

Abstract
Infection with high-risk human papillomavirus (HPV) type 16 is an independent risk factor for the development of oropharyngeal squamous cell carcinomas (OSCC). However, it is unclear whether viral integration is an essential hallmark in the carcinogenic process of OSCC and whether HPV integration correlates with the level of viral gene transcription and influences the expression of disrupted host genes. We analyzed 75 patients with OSCC. HPV16-positivity was proven by p16INK4A immunohistochemistry, PCR and FISH. Viral integration was examined using DIPS- as well as APOT-PCR. Viral E2, E6 and E7 gene expression levels were quantified by quantitative reverse transcriptase (RT-q)PCR. Expression levels of 7 human genes disrupted by the virus were extracted from mRNA expression profiling data of 32 OSCCs. Viral copy numbers were assessed by qPCR in 73 tumors. We identified 37 HPV16-human fusion products indicating viral integration in 29 (39%) OSCC. In the remaining tumors (61%) only episome-derived PCR products were detected. When comparing OSCC with or without an integration-derived fusion product, we did not find significant differences in the mean RNA expression of viral genes E2, E6 and E7 or the viral copy numbers per cell, nor did the RNA expression of the HPV-disrupted genes differ from either group of OSCC. In conclusion, our data do not support the hypothesis that integration affects the levels of viral and/or HPV-disrupted human gene transcripts. Thus constitutive, rather than a high level, of expression of oncogene transcripts appears to be required in HPV-related OSCC.
Introduction
Approximately 600.000 new cases of head and neck squamous cell carcinoma (HNSCC) have been estimated to occur worldwide in 2011, ranking them in sixth position of all carcinomas [1]–[3]. Risk factors for the development of HNSCC include environmental factors, excessive tobacco and alcohol use, as well as human papillomavirus (HPV) infections. Particularly oropharyngeal squamous cell carcinomas (OSCC) are associated with HPV16 [4]. This group of carcinomas shows clinicopathological and molecular characteristics that differ from alcohol- and tobacco-induced carcinomas [4]–[6]. Studies that have assessed the prevalence of HPV-induced OSCC report frequencies ranging from 20% to up to 90% [5],[7]–[9].
Although integration of the viral DNA into the host genome is not part of the normal viral life cycle, studies in anogenital carcinomas have shown a significant correlation between integration and progression of dysplastic lesions to invasive carcinomas [10],[11]. For example in uterine cervical carcinomas, it has been shown that oncogene transcripts indicating viral integration can be identified in 55% of HPV16 positive cases and 92% of the HPV18 positive cases, and that particularly for HPV16 the integration events have been found to occur already in cervical intraepithelial neoplasia (CIN) [11]. We recently also detected viral integration in head and neck oropharyngeal dysplasia adjacent to squamous cell carcinoma by FISH, however these dysplasia are a rare finding in the oropharynx [12].
Using Amplification of Papillomavirus Oncogene Transcripts PCR (APOT-PCR), so far only two studies report HPV16 integration in 2 out of 4 and 6 out of 9 tumors in HPV-DNA positive OSCC [13],[14].
However, there is controversy with respect to the relation between viral integration and viral gene expression. Integration of HPV DNA in uterine cervical squamous cell carcinomas (UCSCC) has been correlated to disruption of the viral regulatory gene E2 [15],[16]. Studies in cell lines have shown that E2 represses the viral E6 and E7 expression [17]. In the uterine cervical cell line W12, integration of HPV was shown to result in higher levels of the oncogenes E6/E7 and a selective growth advantage over cells harboring extrachromosomal HPV DNA [18]. This had led to the hypothesis that the levels of viral E6 and E7 transcripts are higher in lesions in which viral integration resulted in E2 disruption, which is thought to lead to deregulation of cell cycle control [19]–[21].
On the other hand, a study in primary keratinocytes immortalized with HPV16 genomes has shown that disruption of the E2 gene sequence upon viral integration does not result in increased expression of the viral E6 and E7 oncogenes [13]. In addition, a publication by Häfner et al. using APOT-PCR has shown no correlation between the integration state of the viral genome and the expression of the viral gene E6 in a collection of 55 HPV16-positive cervical carcinoma samples [22]. It would be interesting to examine viral physical status and E2, E6 and E7 gene expression in primary OSCC since this information is lacking.
Here we present the HPV16 integration status for a collection of 75 HPV16-DNA-positive and p16INK4A-positive OSCC using APOT- and Detection of Integrated Papillomavirus Sequences PCR (DIPS-PCR), and its relation to the level of gene expression for the viral genes E2, E6 and E7, gene expression analysis of a number of human genes disrupted by viral integration and viral DNA-load.
Materials and Methods
Subjects and Material
Fresh frozen clinical OSCC samples from 75 patients treated at the Departments of Otorhinolaryngology and Head and Neck Surgery of the University Hospitals of Cologne and Maastricht between 1994 and 2009 were collected from the archives of the Departments of Pathology of both hospitals. Inclusion criteria were the availability of sufficient fresh frozen tumor tissue containing ≥70% tumor cells, high quality tumor DNA and RNA and HPV16 infection, as detected by HPV-specific PCR and FISH analysis [4],[5],[23],[24] and overexpression of the surrogate marker p16INK4A as detected by immunohistochemistry [25],[26].
