

Abstract
Context
Hereditary vitamin D resistant rickets (HVDRR), also known as vitamin D-dependent rickets type II, is an autosomal recessive disorder characterized by the early onset of rickets with hypocalcemia, secondary hyperparathyroidism and hypophosphatemia and is caused by mutations in the vitamin D receptor (VDR) gene. The human gene encoding the VDR is located on chromosome 12 and comprises eight coding exons and seven introns.
Objectives, Patients, and Methods
We analyzed the VDR gene of 5 previously unreported patients, two from Singapore and one each from Macedonia (former Yugoslav Republic), Saudi Arabia and Turkey. Each patient had clinical and radiographic features of rickets, hypocalcemia, and the 4 cases that had the measurement showed elevated serum concentrations of 1,25-dihydroxyvitamin D (1,25(OH)2D). Mutations were re-created in the WT VDR cDNA and examined for 1,25(OH)2D3-mediated transactivation in COS-7 monkey kidney cells.
Results
Direct sequencing identified four novel mutations and two previously described mutations in the VDR gene. The novel mutations included a missense mutation in exon 3 causing the amino acid change C60W; a missense mutation in exon 4 causing the amino acid change D144N; a missense mutation in exon 7 causing the amino acid change N276Y; and a 2 bp deletion in exon 3 5’-splice site (IVS3Δ+4–5) leading to a premature stop.
Conclusions
These 4 unique mutations add to the previous 45 mutations identified in the VDR gene in patients with HVDRR.
Keywords: Vitamin D, rickets, hypocalcemia, mutations, vitamin D receptor, HVDRR
INTRODUCTION
The biological actions of 1,25-dihydroxyvitamin D3 [1,25(OH)2D3] including regulation of calcium homeostasis, cellular differentiation, and immune function, are mediated by the vitamin D receptor (VDR) a member of the steroid/nuclear receptor superfamily of ligand activated transcription factors [1, 2]. Inherited or spontaneous mutations in the VDR gene cause hereditary vitamin D resistant rickets (HVDRR), also known as vitamin D dependent rickets type II (VDDR II) [3–5]. Patients with HVDRR exhibit a constellation of features including early onset rickets, hypocalcemia, secondary hyperparathyroidism and hypophosphatemia. Some HVDRR patients also have total body alopecia [6]. Patient’s with HVDRR have significantly elevated serum levels of 1,25(OH)2D (written without the subscript when it connotes D3 and/or D2). Elevated 1,25(OH)2D distinguishes HVDRR from patients with 1α-hydroxylase deficiency, also known as vitamin D dependent rickets type I (VDDR I). 1α-Hydroxylase deficiency is caused by mutations in the CYP27B1 gene that have absent to low serum levels of 1,25(OH)2D [7]. Several types of heterogeneous genetic abnormalities have been found in the VDR gene mainly missense mutations, nonsense mutations and splicing mutations [3]. Mutations in the DNA binding domain (DBD) of the VDR interfere with VDR-DNA interactions but not ligand-binding and result in loss of VDR function and are usually associated with alopecia [8–12]. Mutations in the VDR ligand binding domain (LBD) variously alter the ligand binding affinity, affect heterodimerization with RXR, or inhibit coactivator interactions. These LBD mutations may result in partial or total hormone unresponsiveness and may or may not have alopecia [13–17]. In this report, we identify several novel mutations in the VDR in five patients with HVDRR and summarize the reported mutations in this disease.
MATERIALS AND METHODS
Informed consent
Informed consents were obtained by the local physicians from the patients and parents for minors under a Stanford University IRB approved protocol.
Cell culture
Dermal skin fibroblasts from patient 2 and normal control fibroblasts were grown in DMEM containing 4.5 g glucose, 10 mM sodium pyruvate and 10% fetal bovine serum (FBS) at 37°C in an atmosphere of 5% CO2/95% air.
Gene amplification and DNA sequencing
Exons 2–9 of the VDR gene from the patient’s and parent’s DNA were amplified by PCR and directly sequenced at the Stanford Protein and Nucleic Acid core lab.
Site-Directed Mutagenesis
Site-directed mutagenesis of the WT VDR cDNA in pSG5 was performed using QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies, La Jolla, CA). Clones were sequenced to confirm the presence of the mutation.
Real-time RT-PCR
Fibroblasts from patient 2 and a normal control were treated for 24 hr with vehicle (0.1% ethanol) or 0.1 to 1000 nM 1,25(OH)2D3. RNA was isolated using RNeasy Plus Mini Kit (Qiagen, Valencia, CA). cDNA was prepared using Maxima Universal First Strand cDNA Synthesis kit (Fisher Scientific, Pittsburg, PA). Real time PCR was performed using the DyNAmo ColorFlash qPCR kit (Fisher Scientific). Relative changes in mRNA expression were assessed by the 2-ΔΔC(T) method and normalized to that of the reference gene glyceraldehyde phosphate dehydrogenase (GAPDH). Assays were performed in triplicate. The primers used were CYP24A1 (forward) 5’-GGCTCTTTGTTGGATTGTCC, CYP24A1 (reverse) 5’-AAACCAGCAGTGAACCCTGT and GAPDH (forward) 5’-GAAGGTGAAGGTCGGAGTCA, GAPDH (reverse) 5’-GATCTCGCTCCTGGAAGATG.
Transactivation Assays and Immunoblotting
Transactivation assays and immunoblotting were performed as previously described [18]. Luciferase activities were determined using the Dual Luciferase Assay (Promega) and a Turner Design luminometer (Turner Design, Sunnyvale, CA). Samples from the transactivation assays were denatured in LDS-sample buffer for 10 min at 70°C and electrophoresed on 10% NuPAGE gels in MOPSSDS running buffer (Invitrogen). Proteins were transferred to nitrocellulose and incubated with a mouse anti- VDR monoclonal antibody (D-6, Santa Cruz Biotechnology, Santa Cruz, CA).
RESULTS
Case Reports
Initial lab data and uniform common normal ranges for all 5 subjects are shown in Table 1 where the values are all converted to similar units. In the case reports individual lab tests and the normal ranges are mentioned in parentheses for the different hospitals.
Table 1.
Summary of Initial Lab Data and Mutations in the Five HVDRR Patients
| info | ~nl range*/units | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 |
|---|---|---|---|---|---|---|
| gender | male | male | male | female | male | |
| origin | Saudi | Macedonia | Chinese | Indian | Turkish | |
| consanguinity | yes | no | no | yes | yes | |
| alopecia | no | no | no | yes | yes | |
| ionized Ca | 1.2–1.38 mmol/L | 0.85 | 1.11 | |||
| total Ca | 2.2–2.6 mmol/L | 1.27 | 1.79 | 1.85 | 2.01 | 1.6 |
| phos | 1.36–2.26 mmol/L | 0.92 | 0.86 | 0.78 | 0.63 | 1.25 |
| alk phos | 120–300 IU/L | 1541 | 1506 | 1057 | 2150 | 704 |
| PTH | 10–65 pg/mL | 507 | 578 | 434 | 301 | 193 |
| 25D | 10–50 ng/mL | 39 | 10.3 | 39.2 | 185 | |
| 1,25D | 15–60 pg/mL | 912 | 372 | 320 | 190 | |
| mutation | D144N | R158C | R80Q/N276Y | 2bp deletion | C60W | |
| Recreated mutation response | ↓33-fold | ↓33-fold | ↓76-fold | no activity | ||
| Transactivation of Pt’s cells | no response | |||||
The normal ranges vary from lab to lab.
