrometry as described21. LCLs were normalized to a uniform cell density and exposed to 2μM SVA (simvastatin-exposed) or control buffer (control-exposed) for twenty-four hours as described21. This concentration was selected by assessing dose-response effects on expression profiles (n=8 LCLs at 4 doses), wherein a more robust change in expression profiles was observed with 2μM simvastatin exposure (7.8% of genes, q=0.001) than lower doses (<0.1% of genes for 0.02μM or 0.2μM, q=0.001, data not shown). Pre-experiment cell density was recorded as a surrogate for cell growth rate. Following exposure, cells were lysed in RNAlater (Ambion), and RNA was isolated using the Qiagen miniprep RNA isolation kit with column DNAse treatment.
Expression profiling and differential expression analysis
RNA quality and quantity were assessed by Nanodrop ND-1000 spectrophotometer and Agilent bioanalyzer, respectively. Paired RNA samples, selected based on RNA quality and quantity, were amplified and biotin labeled using the Illumina TotalPrep-96 RNA amplification kit, hybridized to Illumina HumanRef-8v3 beadarrays (Illumina), and scanned using an Illumina BeadXpress reader. Data were read into GenomeStudio and samples were selected for inclusion based on quality control criteria: (1) signal to noise ratio (95th:5th percentiles), (2) matched gender between sample and data, and (3) average correlation of expression profiles within three standard deviations of the within-group mean (r=0.99±0.0093 for control-exposed and r=0.98±0.0071 for simvastatin-exposed beadarrays). In total, viable expression data were obtained from 1040 beadarrays including 480 sets of paired samples for 10195 genes. Genes were annotated through biomaRt from ensMBL Build 54 (http://may2009.archive.ensemble.org/biomart/martview). Treatment specific effects were modeled from the data following adjustment for known covariates using linear regression32. False discovery rates were calculated for differentially expressed transcripts using qvalue33. Ontological enrichment in differentially expressed gene sets was measured using GSEA (1000 permutations by phenotype) using gene sets representing Gene Ontology biological processes as described in the Molecular Signatures v3.0 C5 Database (10-500 genes/set)34.
Expression QTL mapping
For association mapping, we use a Bayesian approach23 implemented in the software package BIMBAM35 that is robust to poor imputation and small minor allele frequencies36. Gene expression data were normalized as described in the Supplementary Methods for the control-treated (C480) and simvastatin-treated (T480) data and used to compute D480 = T480 - C480 and S480 = T480 + C480, where T480 is the adjusted simvastatin-treated data and C480 is the adjusted control-treated data. SNPs were imputed as described in the Supplementary Methods. To identify eQTLs and deQTLs, we measured the strength of association between each SNP and gene in each analysis (control-treated, simvastatin-treated, averaged, and difference) using BIMBAM with default parameters35. BIMBAM computes the Bayes factor (BF) for an additive or dominant response in expression data as compared with the null, which is that there is no correlation between that gene and that SNP. BIMBAM averages the BF over four plausible prior distributions on the effect sizes of additive and dominant models. We used a permutation analysis (see Supplementary Methods) to determine cutoffs for eQTLs in the averaged analysis (S480) at an FDR of 1% for cis-eQTLs (log10 BF > 3.24) and trans-eQTLs (log10 BF > 7.20). For cis-eQTLs, we considered the largest log10BF above the cis-cutoff for any SNP within 1MB of the transcription start site or the transcription end site of the gene under consideration. For trans-eQTLs, we considered the largest log10BF above the trans-cutoff for any SNP, and if that SNP was in the cis-neighborhood of the gene being tested, we ignored any potential trans-associations; there were 6130 for which the SNP with the largest log10BF was not in cis with the associated gene. Correspondingly, we only considered those 6130 genes when computing the permutation-based FDR for the trans-associations.
Differential expression QTL mapping
We define cis-SNPs as being within 1 Mb of the transcription start site or end site of that gene. To identify differential eQTLs, we first computed associations between all SNPs and the log fold change using BIMBAM as above.