Patient age ranged from 44–83 years (median 62.7 years). Fifty-seven (76.0 %) patients were male and eighteen (24.0 %) were female.
Ethics Statement
Patient material was used according to the code for proper secondary use of human tissue. The ethics committees of the Universities of Cologne and Maastricht medical faculties approved this study. Written, informed consent had been obtained from all patients.
Amplification of Papillomavirus Oncogene Transcripts PCR (APOT-PCR)
Total RNA was extracted from five 10 µm-thick snap frozen tissue sections using the RNeasy mini kit (Qiagen, Hilden, Germany) and DNase treatment. RNA concentration and quality were determined by RNA StdSens Chips on a BioRad Experion system (BioRad, Munich, Germany). HPV oncogene transcripts were amplified as described before [27]. Briefly, reverse transcription was performed using 25 µM oligo-(dT)17 primer coupled to a linker sequence (dT)17-p3, 10 mM dNTPs each, 0.1 M DTT, 5× RT-buffer and SuperScript reverse transcriptase (Invitrogen, Karlsruhe, Germany) [28]. Quality of transcribed cDNA was determined by a standard GAPDH gene PCR (441 bp product). First-strand cDNAs containing viral oncogene sequences were subsequently amplified with semi-nested PCR using HPV-E7 specific 5′-primers and oligo(dT) and adaptor primers (3′). PCR products were separated on a 1.2% agarose gel (see Figure S1 for representative PCR results). Both bands typical for episomal and integration status were cut out, purified using the QIAGEN Gel extraction kit (QIAGEN, Hilden, Germany) and sequenced (GATC Biotech, Konstanz, Germany). Sequence results were analyzed using the BLASTN program and further mapped using map viewer (both NCBI) [29],[30].
Detection of Integrated Papillomavirus Sequences PCR (DIPS-PCR)
Integrated papillomavirus sequences were detected using the Detection of Integrated Papillomavirus Sequences-PCR (DIPS-PCR) assay, as described earlier [31]. Briefly, genomic DNA was digested using the Sau3AI restriction enzyme and an enzyme-specific adapter was ligated to the restriction-digested DNA using T4 DNA ligase (Roche Diagnostics, Mannheim, Germany). Linear PCR was performed using 5 HPV16 specific forward primers in independent setups, all using the same specific adapter primer 1 (AP1). All independent PCRs were followed by individual exponential PCRs using further virus-specific forward primers and the AP1 reverse primer. PCR products were separated on a 1.2% agarose gel and products of interest were excised, purified, sequenced and analyzed as described before (see Figure S1 for representative PCR results).
Gene Expression Analysis
mRNA Expression Profiling
Total RNA was collected from a subset of 32 samples, randomly selected from the 75 patients in this study. Samples were analysed using Agilent Whole Human Genome 4×44K Microarrays, which represent more than 41,000 unique human transcripts. Labelling and hybridizations were performed according to the manufacturer's instructions (Agilent Technologies). Hybridized arrays were scanned using an Axon GenePix 4000B or 4200A scanner. Microarray analysis was performed using GenePix Pro 6.0.1.25. For normalization processing, the median array intensity was calculated based on the background-subtracted intensity value for all spots excluding control type spots on the array. The background-subtracted intensity value of each spot was then divided by the median array intensity of each microarray.
In the 32 tumor samples, 6 samples showed fusion products that were located within 7 genes. Normalized expression data for these genes were collected for all 32 samples. Per gene the expression in the sample with integration in that gene was compared to samples with or without an identified fusion product. Graphs were made using Graph Pad Prism 5.
HPV16 Oncogene Expression by qPCR
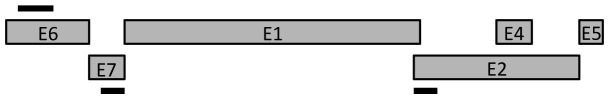
RNA isolated from 61 samples of which sufficient RNA was available after APOT analysis, was reverse transcribed using the iScript cDNA Synthesis Kit (BioRad Laboratories, Hercules, CA, USA). qPCR reactions were performed using SensiMix SYBR & Fluorescein (GC Biotech, Alphen a/d Rijn, the Netherlands). The following HPV-specific primers were used (see Figure 1): E2 (87 bp product) forward primer 5′- TGATAGTACAGACCTACGTGACCATATAGA-3′ (Primer Express v2.0 (Applied Biosystems, Carlsbad, USA)) and reverse primer 5′- ATTACAAGGCCAGAGAAATGGG-3′ (Primer Express); E6 (106 bp product) forward primer 5′-CAGTTATGCACAGAGCTGCAA-3′ [32] and reverse primer 5′- ATGACTTTGCTTTCGGGATT-3′ [32] ; E7 (86 bp product) forward primer 5′-AGAGGAGGAGGATGAAATAGATGGT-3′and reverse primer 5′-CAATATTGTAACCTTTTGTTGCAAGTG-3′ (designed using Primer-BLAST (National Center for Biotechnology Information (NCBI)) [33]. Because of limited material, E7 expression analysis was performed on 45 samples. The detection of the housekeeping gene Hypoxanthine Phosphoribosyltransferase (HPRT) was used for normalization of mRNA levels: forward primer 5′-CACTGGCAAAACAATGCAGACT -3′ and reverse primer 5′-GTCTGGCTTATATCCAACACTTCGT -3. Cell lines SiHa and CaSki were used as positive controls. The HPV18-positive cell line HeLa was used as a control for HPV16 specificity and expression of viral genes did not yield values higher than the background signal.