Patient 1 is a Saudi Arabian boy. His parents originate from the same tribe indicating likely consanguinity even though they are not close relatives. This is their second born child. His older brother died at the age of 22 months with severe rickets and recurrent pneumonias that resulted in respiratory failure. The patient was diagnosed with rickets at the age of 11 months and was given calcium and vitamin D supplementation. He did not respond to this treatment and was referred to King Faisal Specialist Hospital and Research Centre-Jeddah at the age of 18 months. At that time, he had severe respiratory distress, very poor muscle power (unable to sit, poor head support), and significant signs of rickets without alopecia. His initial evaluation lab tests are shown in Table 1 and revealed an ionized calcium level of 1.27 mmol/L (1.60–2.10), and a phosphate level of 0.92 mmol/L (1.36–2.26). Alkaline phosphatase was 1541 IU/L (120–300) and PTH levels were 507 ng/L (15–65). 25-hydroxyvitamin D [25(OH)D] level was 15.6 ng/mL (9–47) and 1,25(OH)2D level was 912 pg/mL (15–60). The elevated 1,25(OH)2D concentration in the face of hypocalcemia and rickets suggested target organ resistance to 1,25(OH)2D and indicated the likely diagnosis of HVDRR. Despite treatment with high doses of alfacalcidiol and oral calcium, he continued to develop recurrent pneumonias that required admission to the intensive care unit where he initially failed to show clinical improvement. He was therefore started on high dose intravenous (IV) calcium infusion therapy. Within a few weeks, there was marked improvement in his clinical, biochemical, and radiological status. By 4 years of age, he had normal growth and motor development and he continued on treatment with intermittent high dose IV calcium infusions and high dose oral calcium in between periods of IV infusion.
Patient 2 is a young boy from Macedonia (former Yugoslav Republic). The child was born in April 1999 after an uneventful pregnancy. There was no known consanguinity between the parents. In May 2001 he was investigated because of florid rickets, which ensued despite regular oral vitamin D prophylaxis (1000 IU/day). He had rachitic rosary, deformed thorax with Harrison’ groove and gibus of the spine. He exhibited severe motor retardation and could not walk. He also had severe hypotonia and a very low serum creatinine (8 micromol/L). There was no alopecia. There were no clinical and laboratory data indicating malabsorption syndrome. His renal function was normal. The laboratory data and the clinical course with a clinical response to extremely high doses of calcitriol favored a diagnosis of HVDRR. The pretreatment laboratory investigation shown in part in Table 1 revealed: total Ca 1.79, Pi 0.86, Mg 0.7 (all mmol/L), alkaline phosphatase 1506 IU/L. There was generalized hyperaminoaciduria and iPTH was very elevated at 578 pg/ml (11–62). Vitamin D metabolites were as follows: 25(OH)D3 10.3 ng/ml (9–49) and 1,25(OH)2D 372 pg/ml (17–53). These pretreatment results raised the suspicion for HVDRR and led to treatment with high doses of activated vitamin D. The treatment was started with low doses of calcitriol (0.25 µg/day) and gradually increased at weekly intervals. There was no clinical and laboratory improvement, until a dose of 20 µg/day was reached. At the 6 month mark after diagnosis the serum biochemistry was as follows: Ca 2.35 mmol/l, Mg 0.9 mmol/l, Pi 1.42 mmol/l, alkaline phosphatase 518 IU/L, An ultrasound scan of the kidneys was normal without nephrocalcinosis. After reaching the calcitriol dose of 20 µg/day there was a significant clinical improvement; the boy could now walk without assistance, there was a gain of muscle mass and improved muscle strength; his appetite improved and he was more active. His height of 82 cm was still under the 3rd percentile. More recently, his Ca and Pi reached the normal range, alkaline phosphatase significantly decreased from the pretreatment value (>1500 IU/L) and serum creatinine increased to rormal. For the first time calcium appeared in the urine 0.59 mmol/l (otherwise it was not detected by the current assay <0.12 mmol/l). After another 2–3 months of therapy the boy appeared clinically well, his biochemistries were stable and his alkaline phosphatase further decreased to 284IU/L.
Patient 3 is a boy of Chinese descent. He is the third child of non-consanguineous parents. His 2 sisters are unaffected. He was born full term with a birth weight of 2.45 kg. There was mild motor delay, and he walked at the age of 17 months. He presented at the age of 2 years 3 months with signs of rickets. On examination, his height measured 79.0 cm (<3rd percentile) and his head circumference was 48.5 cm (50th percentile). He had mild frontal bossing with flaring of the wrists and ankles. He also had genu valgum, as well as a mild waddling gait. There was no rachitic rosary and he did not have alopecia. His anterior fontanelle was closed. Investigations confirmed the diagnosis of rickets, with hypocalcemia: with a low total serum calcium of 1.85 mmol/L (2.15–2.55), low serum ionized calcium of 0.85 mmol/L (1.15–1.35), low serum phosphate of 0.78 mmol/L (0.85–1.45) and markedly elevated serum alkaline phosphatase of 1057 IU/L (40–130). The serum intact PTH was elevated at 434 pg/ml (13–76). The renal and liver function tests were normal. The 25(OH)D was normal at 15.7 ng/mL (10.1–40.3), but the 1,25(OH)2D concentration was markedly elevated at 320 pg/ml (22–67), which indicated the likely diagnosis of HVDRR.
He was treated with supraphysiological doses of calcium (maximum dose required was elemental calcium 184 mg/kg/day) and alfacalcidiol drops (maximum dose required was 6 µg/day). There was progressive improvement in his muscle strength associated with a reduction in the serum alkaline phosphatase levels. He required insertion of a metal plate over his tibia to stabilize his progressive genu valgum, but the plate was successfully removed 2 years later as his biochemical profile normalized. His urinary calcium creatinine ratio remained normal during the treatment period and renal ultrasound showed no nephrocalcinosis. He is currently 9 years old with no clinical signs of rickets. His muscle strength is normal and he is able to participate in all physical activities. He remains on calcium and calcitriol and the latest biochemical profile and bone mineral density results are within the normal for his age.
Patient 4 is a girl of Indian descent and the only child of a consanguineous marriage in which her grandparents and great-grandparents were siblings. She was born prematurely at 8 months gestation, of a lower segment cesarean section for a breech presentation and her birth weight was 2.5 kg. She required mechanical ventilation for respiratory distress syndrome and she stayed in hospital for 2 weeks. From 2 months of life, she gradually lost all her relatively sparse hair and developed alopecia totalis. Shortly after she started weight bearing, she developed progressive bilateral genu valgus deformity. She started walking after 18 months and from 2 years of age, her wrists and ankles became progressively more swollen and deformed. She started running only at the age of 3 years and began hopping and jumping only at 4 years 8 months. However, she continued to experience pain in her legs on walking long distances. Her dentition was delayed and only started appearing at 1 year. She first presented to the pediatric endocrine clinic at the National University Hospital, Singapore, at 4 years 8 months with severe short stature, associated with clinical features of rickets and alopecia totalis.