We then considered a larger set of models for differential eQTLs. The associations for the genes in Supplementary Fig. 3 indicate that there are a few possible patterns of differential association. While these patterns may have different mechanistic or phenotypic interpretations, they are not distinguished by a test of log fold change. We used the interaction models introduced in Maranville et al.14 to compute the statistical support (assessed with Bayes factors, or BFs) for the four alternative eQTL models described in Results versus the null model (no association with genotype). These methods are based on a bivariate normal model for the treated data (T) and control-treated data (U). Note that simply quantile transforming T and U to a standard normal distribution is not sufficient to ensure that they are jointly bivariate normal, and so we employed the following more extensive normalization procedure. Let D = qT-qU and S = qT+qU, where q indicates that the vector following it has been quantile normalized. We then quantile normalize and scale D and S to produce S = (σSqS) and D = (σDqD), where σS, σD are robust estimates of the standard deviations of S and D respectively (specifically, they are the median absolute deviation multiplied by 1.4826). Note that this transformation ensures that S and D are univariate normal. Further, they are approximately independent which ensures that they are also bivariate normal. Finally let U = ½ (S − D) and T = ½ (S + D).
The BF when the eQTL effect is identical in the two conditions (model 1) uses the linear model L(S ~ D + g), where g is the vector of genotypes at a single SNP. The BF when the eQTL is only present in the control-treated samples (model 2) uses the model L(U ~ T + g). The BF when the eQTL is only present in the simvastatin-treated samples (model 3) uses the model L(T ~ U + g). The BF when the eQTL effect is in the same direction but unequal in strength (model 4) uses the model L(D ~ S + g). We averaged each BF for each gene and each cis-SNP over four plausible effect size priors (0.05, 0.1, 0.2, 0.4).
To find eQTLs that interact with treatment (i.e., conform best to one of the differential models 2-4, rather than the null model or the stable model) we defined an interaction Bayes factor (IBF) as IBF = 2(BF2 + BF3 + BF4) / 3(BF1+1), where BFi denotes the BF for model i compared with the null model (the 1 in the denominator represents the null model BF0). Large values of the IBF represent strong support for at least one interaction model (2-4) compared with the two non-interacting models (0-1), and hence strong support for a differential association.
Association with statin-induced myopathy
Marshfield Cohort31: Cases of myopathy were identified from electronic medical records of patients treated at the Marshfield Clinic (Wisconsin, USA) using a combination of automated natural language processing and manual review as described27. 72 cases of incipient myopathy (creatine kinase concentrations > 3-fold normal with evidence in the charts of muscle complaints) were identified for which patients were not also undergoing treatment with concomitant drugs known to increase incidence of statin-induced myopathy (fibrates or niacin). Controls were matched based on statin exposure, age and gender. This study was approved by the Marshfield Clinic institutional review board. The study population included residents living in Central and Northern Wisconsin, served by the Marshfield Clinic, a large multispecialty group practice.27 SEARCH and Heart Protection Study Collaborative Groups10,38: A total of 100 myopathy cases were identified from participants with genotyping data in the SEARCH trial, including 39 definite myopathy cases (creatine kinase >10 × ULN with muscle symptoms) and 61 incipient myopathy cases (defined as creatine kinase ≥5.0 times baseline value and alanine transaminase ≥1.7 times baseline value and creatine kinase >3.0 × ULN). Genotypes were available from the Illumina Human610-Quad Beadchip for 25 myopathy cases (12% of which had definite myopathy) and from the Illumina HumanHap300-Duo BeadChip for 75 myopathy cases (48% of which had definite myopathy). Genotypes for rs9806699 were only available in individuals genotyped on the Illumina Human610-Quad Beadchip so proxy SNPs were used. All myopathy cases were compliant with statin therapy (95 myopathy cases occurred whilst the patient was taking simvastatin 80mg daily, and 5 cases whilst taking simvastatin 20mg daily). Controls were identified from the SEARCH Study and the Heart Protection Study as well as from the Heart Protection Study (where considerably more participants had been genotyped). Controls from the Heart Protection Study had similar baseline characteristics to those in the SEARCH Study and inclusion of this large number of additional controls improved statistical power. Multicentre ethics approval was obtained from the South East Research Ethics Committee for the SEARCH study, and from the local ethics committees covering each of the 69 UK hospitals involved in the Heart Protection Study. Genetic associations were determined by chi-squared analysis using an additive model. Meta-analysis was performed using a random effects model and, for Bayesian analysis, considering an expected effect size to 0.2. Associations of rs9806699 with plasma creatine kinase in the CAP2 and JUPITER3 trials were also assessed using linear regression. The CAP trial (ClinicalTrials.gov number, NCT00451828) was approved by the institutional review boards located at Children’s Hospital Oakland Research Institute (Oakland, CA) and all enrollment sites. The JUPITER trial (ClinicalTrials.gov number, NCT00239681) was approved by the Institutional Review Board of Brigham and Women’s Hospital. Informed consent was obtained from all participants in all trials.