Figure 1. HPV16 genome showing the localization of the RT-qPCR-products obtained for E2, E6 and E7 viral oncogenes.
Fluorescence In Situ Hybridisation (FISH) for Co-localization of HPV16 and Human DNA Sequences
FISH on 4 µm thick tissue sections of formalin-fixed, paraffin-embedded tumour blocks was performed as described previously [5],[25],[34]. Briefly, sections were deparaffinised, pretreated with 85% formic acid/0.3% H2O2, 1 M NaSCN and 4 mg/ml pepsin, postfixed in 4% formaldehyde in PBS and dehydrated in an ascending ethanol series. For the co-localization experiments, we used biotin-labelled HPV16 probes (PanPath, Amsterdam, the Netherlands) together with digoxigenin-labelled BAC-clones containing human DNA sequences also identified in the viral-human fusion PCR products obtained by APOT/DIPS PCR. These BAC clones were grown according to the manufacturer's instructions (BACPAC Resources Centre, Childrens Hospital Oakland Research Institute, Oakland, USA). DNA was isolated using the Nucleobond BAC-100 kit (BioKé, Leiden, the Netherlands). Both probes were applied under a coverslip in a hybridization mixture containing 2 ng/µl HPV16 probe, 10 ng/µl BAC probe, 50% formamide, 2×SSC pH 7.0, 50× excess salmon sperm DNA (Sigma) and 10× excess human CoT DNA. Probes and tissue DNA were denatured simultaneously for 5 minutes at 80°C prior to hybridization overnight at 37°C in a humid chamber. After hybridization the preparations were washed stringently in 2× SSC+0.05% tween-20 at 42°C (2 times 5 min), 0.1× SSC at 61°C (2 times 5 min) and 4× SSC+0.05% tween-20 at RT. Biotin-labelled probe was detected using peroxidase-conjugated-avidine (Dako, Glostrup, Denmark; 1∶200 diluted in 4× SSC containing 5% non-fat dry milk; 30 minutes at 37°C) and biotin-conjugated goat-anti-avidine (Vector laboratories, Burlingame, CA; 1∶100 diluted in 4× SSC containing 5% non-fat dry milk; 30 minutes at 37°C). Thereafter, a tyramide signal amplification reaction was performed under a coverslip by applying 50 µl Cy3-labelled tyramide in PBS containing 0.1 M imidazole, pH 7.6 and 0.001% H2O2 for 10 minutes at 37°C. The digoxigenin-labelled probe was detected using mouse-anti-digoxigenin (Dako; 1∶200), followed by TRITC-conjugated rabbit-anti-mouse (Dako; 1∶1000) and finally TRITC-conjugated swine-anti-rabbit (Dako; 1∶100); incubated for 30 minutes at 37°C. Finally, slides were washed and dehydrated in an ascending ethanol series and mounted in Vectashield (Vector laboratories, Burlingame, CA) containing 4′,6-diamidino-2-phenylindole (DAPI; Sigma; 0.2 µg/µl). See Figure 2 for representative samples.
Figure 2. Representative examples of three different (Co-)FISH staining patterns: Representative sample of a HPV-positive FISH staining (A).
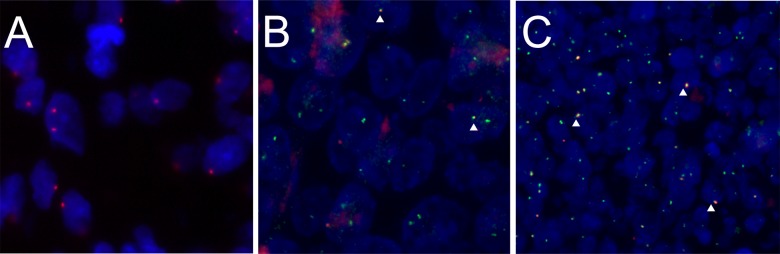
Representative examples of co-hybridization using both a specific BAC-clone and the HPV16 probe (B–C). (B) BAC-clone RP11-731I19 (containing BRE), case 2, table S1. (C) BAC-clone RP11-299P2 (containing BCL2), case 21. Arrowheads indicate co-localization.
Viral Load
Viral load of HPV16 was determined using real-time fluorescence PCR with type-specific primers and probes as described earlier [26]. Briefly, viral load was expressed as the number of HPV16 DNA copies per β-globin-gene copy. Gene copy numbers of β-globin were determined using the LightCycler-Control Kit DNA (Roche Molecular Biochemicals) according to the manufacturer's instructions as previously described [35]. Calculation of initial copy numbers in samples was performed by the LightCycler 480 software (Version 1.5) using a standard curve generated with exactly quantified HPV DNA standards (ten-fold dilution series of full length HPV16 plasmid) that were amplified in the same PCR run [35]–[37]. The analytical sensitivity of the assay was ten copies of HPV16 standard DNA. A negative control (water or DNA extracted from RTS3B cells that are negative for HPV) was included in each run and never yielded fluorescence signals above the background [35].