On initial clinical examination, her height was 89.3 cm (<3rd percentile, −5SD), her weight was 11.3 kg (<3rd percentile) and her head circumference was 46.3 cm (<3rd percentile). Her upper to lower segment ratio was 1.2, which was increased, indicating shortening of her lower limbs. Her BP was 115/70 mm Hg and she had alopecia totalis, with no scalp or limb hair and no eyebrows or eyelashes. She also had lumbar lordosis with very prominent metaphyses at both wrists, lower femoral, upper tibial regions and ankles. She had marked severe genu valgus deformity (worse on the left), with the distance between the medial malleoli of 13.0 cm associated with bilateral pes planus.
Initial investigations confirmed the diagnosis of rickets, based on mild hypocalcemia with a total serum calcium of 2.01 mmol/L (2.15–2.55), serum ionized calcium of 1.11 mmol/L (1.15–1.35), low serum phosphate of 0.63 mmol/L (0.85–1.45) and markedly elevated serum alkaline phosphatase of 2150 IU/L (40–130). The serum intact PTH was markedly and appropriately elevated at 301 pg/ml (10–65). The renal, liver and thyroid function tests were normal. A skeletal survey demonstrated generalized osteopenia with symmetrical deformed, cupped and flared metaphyses present and deformities of the epiphyses. An X-ray left hand for bone age was 3 years at a chronological age of 4 years 8 months. The 25(OH)D was 19 ng/mL and 1,25(OH)2D concentration was markedly elevated at 190 pg/ml (15–60), which suggested the diagnosis of HVDRR.
She was initially commenced on supraphysiological doses of calcium (elemental calcium 160 mg/kg/day) and alfacalcidiol (9 µg/day), which were subsequently titrated to maintain normal serum calcium levels, with a progressive reduction in the serum alkaline phosphatase levels. Concomitantly, the genu valgus deformity improved significantly, with resolution of the metaphyseal flaring of the wrists and ankles. However, she developed mild nephrocalcinosis with tiny echogenic foci in the medullary regions of both kidneys. Her urinary calcium creatinine ratio has remained normal. Over the next 12 years, she has been regularly followedup as an outpatient. At 17 years 6 months, her height was 142.1 cm (<3rd percentile) and her weight was 51.3 kg (50th–75th percentile). She had alopecia, with no eyebrows, but used a custom-made wig. Her right leg was 3.0 cm longer than her left leg, but there was no genu valgus deformity. She had low normal serum total calcium of 2.16 mmol/L (2.15–2.55) with a low normal ionized calcium of 1.17 mmol/L (1.15–1.35). The serum alkaline phosphatase was 236 IU/L (40–130).
Patient 5 is a 6 4/12 year-old Turkish boy from Northwestern Turkey who was referred to the pediatric endocrinology unit of Trakya University Faculty of Medicine for severe deformities of both the legs and alopecia. He was the fifth child of consanguineous parents, born at 38 weeks of gestation after an uneventful pregnancy (birth weight 2.75 kg). He had progressive loss of hair from the scalp, eyebrows and eyelashes in the first few months of life. He had received prophylactic vitamin D supplementation from 1 month to 6 months of life followed by calcium supplementation upon development of a seizure-like event caused by hypocalcemia, however, no further investigation was carried out. At 9 months of age, seizure-like events recurred, and he was hospitalized for evaluation. He presented with classical rickets with alopecia and was diagnosed as HVDRR and treated with intravenous calcium in addition to high dose calcitriol for 2 months. He was lost to follow-up after discharge, and he was not given his treatment regularly. He was readmitted for bowing of the legs at 2 years of age. Tooth eruption started at the normal age, however he developed caries and underwent tooth extraction. An elder brother had similar findings and died at the age of eight years due to pneumonia. His parents and elder sister were normal. On admission his height and weight were 94 cm and 13.9 kg, respectively (both <3rd percentile). He had total alopecia with no eyebrow or eyelashes. He had severe genu valgum deformity bilaterally, as well as widening of the wrist and ankles. Features of hypocalcemia (Chvostek’s sign and carpopedal spasm) were negative. Investigations revealed a serum total calcium of 6.2 mg/dl (8.6–10 mg/dl), inorganic phosphorus of 3.9 mg/dl (2.4–4.7), alkaline phosphatase of 704 IU/L (32–91), intact PTH of 192.7 pg /ml (12–88 pg/ml) and 25(OH)D of 72.6 ng/ml (7.6–75) as well as 24 h urinary calcium excretion of 5 mg/d with a urinary calcium/creatinine ratio of 0.003. X-ray of the wrist and knee revealed cupping and fraying of the metaphysis, widening of epiphysis and generalized osteopenia and rickets. Bone age was 5 10/12 years (Greulich & Pyle). DEXA scan showed a z score of −1.3 for the lumbar spine consistent with osteopenia. A clinical diagnosis of HVDRR was made and a blood sample drawn for analysis of the VDR gene after informed consent was obtained from the parents. The patient was treated with supraphysiological oral doses of calcium (150 mg/kg/d elemental calcium) and calcitriol (4 µg/day). However there was no improvement in the serum calcium level and hypocalcemia continued for the next five days. On day 6, intravenous calcium was initiated. IV calcium gluconate was administered at a dose of 1000 mg of elemental calcium (75 mg/kg/d) over a period of 18 hours which was subsequently titrated to his serum calcium levels. He responded to IV calcium therapy with a rapid correction of the serum Ca (6.1 to 8.2 mg/dL after one week of therapy). However IV calcium could be continued for only one week since the family requested to discharge the patient. Therapy was changed to oral calcium at a dose of 150 mg/kg/day and calcitriol 1 µg 3 times/day. At the time of discharge the patient was advised to return for IV calcium infusions once a week while oral calcium and calcitriol were continued. Over the following 3 years clinical and laborartory findings improved and the calcium and calcitriol doses were tapered. Recent labs included Ca 8.9 mg/dL, phos 3.4 mg/dL alkaline phosphatase of 75 IU/L and PTH 36.5 pg/ml.
Molecular analyses
To determine whether the patients harbored mutations in the VDR gene we amplified and sequenced exons 2–9 from genomic DNA samples. Patient 1 was homozygous for G to A missense mutation in exon 4 creating a novel aspartic acid to asparagine substitution at amino acid 144 (D144N). Both parents were heterozygous for the mutation as demonstrated by the co-eluting bases on the chromatogram. The D144N mutation is located in helix 1 in the ligand-binding domain (LBD) (Fig. 1).
Fig. 1. Mutations in the VDR causing HVDRR.

A. Location of mutations in the DNA-binding domain (DBD). Conserved amino acids are shaded.
B. Location of mutations in the ligand-binding domain (LBD).
The α-helices are shown as black boxes and the β-turns as hatched box. Missense mutations are on top and nonsense mutations on bottom. E1 and AF-2 (activation function 2) represent helices important for transactivation. Adapted from Feldman et al [7]. Mutations from the current patients are indicated with an *.
Patient 2 was homozygous for a C to T base change in exon 5 that changed arginine to cysteine at amino acid 158 (R158C). Both parents were heterozygous for the mutation. The R158C mutation is located in the connecting loop between helix 2 and helix 3n in the LBD (Fig. 1).