Functional analysis of candidate genes
GATM knockdown was achieved by 48-hour transfection of Ambion Silence Select siRNA or non-targeting control into 80,000 HepG2 or Huh7 cells/well in 12-well plates. To assess the influence of sterol depletion, cell culture media was replaced with media containing 10% lipoprotein deficient serum (Hyclone) or fetal bovine serum (Omega Scientific) at 24-hr transfection. All samples were harvested 48-hr post-transfection. Transcript levels were quantified by qPCR and normalized to CLPTM. Cell culture media was collected from all samples at time of harvest, and ApoB (MP Biomedicals), ApoAI (Meridian Life Sciences), and ApoE (Biodesign) were quantified in triplicate by sandwich-style ELISA. Samples with a coefficient of variation greater than 15% were subjected to repeat measurement.
Supplementary Material
Acknowledgments
This project was funded by a grant from the National Institutes of Health, U01 HL69757. BE was funded through the Bioinformatics Research Development Fund, supported by Kathryn and George Gould and NIH K99/R00 HG006265. MS was additionally funded by HG002585. We acknowledge the efforts of Terrie Kitchner and Ravi Mareedu for case validation in the Marshfield cohort. SEARCH was supported by the Medical Research Council, British Heart Foundation, National Health Service Genetic Knowledge Park, Centre National de Génotypage and Merck. The Heart Protection Study was funded by grants from the Medical Research Council, British Heart Foundation, Roche Vitamins and Merck. JCH acknowledges support from the BHF Centre of Research Excellence, Oxford. Genetic analysis in JUPITER was funded by a grant from AstraZeneca to DIC and PMR.
Footnotes
Author Contributions L.M.M. designed experiment and analyses, generated samples, performed analyses, and wrote the manuscript. B.E.E. designed and performed analyses and wrote the manuscript. C.D.B. performed analyses of ENCODE data. B.H.M. designed and performed correlation analyses. J.D.S., M.J.R., and D.A.N. generated expression and genotype data. M.W.M. and D.N. designed, performed and analyzed functional experiments. B.H. and H.S. developed and performed the imputation methodology, R.A.W, Q.F, J.D.S, M.J.R. and D.A.N collected and genotyped the myopathy cohort from the Marshfield clinic and performed association analyses, J.C.H., S.P, J.A. and R.C. collected and genotyped myopathy cohort from the SEARCH consortium and performed association analyses in that cohort along with the Heart Protection Study. J.I.R. and Y.I.C. measured creatine kinase in CAP. D.I.C. and P.M.R. measured creatine kinase and performed related analyses in JUPITER. M.S. supervised, designed, and contributed to analyses and participated in manuscript development. R.M.K. supervised the project and participated in experimental design and manuscript development.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
The gene expression data has been deposited in the Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE36868 and in Synapse (synapse.sagebase.org) under accession number syn299510. Code and analytical output complementary to this analysis are also provide through Synapse at: https://www.synapse.org/#!Synapse:syn299510. The genotype data has been deposited in the database for genotypes and phenotypes (dbGaP, http://www.ncbi.nlm.nih.gov/gap) under accession number phs000481. The full set of eQTLs identified in our study (log10BF > 1.0) is available at http://eqtl.uchicago.edu.