Statistics
Differences in viral and human gene expression levels were analyzed using a 2-tailed Fisher's exact test after testing for equality of variances. A significance level of p<0.05 was chosen for all analyses. To test whether a single sample deviated from a group of samples, SPSS was used to identify outliers. This was defined as any value that lays more than 1.5 times the interquartile range below the first quartile in a Box-and-Whisker Plot from all samples, or more than 1.5 times the interquartile range above the third quartile. All calculations were performed using IBM SPSS Statistics 19.
Results
Detection of Viral Integration by DIPS- and APOT-PCR
Table S1 and Figure S2 summarize the integration sites of HPV16 into the genome of 29 of the 75 HPV16 DNA- and p16INK4A-positive OSCC (39%) as identified by DIPS- and APOT-PCR. Exclusively episomal PCR products were detected in the remaining 46 tumors (61%).
In the 29 tumors with viral integration a total of 37 fusion products were identified, of which 10 harbored cellular sequences corresponding to intergenic regions and 27 to known or predicted genes, including 12 tumor-related genes (BCL2, BRE, EPHA7, FANCC (2×), HDAC2, INO80C, LEPREL1, SYNPO2, TP63, TRAF3, TUBD1), 5 genes involved in deregulated tumor-related pathways (ERC2, GARS, SLC7A1, SYN3, ZMAT4), and 10 genes with no known role in tumorigenesis. All genes were verified using the ATLAS of genetics and cytogenetics in oncology and hematology Database [38] and the UniProtKB Database [39]. Figure S3 summarizes viral-cellular splicing observed in this study, including a new type D splicing not described before.
Detection of Gene Expression
Subsequently, we analyzed whether HPV16 integration as detected by PCR, correlated with the level of expression of the disrupted gene. In addition, we determined whether integration correlated with the expression of the viral genes E2, E6 and E7.
Expression of Genes Disrupted by HPV
We extracted the level of expression of HPV-disrupted genes from expression profiling data of a subset of 32 OSCC, in which HPV16 integrated within a gene in 6 out of 32 OSCC, one of which contained two different integration sites (sample 10, Table S1). For each gene, its expression was compared between the single sample with HPV integration in the affected gene, the group of samples showing exclusively episomal PCR products (n = 20) and the group of samples with fusion products harboring sequences derived from other chromosomal loci (n = 11) (Figure 3). In all cases there was no significant change in the mean mRNA expression levels of the HPV-disrupted genes between the subgroups with or without a fusion product. In the tumor with the HPV integration in the particular gene, the expression did not surpass the 1.5 interquartile range (IQR) of the group of samples with integration-derived fusion products, as calculated using SPSS, in 6 of the 7 genes. In the tumor with integration in the FANCC gene, the expression of the gene fell between 1.5 and 3 times the IQR and was considered a mild outlier. However, one additional sample without a fusion product surpassed the IQR more than 3 times and was considered an extreme outlier.
Figure 3. Expression intensities for genes affected by HPV integration.

The expression of a gene, affected by HPV integration in one sample, is compared to the expression of that gene in samples where exclusively episomal PCR products could be detected using APOT- and/or DIPS-PCR, and to the expression in samples where fusion products could be identified. Bars: Mean with standard deviation.
In conclusion, our data suggest that the mRNA expression as detected by the array does not differ between a gene disrupted by HPV16 and the expression of that gene in samples where it has not been disrupted by the virus.
Viral Gene Expression
Viral gene expression could be assessed in 63 cases. APOT-PCR was able to detect a PCR product in 59 of these cases, however, the expression levels of E2, E6 and E7 as detected using RT-qPCR, varied widely. The viral gene expression of the 4 cases without detectable APOT-PCR product was nearly zero, indicating that the viral genome is not transcribed.
When comparing cases in which a fusion transcript was detected using APOT-PCR (i.e. actively transcribed fusion product) with the remaining cases, no significant differences were seen in the mean log2 expression levels of either E2 (1717 vs. 97; p = 0.308), E6 (1859 vs. 195, p = 0.344) or E7 (1724 vs. 8, p = 0.2943) (see Figure 4). Rather, a large variation in expression levels of these viral E2, E6 and E7 genes was observed, independent of HPV integration status.
Figure 4. Expression of the viral genes E2, E6 and E7 and viral load.

(A–C) Expression of the viral genes has been normalized to HPRT expression. (D) Viral load has been normalized to β-globin. E = episomal, E+I = episomal and integrated, I = integrated. Bars: Median with interquartile range.
Viral Load
To examine whether tumors with episomal virus have a higher viral load than those with integration as determined by APOT- and/or DIPS-PCR, we have performed qPCR in 73 OSCC samples. Viral load ranged from 3.4*10−6 up to 97 HPV DNA copies per cell. When comparing the average viral load in cases in which a fusion product was detected using APOT- and/or DIPS-PCR with the remaining cases, no significant differences were seen (7 vs. 8.5 HPV DNA copies/cell; p = 0.683). No correlation was seen between the mean log2 expression levels of the viral genes E2, E6 or E7 and the viral load.