Patient 3 was heterozygous for two missense mutations in the VDR gene. One mutation, a G to A substitution in exon 3, changed arginine to glutamine at amino acid 80 (R80Q). The patient’s mother was heterozygous for the R80Q mutation (the base caller recorded an A and not an N for the G/A sequence). The second mutation, a novel A to T base substitution in exon 7 (the base caller recorded a T and not an N for the A/T sequence), created a novel asparagine to tyrosine change at amino acid 276 (N276Y). The mother and father’s exon 7 sequences were normal suggesting that the N276Y mutation likely arose spontaneously in the patient. The R80Q mutation is located in the second zinc-finger of the DNA-binding domain (DBD) and the N276Y mutation is in helix 5 in the LBD (Fig. 1).
Patient 4 was found to have a unique 2 bp deletion in the 5’-donor splice site at the exon 3-intron E boundry [19] in the VDR gene. The normal VDR exon 3-intron E sequence is GAGT|GTGAGTGTCC. Deletion of either the GA or AG base pair in the splice site would generate the sequence GAGT|GTGTGTCC. Evidence to demonstrate that it was the AG base pair that was deleted and not the GA base pair was determined by examining the exon 3-intron E sequence of the parents DNA that were heterozygous for the 2 bp deletion. The sequence analyses showed that the sequence became mixed beginning at the A base indicating that the AG base pair was deleted.
Patient 5 had a missense mutation in exon 3. The C to G mutation created a novel cysteine to tryptophan substitution at amino acid 60 (C60W). The C60W mutation is located second zinc-finger structure in the VDR DBD (Fig. 1).
Effects of mutations on VDR transactivation
The C60W, D144N, R158C and N276Y mutations were then recreated in the VDR cDNA and their effects on VDR transactivation examined using a CYP24A1 promoter luciferase reporter. In these assays, the D144N (EC50 = 10.98 nM) and R158C (EC50 = 11.25 nM) mutants were ~33-fold less responsive to 1,25(OH)2D3 compared to the WT VDR (EC50 = 0.33 nM) (Fig. 2, panels A and B). The N276Y (EC50 = 26.2 nM) mutant was ~78-fold less responsive to 1,25(OH)2D3 compared to the WT VDR and the C60W mutant exhibited no induction of luciferase activity (Fig. 2, panels C and D). Immunoblotting shows that the VDR mutant proteins were expressed in COS-7 cells (Fig. 2E). These results demonstrate that the C60W, D144N, R158C and N276Y mutations cause resistance to 1,25(OH)2D3 and are the molecular cause of HVDRR in these patients.
Fig. 2. Transactivation of mutant VDRs by 1,25(OH)2D3 in COS-7 cells.
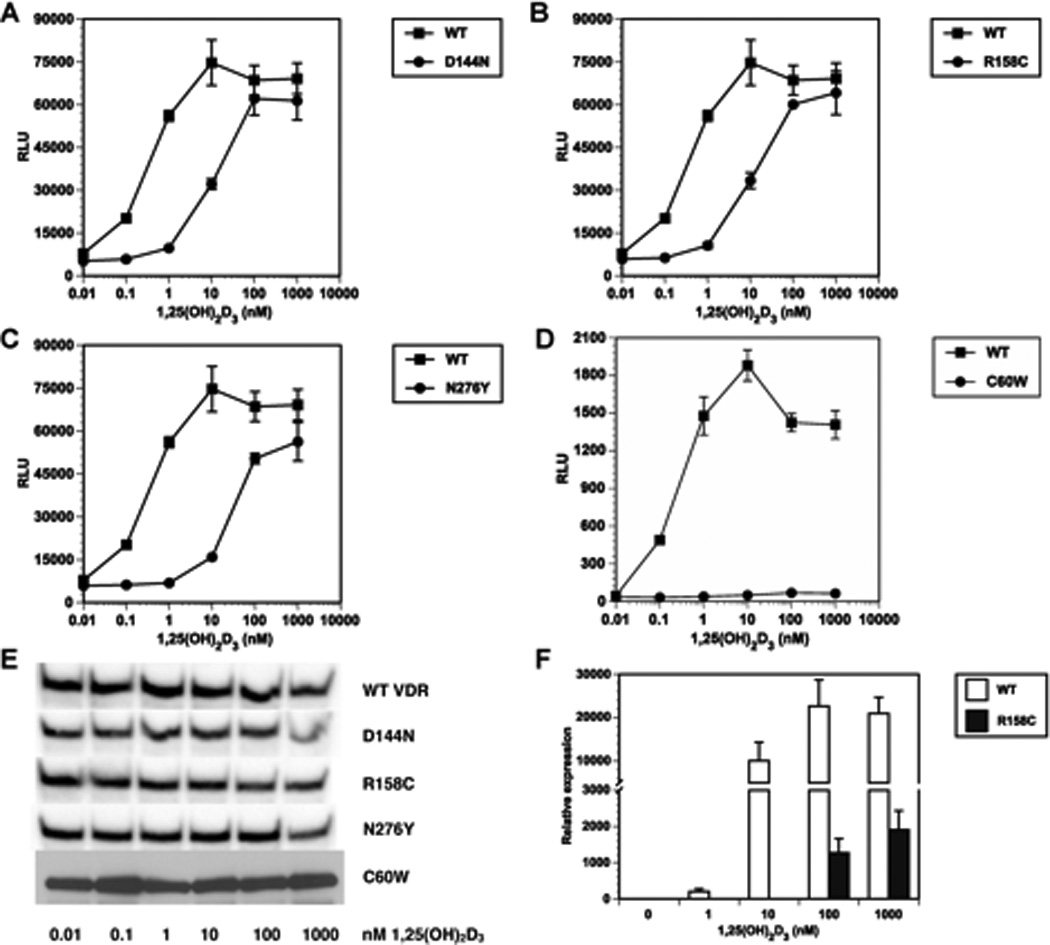
Panels A–D, COS-7 cells were transfected with VDR expression vectors and CYP24A1 promoter luciferase reporter plasmids. Cells were treated with vehicle (0.1% ethanol) or graded concentrations of 1,25(OH)2D3 for 24 hrs then assayed for luciferase activity. Panel E, immunoblots of VDR mutant expression in COS-7 cells. Panel F, patient 2 fibroblasts and normal control fibroblasts were treated with vehicle (0.1% ethanol) or graded concentrations of 1,25(OH)2D3 for 24 hrs then assayed for CYP24A1 gene expression by real-time RT-PCR. Error bars represent standard deviation from the mean.
Cultured skin fibroblasts were only available from patient 2 with the R158C mutation in the LBD. To determined whether the cells were resistant to 1,25(OH)2D3 we examined induction of CY24A1 (24-hydroxylase) gene expression, a classic marker of 1,25(OH)2D3 responsiveness. As shown in Fig. 2F, when 1,25(OH)2D3 was added to normal control fibroblasts the cells induced CYP24A1 gene expression in a dose-dependent manner. In contrast, patient 2 cells had no response to l and 10 nM 1,25(OH)2D3 and exhibited only a minimal response to 100 nM 1,25(OH)2D3 and the very high dose of 1 µM 1,25(OH)2D3 (Fig. 2F) compared to the WT control cells.
DISCUSSION
We describe five new unrelated cases of HVDRR. Patient 1 had a unique D144N mutation in the LBD. The D144N mutation resulted in a VDR that was 33-fold less responsive to 1,25(OH)2D3 compared to WT VDR in cultured COS-7 cells. The lower transactivation activity exhibited by the D144N mutant VDR suggests that the D144N mutation lowered the VDR’s affinity for 1,25(OH)2D3. Although D144 is not a contact site for 1,25(OH)2D3 its neighboring residue Y143 forms a hydrogen bond with the 3-OH group of 1,25(OH)2D3 [20]. The D144N mutation may interfere with the hydrogen bonding activity of Y143 and therefore lower mutant VDR’s affinity for 1,25(OH)2D3.