References
- 1.Baigent C, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–78. doi: 10.1016/S0140-6736(05)67394-1. [DOI] [PubMed] [Google Scholar]
- 2.Simon J, et al. Phenotypic predictors of response to simvastatin therapy among African-Americans and Caucasians: the Cholesterol and Pharmacogenetics (CAP) Study. The American Journal of Cardiology. 2006;97:843–50. doi: 10.1016/j.amjcard.2005.09.134. [DOI] [PubMed] [Google Scholar]
- 3.Ridker PM, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. The New England Journal of Medicine. 2008;359:2195–207. doi: 10.1056/NEJMoa0807646. [DOI] [PubMed] [Google Scholar]
- 4.Fernandez G, Spatz ES, Jablecki C, Phillips PS. Statin myopathy: a common dilemma not reflected in clinical trials. Cleveland Clinic journal of medicine. 2011;78:393–403. doi: 10.3949/ccjm.78a.10073. [DOI] [PubMed] [Google Scholar]
- 5.Rajpathak SN, et al. Statin therapy and risk of developing type 2 diabetes: a meta-analysis. Diabetes care. 2009;32:1924–9. doi: 10.2337/dc09-0738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Teslovich TM, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–13. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barber MJ, et al. Genome-wide association of lipid-lowering response to statins in combined study populations. PLoS One. 2010;5:e9763. doi: 10.1371/journal.pone.0009763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chasman DI, et al. Pharmacogenetic study of statin therapy and cholesterol reduction. JAMA : the Journal of the American Medical Association. 2004;291:2821–7. doi: 10.1001/jama.291.23.2821. [DOI] [PubMed] [Google Scholar]
- 9.Trompet S, et al. Replication of LDL GWAs hits in PROSPER/PHASE as validation for future (pharmaco)genetic analyses. BMC Medical Genetics. 2011;12:131. doi: 10.1186/1471-2350-12-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Link E, et al. SLCO1B1 variants and statin-induced myopathy--a genomewide study. The New England journal of medicine. 2008;359:789–99. doi: 10.1056/NEJMoa0801936. [DOI] [PubMed] [Google Scholar]
- 11.Brem RB, Storey JD, Whittle J, Kruglyak L. Genetic interactions between polymorphisms that affect gene expression in yeast. Nature. 2005;436:701–3. doi: 10.1038/nature03865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grundberg E, et al. Global analysis of the impact of environmental perturbation on cis-regulation of gene expression. PLoS Genetics. 2011;7:e1001279. doi: 10.1371/journal.pgen.1001279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Romanoski CE, et al. Systems genetics analysis of gene-by-environment interactions in human cells. American Journal of Human Genetics. 2010;86:399–410. doi: 10.1016/j.ajhg.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maranville JC, et al. Interactions between glucocorticoid treatment and cis-regulatory polymorphisms contribute to cellular response phenotypes. PLoS Genetics. 2011;7:e1002162. doi: 10.1371/journal.pgen.1002162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smirnov DA, et al. Genetic variation in radiation-induced cell death. Genome research. 2011:332–9. doi: 10.1101/gr.122044.111. at http://www.ncbi.nlm.nih.gov/pubmed/21844125. [DOI] [PMC free article] [PubMed]
- 16.Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–40. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 17.Morley M, et al. Genetic analysis of genome-wide variation in human gene expression. Nature. 2004;430:743–7. doi: 10.1038/nature02797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stranger BE, et al. Population genomics of human gene expression. Nature Genetics. 2007;39:1217–24. doi: 10.1038/ng2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caliskan M, Cusanovich DA, Ober C, Gilad Y. The effects of EBV transformation on gene expression levels and methylation profiles. Human Molecular Genetics. 2011;20:1643–52. doi: 10.1093/hmg/ddr041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choy E, et al. Genetic analysis of human traits in vitro: drug response and gene expression in lymphoblastoid cell lines. PLoS Genetics. 