Discussion
In this study we have comprehensively analyzed a large collection of 75 HPV16 positive OSCC for their HPV16 physical status (episomal vs. integrated) and its relation to viral oncogene expression and virally disrupted human genes. In particular we were interested to see if cases with proven integration would show higher E6/E7 viral oncogene expression than E2 expression as suggested by studies with cervical cancer cell lines [17],[19]–[21]. By detecting viral-human fusion products with APOT- and/or DIPS-PCR in 39% of these cases we provided direct evidence for viral integration. The so-called episomal products obtained by DIPS- and/or APOT-PCR in the remaining cases are indicative for the presence of episomal HPV DNA, although they by themselves provide no proof for this assumption, because they could eventually also arise from integrated head to tail repeats of the viral genome. In this respect, two recently published studies have shown that using DIPS-PCR with other primer combinations or multiplex PCR followed by massive parallel sequencing may detect additional sites of HPV integration which is in agreement with our findings comparing DIPS- and APOT-PCR [40],[41]. The expression of HPV16 interrupted genes as well as viral genes E2, E6 and E7 in the tumors analyzed here, however, did not differ significantly from cases where no fusion product was detected. Furthermore, the cases with integration showed no notable differences in viral load in comparison with the remaining tumors. These data indicate that HPV16 integration in these tumors does not necessarily affect the levels of HPV-disrupted human gene transcripts as detected by mRNA expression arrays and/or viral gene transcripts. Thus constitutive rather than a high level of expression of oncogene transcripts appears to be required in HPV-related OSCC.
We identified integration sites by APOT- and/or DIPS-PCR in 27 out of 75 OSCC, of which 21 showed one, 5 showed two and 1 case showed four integration sites. In addition, 8 of these 27 tumors also harbored episomal viral DNA. Exclusively viral HPV16 DNA or RNA sequences indicating the presence of episomal virus were identified in the remaining 48 OSCC. This finding is in agreement with results on a series of HPV16 positive cervical squamous cell carcinomas in which 55% showed viral integration by APOT-PCR [11]. In the OSCC, integration sites showed to be distributed all over the human genome with half of them near fragile sites and some of them in previously detected clusters of viral integration (3q28, 8q24.21, 13q22.1 and 17q21) [42]. Interestingly, in 27 out of 37 detected sequences HPV16 directly interrupted known or predicted genes. Taken together, these data suggest that HPV16 integration is not simply a random event, but rather has a preference for less protected and more accessible chromosomal regions like transcribed tumor-genes and fragile sites. It can be speculated that integration takes place in genes which are highly expressed during carcinogenesis rather than that the integration itself affecting the genomic sites is the driving force. Nevertheless, another hypothesis might be that integration occurs randomly and cells with integration in particular genes preferably develop into a carcinoma. However, this is difficult to study, because premalignant lesions with HPV-infection developing in a carcinoma are seldom found.
We had access to mRNA expression profiling data of a subset of the OSCC used in this study including 6 cases with proven integration (7 sites in total). In these cases integration of HPV16 occurred within gene sequences, including the known tumor related genes FANCC, HDAC2, SYNPO2 and TRAF3. Indeed, expression of FANCC and HDAC2 genes has been reported to play a role in HNSCC [43]–[46]. Viral integration, however, did not lead to significantly different expression of the interrupted gene in comparison to OSCC having integration in another DNA sequence or showing solely episomal virus. This is in contrast to a recent study of our group showing that integration of low-risk HPV6 in the AKR1C3 gene resulted in loss of gene expression in a laryngeal carcinoma [47]. In this case, however, the other gene copy was lost in the tumor as shown by array CGH analyses. In the 6 OSCCs studied here, no loss of the chromosomal regions containing the virally interrupted genes has been detected by array CGH (Olthof, Lam, unpublished results). This indicates that one or more expressed gene copies are still present in these tumors, which can mask a possible effect of the integration on gene expression. On the other hand, this might also point to the fact that viral integration is not per se meant to deregulate the interrupted gene in the cell, as also can be concluded by the finding of HPV16 integrated in intergenic sequences of 10 OSCC in this study. In conclusion, these data suggest that if there is an effect of viral integration on carcinogenesis, affecting the genomic site is unlikely to be the driving force in OSCC. Nevertheless, this has to be confirmed on the protein level in further studies.
Alternatively, integration might have an effect on viral oncogene E6 and/or E7 expression. In this respect it has been hypothesized that integration leads to disruption of the viral E2 gene, which as a consequence cannot regulate E6 and E7 gene expression anymore from the LCR promoter region. Our DIPS-PCR data show that integration always affected the E2 gene, either by disrupting the viral E2 gene itself (38%) or the upstream E1 gene (62%), also leading to E2 loss. Nevertheless, in most of these tumors E2 mRNA transcripts were detectable at different levels of expression and these transcript levels did not differ significantly from those detected in OSCC with episomal virus. This is in contrast to the results of Häfner et al., which showed a decrease in E5 transcript levels (downstream of E2) in uterine cervical lesions with integrated HPV16 [22]. Nevertheless, a rather constant transcription level of E6 oncogene transcripts was detected independent of the physical status of the virus in these lesions. In OSCC, we also observed a broad distribution for E6 and E7 transcript levels independent of a detected viral integration event. This points to the fact that a constitutive expression of viral transcripts seems to be required within tumors. Only in a few cases very high levels of viral gene transcripts (E2 as well as E6 and E7) were detected, indicating that mechanisms other than E2 binding to the viral LCR promoter region might influence transcription levels such as methylation of the LCR region [48],[49] [50].