Patient 2 had an R158C mutation in the LBD. The R158C mutation was previously identified in a Korean girl with HVDRR without alopecia [21]. The Korean patient had a compound mutation; she was heterozygous for the R158C mutation and the other VDR allele had a Thr146Ile mutation in the LBD. In our transactivation assays, the recreated R158C mutant VDR exhibited a 33-fold decrease in transactivation activity compared to the WT VDR in COS-7 cells. On the other hand, when we examined 1,25(OH)2D3 responsiveness in fibroblasts from patient 2 we observed a significant lack of induction of CYP24A1 gene expression indicating that the R158C mutation may have additional effects on VDR function in the patient’s cells (Fig. 2). Since the R158C mutation is located in one of the two nuclear localization signals in the VDR [22, 23], a reduction in nuclear localization would likely further contribute to the low induction of CYP24A1 gene expression observed in the patient’s cells.
Patient 3 was a compound heterozygote with mutations R80Q in the VDR DBD and N276Y in the VDR LBD. The missense mutation in exon 3 in the patient was inherited from the mother as she was heterozygous for the R80Q mutation. The R80Q mutation was previously described in four patients from three unrelated families all with origins in North Africa [3, 10, 11]. Mutations in the DBD disrupt DNA binding and prevent gene transactivation by the VDR [24]. The N276Y mutation appears to have arisen spontaneously as neither the patient’s mother nor father harbors the mutation (Fig. 1). However, it can’t be ruled out that N276Y mutation was inherited from a different father. The N276Y mutation is located in helix H5 in the LBD (Fig. 3). N276 is not a contact point for 1,25(OH)2D3 but a number of neighboring amino acids do contact the ligand including R274, S275 and S278. It is thus likely that the N276Y mutations interferes with ligand-binding as our studies showed that the R276Y mutant VDR required ~78-fold higher concentrations of 1,25(OH)2D3 for transactivation compared to the WT VDR.
Patient 4 had a unique 2 bp deletion that altered the exon 3-intron E 5’-splice site from GTGAGT to GTGTGT. The 2 bp deletion altered the 5’-donor splice site that would either cause exon 3 to be skipped or intron E to be incorporated into the VDR mRNA. It is more likely that exon 3 is skipped as a previous report of a single G to C base substitution in the exon 3-intron E 5’-splice site that changed the sequence from GTGAGT to GTGACT in a patient with HVDRR showed that patient’s VDR cDNA had a fusion of exon 2 and exon 4 demonstrating that the splice-site mutation caused exon 3 to be skipped [25]. This resulted in a premature stop signal that produced a mutant protein that contained 48 amino acids of the wild-type sequence plus 5 additional amino acids due to the frameshift in exon 4 (Phe48fs). No specific [3H]-1,25(OH)2D3 binding was found in cell extracts from that patient’s fibroblasts and the cells exhibited no 24-hydroxylase activity in response to 1,25(OH)2D3 treatment demonstrating resistance to 1,25(OH)2D3 [25].
Patient 5 had a unique C60W mutation in the second zinc-finger structure in the DBD (Fig. 1). The C60W mutant was totally inactive in transactivation assays most likely due to a failure to bind to DNA.
As of this writing there are 50 unique mutations in the VDR gene consisting of 5 deletions (including 1 in a Pomeranian dog), 4 splice-site mutations, 1 deletion/substitution, 1 insertion/duplication, 6 nonsense mutations and 33 missense mutations (Tables 2 and 3). Of the missense mutations, 12 mutations were in the DBD and 21 mutations were in the LBD (Fig. 1). All patients with mutations in the DBD including the patient described here with the C60W mutation had HVDRR with alopecia indicating that DNA interaction is a critical function of the VDR in regulating hair growth [6]. On the other hand, HVDRR patients with mutations in the LBD that affect ligand-binding or coactivator-binding do not have alopecia including patient’s 1 and 2 described here with the D144N and R158C mutations. Interestingly, patient 3 with compound heterozygous mutations R80Q in the DBD and N276Y in the LBD did not have alopecia indicating that the N276Y mutant VDR allele prevented alopecia even though it was not able to prevent hypocalcemia and rickets [6]. Of the other 4 patients previously reported with compound heterozygous mutations R50X/ΔK246 [26], 366delC/E329K [27], T146I/R158C [21], and R263L/R391S [28] only the patient with the T146I/R158C mutations did not have alopecia [6].
Table 2.
Missense mutations in the VDR gene causing HVDRR
| Missense Mutation | Exon | Alopecia | Reference |
|---|---|---|---|
| V26M | 2 | Y | [29] |
| G33D | 2 | Y | [8, 30] |
| H35Q | 2 | Y | [31] |
| C41Y | 2 | Y | [32] |
| K45E | 2 | Y | [33] |
| G46D | 2 | Y | [12] |
| F47I | 2 | Y | [33] |
| R50Q | 3 | Y | [9] |
| C60W | 3 | Y | This report |
| R73Q | 3 | Y | [8] |
| R80Q | 3 | Y | [10, 11] |
| C84R | 3 | Y | [34] |
| D144N | 4 | N | This report |
| R158C | 5 | N | [21] This report |
| L227P | 6 | N | [35] |
| F251C | 6 | Y | [16] |
| Q259P | 7 | Y | [36] |
| Q259E | 7 | Y | [37] |
| L263R | 7 | Y | [28] |
| I268T | 7 | N | [38] |
| R274L | 7 | N | [13] |
| R274H | 7 | N | [39] |
| N276Y | 7 | N | This report |
| W286R | 8 | N | [40] |
| H305Q | 8 | N | [15] |
| I314S | 8 | N | [14] |
| G319V | 8 | Y | [37] |
| E329K | 8 | Y | [27] |
| V346M | 9 | N | [41] |
| R391C | 9 | Y | [14] |
| R391S | 9 | Y | [28] |
| E420K | 9 | N | [17] |
| E420A (heterozygous mutation) | 9 | N | [18] |
Table 3.
Other mutations in the VDR gene causing HVDRR
| Nonsense Mutations | Exon | Alopecia | References |
|---|---|---|---|
| R30X | 2 | Y | [26, 42, 43] |
| R50X | 3 | Y | [44, 45] |
| R73X | 3 | Y | [36, 37, 46, 47] |
| Q152X | 4 | Y | [13] |
| Y295X | 7 | Y | [48, 49] |
| Q317X | 8 | Y | [50] |
| Deletion | |||
| delAG (IVS3Δ+4–5) | Exon 3-intron E | Y | This report |
| c.366delC (c.526delC) | 4 | Y | [27] |
| c.716delA (c.879delA) | 6 | Y | [51] |
| ΔK246 (c.896-898delCTT) | 6 | Y | [26] |
| Deletion/substitution | |||
| 5 bp deletion/8 bp substitution | 4 | N | [52] |
| Insertion/duplication | |||
| 102 bp insertion/duplication | 9 | Patchy | [53] |
| Splice-site Mutation | |||
| IVS3+5G>C | Exon 3-intron E | Y | [25, 47] |
| GTCAGT to GTGAGT (c.862C>G) | 6 | Y | [36] |
| IVS4+1G>C | Exon 4-intron F | Patchy | [54] |
| IVS8+1G>T | Exon 8-intron J | Patchy | [55] |
| Mutation in Pomeranian dog | |||
| c.525delG (dog c.654delG) | 4 | Y | [56] |
Intron nomenclature is from Miyamoto et al [19]. Numbers in parentheses represent coordinates based on NCBI reference sequence: NM_000376.2. The coordinates for the dog sequence were based on NCBI Reference Sequence: XM_543714.3.