2008;4:e1000287. doi: 10.1371/journal.pgen.1000287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mangravite LM, et al. Combined influence of LDLR and HMGCR sequence variation on lipid-lowering response to simvastatin. Arterioscler Thromb Vasc Biol. 2010;30:1485–92. doi: 10.1161/ATVBAHA.110.203273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Medina MW, et al. Coordinately regulated alternative splicing of genes involved in cholesterol biosynthesis and uptake. PloS One. 2011;6:e19420. doi: 10.1371/journal.pone.0019420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stephens M, Balding DJ. Bayesian statistical methods for genetic association studies. Nature Reviews Genetics. 2009;10:681–90. doi: 10.1038/nrg2615. [DOI] [PubMed] [Google Scholar]
- 24.Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genetics. 2007;3:1724–35. doi: 10.1371/journal.pgen.0030161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Innocenti F, et al. Identification, replication, and functional fine-mapping of expression quantitative trait Loci in primary human liver tissue. PLoS Genetics. 2011;7:e1002078. doi: 10.1371/journal.pgen.1002078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Köttgen A, et al. Multiple loci associated with indices of renal function and chronic kidney disease. Nature genetics. 2009;41:712–7. doi: 10.1038/ng.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mareedu RK, et al. Use of an electronic medical record to characterize cases of intermediate statin-induced muscle toxicity. Preventive cardiology. 2009;12:88–94. doi: 10.1111/j.1751-7141.2009.00028.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choe C, et al. L-arginine:glycine amidinotransferase deficiency protects from metabolic syndrome. Human molecular genetics. 2013;22:110–23. doi: 10.1093/hmg/dds407. [DOI] [PubMed] [Google Scholar]
- 29.Ide T, et al. GAMT, a p53-inducible modulator of apoptosis, is critical for the adaptive response to nutrient stress. Molecular cell. 2009;36:379–92. doi: 10.1016/j.molcel.2009.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Edvardson S, et al. l-arginine:glycine amidinotransferase (AGAT) deficiency: clinical presentation and response to treatment in two patients with a novel mutation. Molecular Genetics and Metabolism. 2010;101:228–32. doi: 10.1016/j.ymgme.2010.06.021. [DOI] [PubMed] [Google Scholar]
- 31.Pressman S, Rotter JI. Epstein-Barr virus transformation of cryopreserved lymphocytes: prolonged experience with technique. American Journal of Human Genetics. 1991;49:467. [PMC free article] [PubMed] [Google Scholar]
- 32.Mecham BH, Nelson PS, Storey JD. Supervised normalization of microarrays. Bioinformatics. 2010;26:1308–15. doi: 10.1093/bioinformatics/btq118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Storey JD. A direct approach to false discovery rates. Journal of the Royal Statistical Society: Series B (Statistical Methodology) 2002;64:479–498. [Google Scholar]
- 34.Subramanian A, Kuehn H, Gould J, Tamayo P, Mesirov JP. GSEA-P: a desktop application for Gene Set Enrichment Analysis. Bioinformatics. 2007;23:3251–3. doi: 10.1093/bioinformatics/btm369. [DOI] [PubMed] [Google Scholar]
- 35.Servin B, Stephens M. Imputation-based analysis of association studies: candidate regions and quantitative traits. PLoS Genetics. 2007;3:e114. doi: 10.1371/journal.pgen.0030114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guan Y, Stephens M. Practical Issues in Imputation-Based Association Mapping. PLoS Genetics. 2008;4 doi: 10.1371/journal.pgen.1000279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilke R, et al. Identifying genetic risk factors for serious adverse drug reactions: current progress and challenges. Nature reviews. Drug discovery. 2007;6:904–16. doi: 10.1038/nrd2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hopewell JC, et al. Impact of common genetic variation on response to simvastatin therapy among 18 705 participants in the Heart Protection Study. European heart journal. 2012 doi: 10.1093/eurheartj/ehs344. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.