Although more viral-cellular fusion products can be detected by using both DIPS- and APOT-PCR, a limitation of using these two assays simultaneously lies in the fact that they can result in a different outcome. For example, in two cases where we found integration sequences for both DIPS- and APOT-PCR, the viral-cellular fusion transcript sequence turned out to be in the opposite orientation as the sequence detected by DIPS-PCR. In a third case the integration sequences identified by both techniques were 20 Mb apart from each other on chromosome 22 (sample no 11, Table S1). This might be explained by previous studies showing that HPV-DNA integration can lead to both complex rearrangements changing the orientation of the 5′- and 3′ cellular sequences flanking the viral integration site, as well as amplifications and deletions of larger genomic regions starting at the viral integration site [51],[52].
In some cases (e.g. no. 14–16 and 18, Table S1) we detected integration by DIPS-PCR and episomal copies by APOT-PCR. An explanation for this finding could be a transcriptionally silent integration, for instance as a result of methylation, or if many episomal copies are present in a tumor, either in episomes or integrated in head-to-tail tandem repeats, the identification of fusion products might be difficult. We also analyzed tumors (e.g. no. 20–29, Table S1), in which a fusion product was detected by APOT-PCR, and DIPS-PCR resulted in episomal viral copies or no PCR product. This might be due to the detection of head-to-tail tandem repeats integrated into the genome or viral integration at other disruption sites of the viral genome that can not be detected by the used DIPS-PCR approach [41].
In conclusion our data indicate that HPV physical status (extrachromosomal episomes or host DNA integrated) does not affect the levels of viral and/or HPV-disrupted human gene transcripts. Therefore constitutive and not a high level of expression of oncogene transcripts appears to be required in HPV-related OSCC.
Supporting Information
(TIF)
(TIF)
(TIF)
(DOCX)
Acknowledgments
We would like to thank Oliver Siefer, Dirk Vellinga and Zlatan Mujagic for technical assistance. Primer sequences for HPRT were kindly provided by Prof. Dr. L. Hofland from Erasmus Medical Centre, Rotterdam, the Netherlands.
Funding Statement
This study was supported by the Koeln Fortune Program, Faculty of Medicine, University of Cologne, Cologne, Germany (Grant number 40/2010 to CUH) and the Jean Uhrmacher Foundation, Cologne, Germany. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Parkin DM, Bray F, Ferlay J, Pisani P (2005) Global cancer statistics, 2002. CA: A Cancer Journal for Clinicians 55: 74–108. [DOI] [PubMed] [Google Scholar]
- 2. Kamangar F, Dores GM, Anderson WF (2006) Patterns of cancer incidence, mortality, and prevalence across five continents: defining priorities to reduce cancer disparities in different geographic regions of the world. Journal of Clinical Oncology 24: 2137–2150 10.1200/JCO.2005.05.2308 [DOI] [PubMed] [Google Scholar]
- 3. Jemal A, Bray F, Center MM, Ferlay J, Ward E, et al. (2011) Global cancer statistics. CA: A Cancer Journal for Clinicians 61: 69–90 10.3322/caac.20107 [DOI] [PubMed] [Google Scholar]
- 4. Olthof NC, Straetmans JMJAA, Snoeck R, Ramaekers FCS, Kremer B, et al. (2011) Next-generation treatment strategies for human papillomavirus-related head and neck squamous cell carcinoma: where do we go? Rev Med Virol 10.1002/rmv.714 [DOI] [PubMed] [Google Scholar]
- 5. Hafkamp HC, Manni JJ, Haesevoets A, Voogd AC, Schepers M, et al. (2008) Marked differences in survival rate between smokers and nonsmokers with HPV 16-associated tonsillar carcinomas. Int J Cancer 122: 2656–2664 10.1002/ijc.23458 [DOI] [PubMed] [Google Scholar]
- 6. Klussmann JP, Mooren JJ, Lehnen M, Claessen SMH, Stenner M, et al. (2009) Genetic signatures of HPV-related and unrelated oropharyngeal carcinoma and their prognostic implications. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research 15: 1779–1786 10.1158/1078-0432.CCR-08-1463 [DOI] [PubMed] [Google Scholar]
- 7. Mellin H, Dahlgren L, Munck-Wikland E, Lindholm J, Rabbani H, et al. (2002) Human papillomavirus type 16 is episomal and a high viral load may be correlated to better prognosis in tonsillar cancer. Int J Cancer 102: 152–158 10.1002/(ISSN)1097-0215 [DOI] [PubMed] [Google Scholar]