The biochemical and genetic analyses of the VDR in HVDRR patients has yielded important insights into the structure and function of the receptor that mediates 1,25(OH)2D action. A concerted investigative approach of HVDRR at the clinical, cellular and molecular level has proven exceedingly valuable in gaining knowledge about the functions of the domains of the VDR protein and in elucidating a detailed understanding of the mechanism of action of 1,25(OH)2D. These studies have been essential to promote the wellbeing of the families with HVDRR and in improving the diagnostic and especially the clinical management of children with this rare genetic disease.
Research Highlights.
Five new cases of HVDRR are described
Novel mutations are added to the described defects in the Vitamin D Receptor
Diagnostic and clinical findings on each case are reported
The VDR mutations are documented and the extent of the resistance caused by each mutation is assessed
The entire range of known mutations is reviewed
The syndrome of HVDRR and its diagnosis and treatment are discussed
Acknowledgments
Supported by NIH Grant DK42482 (to D.F.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Feldman D, Pike JW, Adams JS. Vitamin D. Third Edition. San Diego: Elsiever; 2011. pp. 1–2144. [Google Scholar]
- 2.Haussler MR, Whitfield GK, Kaneko I, Haussler CA, Hsieh D, Hsieh JC, Jurutka PW. Molecular mechanisms of vitamin D action. Calcif Tissue Int. 2013;92:77–98. doi: 10.1007/s00223-012-9619-0. [DOI] [PubMed] [Google Scholar]
- 3.Malloy PJ, Pike JW, Feldman D. The vitamin D receptor and the syndrome of hereditary 1,25-dihydroxyvitamin D-resistant rickets. Endocr. Rev. 1999;20:156–188. doi: 10.1210/edrv.20.2.0359. [DOI] [PubMed] [Google Scholar]
- 4.Malloy PJ, Feldman D. Genetic disorders and defects in vitamin D action. Endocrinol Metab Clin N Am. 2010;39:333–346. doi: 10.1016/j.ecl.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malloy PJ, Tiosano D, Feldman D. Hereditary 1,25-dihydroxyvitamin D resistant rickets. In: Feldman D, Pike JW, Adams JS, editors. Vitamin D. Third Edition. San Diego: Elsevier; 2011. pp. 1197–1232. [Google Scholar]
- 6.Malloy PJ, Feldman D. The role of vitamin D receptor mutations in the development of alopecia. Mol. Cell. Endocrinol. 2011;347:90–96. doi: 10.1016/j.mce.2011.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feldman D, Malloy PJ, Miller WL. Genetic disorders of vitamin D synthesis and action. In: Thakker RV, Whyte MP, Eisman JA, Igarashi T, editors. Genetics of Bone Biology and Skeletal Disease. San Diego: Elsevier; 2013. pp. 537–552. [Google Scholar]
- 8.Hughes MR, Malloy PJ, Kieback DG, Kesterson RA, Pike JW, Feldman D, O'Malley BW. Point mutations in the human vitamin D receptor gene associated with hypocalcemic rickets. Science. 1988;242:1702–1705. doi: 10.1126/science.2849209. [DOI] [PubMed] [Google Scholar]
- 9.Saijo T, Ito M, Takeda E, Huq AH, Naito E, Yokota I, Sone T, Pike JW, Kuroda Y. A unique mutation in the vitamin D receptor gene in three Japanese patients with vitamin D-dependent rickets type II: utility of single-strand conformation polymorphism analysis for heterozygous carrier detection. Am. J. Hum. Genet. 1991;49:668–673. [PMC free article] [PubMed] [Google Scholar]
- 10.Sone T, Marx SJ, Liberman UA, Pike JW. A unique point mutation in the human vitamin D receptor chromosomal gene confers hereditary resistance to 1,25-dihydroxyvitamin D3. Molecular Endocrinology. 1990;4:623–631. doi: 10.1210/mend-4-4-623. [DOI] [PubMed] [Google Scholar]
- 11.Malloy PJ, Weisman Y, Feldman D. Hereditary 1 alpha,25-dihydroxyvitamin D-resistant rickets resulting from a mutation in the vitamin D receptor deoxyribonucleic acid-binding domain. J. Clin. Endocrinol. Metab. 1994;78:313–316. doi: 10.1210/jcem.78.2.8106618. [DOI] [PubMed] [Google Scholar]
- 12.Lin NU-T, Malloy PJ, Sakati N, Al-Ashwal A, Feldman D. A novel mutation in the deoxyribonucleic acid-binding domain of the vitamin D receptor gene causes hereditary 1,25-dihydroxyvitamin D resistant rickets. J. Clin. Endocrinol. Metab. 1996;81:2564–2569. doi: 10.1210/jcem.81.7.8675579. [DOI] [PubMed] [Google Scholar]
- 13.Kristjansson K, Rut AR, Hewison M, O'Riordan JL, Hughes MR. Two mutations in the hormone binding domain of the vitamin D receptor cause tissue resistance to 1,25 dihydroxyvitamin D3. J. Clin. Invest. 1993;92:12–16. doi: 10.1172/JCI116539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whitfield GK, Selznick SH, Haussler CA, Hsieh JC, Galligan MA, Jurutka PW, Thompson PD, Lee SM, Zerwekh JE, Haussler MR. Vitamin D receptors from patients with resistance to 1,25-dihydroxyvitamin D3: point mutations confer reduced transactivation in response to ligand and impaired interaction with the retinoid X receptor heterodimeric partner. Mol. Endocrinol. 1996;10:1617–1631. doi: 10.1210/mend.10.12.8961271. [DOI] [PubMed] [Google Scholar]
- 15.Malloy PJ, Eccleshall TR, Gross C, Van Maldergem L, Bouillon R, Feldman D. Hereditary vitamin D resistant rickets caused by a novel mutation in the vitamin D receptor that results in decreased affinity for hormone and cellular hyporesponsiveness. J Clin Invest. 1997;99:297–304. doi: 10.1172/JCI119158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Malloy PJ, Zhu W, Zhao XY, Pehling GB, Feldman D. A novel inborn error in the ligand-binding domain of the vitamin D receptor causes hereditary vitamin D-resistant rickets. Mol Genet Metab. 2001;73:138–148. doi: 10.1006/mgme.2001.3181. [DOI] [PubMed] [Google Scholar]
- 17.Malloy PJ, Xu R, Peng L, Clark PA, Feldman D. A novel mutation in helix 12 of the vitamin D receptor impairs coactivator interaction and causes hereditary 1,25-dihydroxyvitamin D-resistant rickets without alopecia. Mol. Endocrinol. 2002;16:2538–2546. doi: 10.1210/me.2002-0152. [DOI] [PubMed] [Google Scholar]