- 8. Koskinen WJ, Chen RW, Leivo I, M Kitie A, B Ck L, et al. (2003) Prevalence and physical status of human papillomavirus in squamous cell carcinomas of the head and neck. Int J Cancer 107: 401–406 10.1002/(ISSN)1097-0215 [DOI] [PubMed] [Google Scholar]
- 9. Begum S, Cao D, Gillison M, Zahurak M, Westra WH (2005) Tissue distribution of human papillomavirus 16 DNA integration in patients with tonsillar carcinoma. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research 11: 5694–5699 10.1158/1078-0432.CCR-05-0587 [DOI] [PubMed] [Google Scholar]
- 10. Kraus I, Driesch C, Vinokurova S, Hovig E, Schneider A, et al. (2008) The Majority of Viral-Cellular Fusion Transcripts in Cervical Carcinomas Cotranscribe Cellular Sequences of Known or Predicted Genes. Cancer Res 68: 2514–2522 10.1158/0008-5472.CAN-07-2776 [DOI] [PubMed] [Google Scholar]
- 11. Vinokurova S, Wentzensen N, Kraus I, Klaes R, Driesch C, et al. (2008) Type-Dependent Integration Frequency of Human Papillomavirus Genomes in Cervical Lesions. Cancer Res 68: 307–313 10.1158/0008-5472.CAN-07-2754 [DOI] [PubMed] [Google Scholar]
- 12. Mooren JJ, Kremer B, Claessen SMH, Voogd AC, Bot FJ, et al. (2013) Chromosome stability in tonsillar squamous cell carcinoma is associated with HPV16 integration and indicates a favorable prognosis. Int J Cancer 132: 1781–1789 10.1002/ijc.27846 [DOI] [PubMed] [Google Scholar]
- 13. Lace MJ, Anson JR, Klussmann JP, Wang DH, Smith EM, et al. (2011) Human papillomavirus type 16 (HPV-16) genomes integrated in head and neck cancers and in HPV-16-immortalized human keratinocyte clones express chimeric virus-cell mRNAs similar to those found in cervical cancers. J Virol 85: 1645–1654 10.1128/JVI.02093-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wiest T, Schwarz E, Enders C, Flechtenmacher C, Bosch FX (2002) Involvement of intact HPV16 E6/E7 gene expression in head and neck cancers with unaltered p53 status and perturbed pRb cell cycle control. Oncogene 21: 1510–1517 10.1038/sj.onc.1205214 [DOI] [PubMed] [Google Scholar]
- 15. Arias-Pulido H, Peyton CL, Joste NE, Vargas H, Wheeler CM (2006) Human papillomavirus type 16 integration in cervical carcinoma in situ and in invasive cervical cancer. J Clin Microbiol 44: 1755–1762 10.1128/JCM.44.5.1755-1762.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ziegert C, Wentzensen N, Vinokurova S, Kisseljov F, Einenkel J, et al. (2003) A comprehensive analysis of HPV integration loci in anogenital lesions combining transcript and genome-based amplification techniques. Oncogene 22: 3977–3984 10.1038/sj.onc.1206629 [DOI] [PubMed] [Google Scholar]
- 17. Rampias T, Sasaki C, Weinberger P, Psyrri A (2009) E6 and e7 gene silencing and transformed phenotype of human papillomavirus 16-positive oropharyngeal cancer cells. CancerSpectrum Knowledge Environment 101: 412–423 10.1093/jnci/djp017 [DOI] [PubMed] [Google Scholar]
- 18. Jeon S, Lambert PF (1995) Integration of human papillomavirus type 16 DNA into the human genome leads to increased stability of E6 and E7 mRNAs: implications for cervical carcinogenesis. Proc Natl Acad Sci USA 92: 1654–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Narisawa-Saito M, Kiyono T (2007) Basic mechanisms of high-risk human papillomavirus-induced carcinogenesis: Roles of E6 and E7 proteins. Cancer Science 98: 1505–1511 10.1111/cas.2007.98.issue-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ganguly N, Parihar SP (2009) Human papillomavirus E6 and E7 oncoproteins as risk factors for tumorigenesis. J Biosci 34: 113–123. [DOI] [PubMed] [Google Scholar]
- 21. Gammoh N, Grm HS, Massimi P, Banks L (2006) Regulation of human papillomavirus type 16 E7 activity through direct protein interaction with the E2 transcriptional activator. J Virol 80: 1787–1797 10.1128/JVI.80.4.1787-1797.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Häfner N, Driesch C, Gajda M, Jansen L, Kirchmayr R, et al. (2008) Integration of the HPV16 genome does not invariably result in high levels of viral oncogene transcripts. Oncogene 27: 1610–1617 10.1038/sj.onc.1210791 [DOI] [PubMed] [Google Scholar]
- 23. Klussmann JP, Weissenborn SJ, Wieland U, Dries V, Kolligs J, et al. (2001) Prevalence, distribution, and viral load of human papillomavirus 16 DNA in tonsillar carcinomas. Cancer 92: 2875–2884. [DOI] [PubMed] [Google Scholar]