- 18.Malloy PJ, Zhou Y, Wang J, Hiort O, Feldman D. Hereditary vitamin D-resistant rickets (HVDRR) owing to a heterozygous mutation in the vitamin D receptor. J. Bone Miner. Res. 2011;26:2710–2718. doi: 10.1002/jbmr.484. [DOI] [PubMed] [Google Scholar]
- 19.Miyamoto K, Kesterson RA, Yamamoto H, Taketani Y, Nishiwaki E, Tatsumi S, Inoue Y, Morita K, Takeda E, Pike JW. Structural organization of the human vitamin D receptor chromosomal gene and its promoter. Mol. Endocrinol. 1997;11:1165–1179. doi: 10.1210/mend.11.8.9951. [DOI] [PubMed] [Google Scholar]
- 20.Rochel N, Wurtz JM, Mitschler A, Klaholz B, Moras D. The crystal structure of the nuclear receptor for vitamin D bound to its natural ligand. Mol Cell. 2000;5:173–179. doi: 10.1016/s1097-2765(00)80413-x. [DOI] [PubMed] [Google Scholar]
- 21.Song JK, Yoon KS, Shim KS, Bae CW. Novel compound heterozygous mutations in the vitamin D receptor gene in a Korean girl with hereditary vitamin D resistant rickets. J Korean Med Sci. 2011;26:1111–1114. doi: 10.3346/jkms.2011.26.8.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsieh JC, Shimizu Y, Minoshima S, Shimizu N, Haussler CA, Jurutka PW, Haussler MR. Novel nuclear localization signal between the two DNA-binding zinc fingers in the human vitamin D receptor. J Cell Biochem. 1998;70:94–109. [PubMed] [Google Scholar]
- 23.Michigami T, Suga A, Yamazaki M, Shimizu C, Cai G, Okada S, Ozono K. Identification of amino acid sequence in the hinge region of human vitamin D receptor that transfers a cytosolic protein to the nucleus. J. Biol. Chem. 1999;274:33531–33538. doi: 10.1074/jbc.274.47.33531. [DOI] [PubMed] [Google Scholar]
- 24.Sone T, Scott RA, Hughes MR, Malloy PJ, Feldman D, O'Malley BW, Pike JW. Mutant vitamin D receptors which confer hereditary resistance to 1,25-dihydroxyvitamin D3 in humans are transcriptionally inactive in vitro. J. Biol. Chem. 1989;264:20230–20234. [PubMed] [Google Scholar]
- 25.Hawa NS, Cockerill FJ, Vadher S, Hewison M, Rut AR, Pike JW, O'Riordan JL, Farrow SM. Identification of a novel mutation in hereditary vitamin D resistant rickets causing exon skipping. Clin. Endocrinol. 1996;45:85–92. [PubMed] [Google Scholar]
- 26.Zhou Y, Wang J, Malloy PJ, Dolezel Z, Feldman D. Compound heterozygous mutations in the vitamin D receptor in a patient with hereditary 1,25-dihydroxyvitamin D-resistant rickets with alopecia. J. Bone Miner. Res. 2009;24:643–651. doi: 10.1359/JBMR.081216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller J, Djabali K, Chen T, Liu Y, Ioffreda M, Lyle S, Christiano AM, Holick M, Cotsarelis G. Atrichia caused by mutations in the vitamin D receptor gene is a phenocopy of generalized atrichia caused by mutations in the hairless gene. J Invest Dermatol. 2001;117:612–617. doi: 10.1046/j.0022-202x.2001.01438.x. [DOI] [PubMed] [Google Scholar]
- 28.Nguyen M, d'Alesio A, Pascussi JM, Kumar R, Griffin MD, Dong X, Guillozo H, Rizk-Rabin M, Sinding C, Bougneres P, Jehan F, Garabedian M. Vitamin D-resistant rickets and type 1 diabetes in a child with compound heterozygous mutations of the vitamin D receptor (L263R and R391S): dissociated responses of the CYP-24 and rel-B promoters to 1,25-dihydroxyvitamin D3. J. Bone Miner. Res. 2006;21:886–894. doi: 10.1359/jbmr.060307. [DOI] [PubMed] [Google Scholar]
- 29.Malloy PJ, Wang J, Srivastava T, Feldman D. Hereditary 1,25-dihydroxyvitamin D-resistant rickets with alopecia resulting from a novel missense mutation in the DNA-binding domain of the vitamin D receptor. Mol Genet Metab. 2010;99:72–79. doi: 10.1016/j.ymgme.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malloy PJ, Hochberg Z, Pike JW, Feldman D. Abnormal binding of vitamin D receptors to deoxyribonucleic acid in a kindred with vitamin D-dependent rickets, type II. J. Clin. Endocrinol. Metab. 1989;68:263–269. doi: 10.1210/jcem-68-2-263. [DOI] [PubMed] [Google Scholar]
- 31.Yagi H, Ozono K, Miyake H, Nagashima K, Kuroume T, Pike JW. A new point mutation in the deoxyribonucleic acid-binding domain of the vitamin D receptor in a kindred with hereditary 1,25-dihydroxyvitamin D-resistant rickets. J. Clin. Endocrinol. Metab. 1993;76:509–512. doi: 10.1210/jcem.76.2.8381803. [DOI] [PubMed] [Google Scholar]
- 32.Shafeghati Y, Momenin N, Esfahani T, Reyniers E, Wuyts W. Vitamin D-dependent rickets type II: report of a novel mutation in the vitamin D receptor gene. Arch Iran Med. 2008;11:330–334. [PubMed] [Google Scholar]
- 33.Rut AR, Hewison M, Kristjansson K, Luisi B, Hughes MR, O'Riordan JL. Two mutations causing vitamin D resistant rickets: modelling on the basis of steroid hormone receptor DNA-binding domain crystal structures. Clin Endocrinol (Oxf) 1994;41:581–590. doi: 10.1111/j.1365-2265.1994.tb01822.x. [DOI] [PubMed] [Google Scholar]
- 34.Asunis I, Marini MG, Porcu L, Meloni A, Cabriolu AL, Cao A, Moi P. A novel missense mutation (C84R) in a patient with type II vitamin d-dependent rickets. Exp Clin Endocrinol Diabetes. 2010;118:177–179. doi: 10.1055/s-0028-1103275. [DOI] [PubMed] [Google Scholar]
- 35.Huang K, Malloy P, Feldman D, Pitukcheewanont P. Enteral calcium infusion used successfully as treatment for a patient with hereditary vitamin D resistant rickets (HVDRR) without alopecia: a novel mutation. Gene. 2013;512:554–559. doi: 10.1016/j.gene.2012.09.078. [DOI] [PubMed] [Google Scholar]
- 36.Cockerill FJ, Hawa NS, Yousaf N, Hewison M, O'Riordan JL, Farrow SM. Mutations in the vitamin D receptor gene in three kindreds associated with hereditary vitamin D resistant rickets. J Clin Endocrinol Metab. 1997;82:3156–3160. doi: 10.1210/jcem.82.9.4243. [DOI] [PubMed] [Google Scholar]
- 37.Macedo LC, Soardi FC, Ananias N, Belangero VM, Rigatto SZ, De-Mello MP, D'Souza-Li L. Mutations in the vitamin D receptor gene in four patients with hereditary 1,25-dihydroxyvitamin D-resistant rickets. Arq Bras Endocrinol Metabol. 2008;52:1244–1251. doi: 10.1590/s0004-27302008000800007. [DOI] [PubMed] [Google Scholar]
- 38.Malloy PJ, Xu R, Peng L, Peleg S, Al-Ashwal A, Feldman D. Hereditary 1,25-dihydroxyvitamin D resistant rickets due to a mutation causing multiple defects in vitamin D receptor function. Endocrinology. 2004;145:5106–5114. doi: 10.1210/en.2004-0080. [DOI] [PubMed] [Google Scholar]
- 39.Aljubeh JM, Wang J, Al-Remeithi SS, Malloy PJ, Feldman D. Report of two unrelated patients with hereditary vitamin D resistant rickets due to the same novel mutation in the vitamin D receptor. J Pediatr Endocrinol Metab. 2011;24:793–799. doi: 10.1515/jpem.2011.341. [DOI] [PubMed] [Google Scholar]
- 40.Nguyen TM, Adiceam P, Kottler ML, Guillozo H, Rizk-Rabin M, Brouillard F, Lagier P, Palix C, Garnier JM, Garabedian M. Tryptophan missense mutation in the ligand-binding domain of the vitamin D receptor causes severe resistance to 1,25-dihydroxyvitamin D. J. Bone Miner. Res. 2002;17:1728–1737. doi: 10.1359/jbmr.2002.17.9.1728. [DOI] [PubMed] [Google Scholar]
- 41.Arita K, Nanda A, Wessagowit V, Akiyama M, Alsaleh QA, McGrath JA. A novel mutation in the VDR gene in hereditary vitamin D-resistant rickets. Br J Dermatol. 2008;158:168–171. doi: 10.1111/j.1365-2133.2007.08232.x. [DOI] [PubMed] [Google Scholar]
- 42.Mechica JB, Leite MO, Mendonca BB, Frazzatto ES, Borelli A, Latronico AC. A novel nonsense mutation in the first zinc finger of the vitamin D receptor causing hereditary 1,25-dihydroxyvitamin D3-resistant rickets. J Clin Endocrinol Metab. 1997;82:3892–3894. doi: 10.1210/jcem.82.11.4384. [DOI] [PubMed] [Google Scholar]
- 43.Zhu W, Malloy PJ, Delvin E, Chabot G, Feldman D. Hereditary 1,25-dihydroxyvitamin D-resistant rickets due to an opal mutation causing premature termination of the vitamin D receptor. J. Bone Miner. Res. 1998;13:259–264. doi: 10.1359/jbmr.1998.13.2.259. [DOI] [PubMed] [Google Scholar]
- 44.Forghani N, Lum C, Krishnan S, Wang J, Wilson DM, Blackett PR, Malloy PJ, Feldman D. Two new unrelated cases of hereditary 1,25-dihydroxyvitamin D-resistant rickets with alopecia resulting from the same novel nonsense mutation in the vitamin D receptor gene. J Pediatr Endocrinol Metab. 2010;23:843–850. doi: 10.1515/jpem.2010.136. [DOI] [PubMed] [Google Scholar]
- 45.Supornsilchai V, Hiranras Y, Wacharasindhu S, Mahayosnond A, Suphapeetiporn K, Shotelersuk V. Two siblings with a novel nonsense mutation, p.R50X, in the vitamin D receptor gene. Endocrine. 2011;40:62–66. doi: 10.1007/s12020-011-9450-9. [DOI] [PubMed] [Google Scholar]
- 46.Wiese RJ, Goto H, Prahl JM, Marx SJ, Thomas M, al-Aqeel A, DeLuca HF. Vitamin D-dependency rickets type II: truncated vitamin D receptor in three kindreds. Mol. Cell. Endocrinol. 1993;90:197–201. doi: 10.1016/0303-7207(93)90152-a. [DOI] [PubMed] [Google Scholar]
- 47.Nicolaidou P, Tsitsika A, Papadimitriou A, Karantana A, Papadopoulou A, Psychou F, Liakopoulou D, Georgouli H, Kakourou T, Chrousos G. Hereditary vitamin D-resistant rickets in Greek children: genotype, phenotype, and long-term response to treatment. J Pediatr Endocrinol Metab. 2007;20:425–430. doi: 10.1515/jpem.2007.20.3.425. [DOI] [PubMed] [Google Scholar]
- 48.Malloy PJ, Hochberg Z, Tiosano D, Pike JW, Hughes MR, Feldman D. The molecular basis of hereditary 1,25-dihydroxyvitamin D3 resistant rickets in seven related families. J. Clin. Invest. 1990;86:2071–2079. doi: 10.1172/JCI114944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ritchie HH, Hughes MR, Thompson ET, Malloy PJ, Hochberg Z, Feldman D, Pike JW, O'Malley BW. An ochre mutation in the vitamin D receptor gene causes hereditary 1,25-dihydroxyvitamin D3-resistant rickets in three families. Proc. Natl. Acad. Sci. USA. 1989;86:9783–9787. doi: 10.1073/pnas.86.24.9783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Malloy PJ, Zhu W, Bouillon R, Feldman D. A novel nonsense mutation in the ligand binding domain of the vitamin D receptor causes hereditary 1,25-dihydroxyvitamin D-resistant rickets. Mol Genet Metab. 2002;77:314–318. doi: 10.1016/s1096-7192(02)00173-7. [DOI] [PubMed] [Google Scholar]
- 51.Kanakamani J, Tomar N, Kaushal E, Tandon N, Goswami R. Presence of a deletion mutation (c.716delA) in the ligand binding domain of the vitamin D receptor in an Indian patient with vitamin D-dependent rickets type II. Calcif Tissue Int. 2010;86:33–41. doi: 10.1007/s00223-009-9310-2. [DOI] [PubMed] [Google Scholar]
- 52.Malloy PJ, Xu R, Cattani A, Reyes L, Feldman D. A unique insertion/substitution in helix H1 of the vitamin D receptor ligand binding domain in a patient with hereditary 1,25-dihydroxyvitamin D-resistant rickets. J. Bone Miner. Res. 2004;19:1018–1024. doi: 10.1359/jbmr.2004.19.6.1018. [DOI] [PubMed] [Google Scholar]
- 53.Malloy PJ, Wang J, Peng L, Nayak S, Sisk JM, Thompson CC, Feldman D. A unique insertion/duplication in the VDR gene that truncates the VDR causing hereditary 1,25-dihydroxyvitamin D-resistant rickets without alopecia. Arch. Biochem. Biophys. 2007;460:285–292. doi: 10.1016/j.abb.2006.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Katavetin P, Wacharasindhu S, Shotelersuk V. A girl with a novel splice site mutation in VDR supports the role of a ligand-independent VDR function on hair cycling. Horm Res. 2006;66:273–276. doi: 10.1159/000095546. [DOI] [PubMed] [Google Scholar]
- 55.Ma NS, Malloy PJ, Pitukcheewanont P, Dreimane D, Geffner ME, Feldman D. Hereditary vitamin D resistant rickets: Identification of a novel splice site mutation in the vitamin D receptor gene and successful treatment with oral calcium therapy. Bone. 2009;45:743–746. doi: 10.1016/j.bone.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.LeVine DN, Zhou Y, Ghiloni RJ, Fields EL, Birkenheuer AJ, Gookin JL, Roberston ID, Malloy PJ, Feldman D. Hereditary 1,25-dihydroxyvitamin D-resistant rickets in a Pomeranian dog caused by a novel mutation in the vitamin D receptor gene. J Vet Intern Med. 2009;23:1278–1283. doi: 10.1111/j.1939-1676.2009.0405.x. [DOI] [PubMed] [Google Scholar]