- 24. Klussmann JP, Weissenborn S, Fuchs PG (2001) Human papillomavirus infection as a risk factor for squamous-cell carcinoma of the head and neck. N Engl J Med 345: 376–authorreply377. [PubMed] [Google Scholar]
- 25. Hafkamp HC, Speel EJ, Haesevoets A, Bot FJ, Dinjens WN, et al. (2003) A subset of head and neck squamous cell carcinomas exhibits integration of HPV 16/18 DNA and overexpression of p16INK4A and p53 in the absence of mutations in p53 exons 5–8. Int J Cancer 107: 394–400 10.1002/(ISSN)1097-0215 [DOI] [PubMed] [Google Scholar]
- 26. Klussmann JP, Gültekin E, Weissenborn SJ, Wieland U, Dries V, et al. (2003) Expression of p16 protein identifies a distinct entity of tonsillar carcinomas associated with human papillomavirus. The American Journal of Pathology 162: 747–753 10.1016/S0002-9440(10)63871-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Klaes R, Woerner SM, Ridder R, Wentzensen N, Duerst M, et al. (1999) Detection of high-risk cervical intraepithelial neoplasia and cervical cancer by amplification of transcripts derived from integrated papillomavirus oncogenes. Cancer Res 59: 6132–6136. [PubMed] [Google Scholar]
- 28. Vinokurova S, Wentzensen N, Einenkel J, Klaes R, Ziegert C, et al. (2005) Clonal history of papillomavirus-induced dysplasia in the female lower genital tract. CancerSpectrum Knowledge Environment 97: 1816–1821 10.1093/jnci/dji428 [DOI] [PubMed] [Google Scholar]
- 29.NCBI BLAST website. Available: http://blast.ncbi.nlm.nih.gov/. Accessed 2013 Sep 9.
- 30.NCBI Map Viewer website. Available: http://www.ncbi.nlm.nih.gov/projects/mapview/map_search.cgi?taxid=9606&build=36.3. Accessed 2013 Sep 27.
- 31. Luft F, Klaes R, Nees M, Dürst M, Heilmann V, et al. (2001) Detection of integrated papillomavirus sequences by ligation-mediated PCR (DIPS-PCR) and molecular characterization in cervical cancer cells. Int J Cancer 92: 9–17. [PubMed] [Google Scholar]
- 32. Chow LT, Broker TR (1997) In vitro experimental systems for HPV: epithelial raft cultures for investigations of viral reproduction and pathogenesis and for genetic analyses of viral proteins and regulatory sequences. Clin Dermatol 15: 217–227. [DOI] [PubMed] [Google Scholar]
- 33.NCBI Primer-BLAST website. Available: http://www.ncbi.nlm.nih.gov/tools/primer-blast/. Accessed 2013 Sep 27.
- 34. Hopman AHN, Kamps MA, Smedts F, Speel E-JM, Herrington CS, et al. (2005) HPV in situ hybridization: impact of different protocols on the detection of integrated HPV. Int J Cancer 115: 419–428 10.1002/ijc.20862 [DOI] [PubMed] [Google Scholar]
- 35. Weissenborn SJ, Wieland U, Junk M, Pfister H (2010) Quantification of beta-human papillomavirus DNA by real-time PCR. Nat Protoc 5: 1–13 10.1038/nprot.2009.153 [DOI] [PubMed] [Google Scholar]
- 36. Seedorf K, Krämmer G, Dürst M, Suhai S, Röwekamp WG (1985) Human papillomavirus type 16 DNA sequence. Virology 145: 181–185. [DOI] [PubMed] [Google Scholar]
- 37. Weissenborn SJ, Funke AM, Hellmich M, Mallmann P, Fuchs PG, et al. (2003) Oncogenic human papillomavirus DNA loads in human immunodeficiency virus-positive women with high-grade cervical lesions are strongly elevated. J Clin Microbiol 41: 2763–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Atlas of genetics and cytogenetics in oncogoly and haematology website. Available: http://atlasgeneticsoncology.org. Accessed 2013 Sep 27.
- 39.UniProt Knowledgebase: a hub of integrated protein data website. Available: http://www.uniprot.org. Accessed 2013 Sep 27.
- 40. Li H, Yang Y, Zhang R, Cai Y, Yang X, et al. (2013) Preferential sites for the integration and disruption of human papillomavirus 16 in cervical lesions. Journal of Clinical Virology: The Official Publication of the Pan American Society for Clinical Virology 56: 342–347 10.1016/j.jcv.2012.12.014 [DOI] [PubMed] [Google Scholar]
- 41. Xu B, Chotewutmontri S, Wolf S, Klos U, Schmitz M, et al. (2013) Multiplex Identification of Human Papillomavirus 16 DNA Integration Sites in Cervical Carcinomas. PLoS ONE 8: e66693 10.1371/journal.pone.0066693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schmitz M, Driesch C, Beer-Grondke K, Jansen L, Runnebaum IB, et al. (2012) Loss of gene function as a consequence of human papillomavirus DNA integration. Int J Cancer 131: E593–E602 10.1002/ijc.27433 [DOI] [PubMed] [Google Scholar]
- 43. Ghosh A, Ghosh S, Maiti GP, Mukherjee S, Mukherjee N, et al. (2012) Association of FANCC and PTCH1 with the development of early dysplastic lesions of the head and neck. Ann Surg Oncol 19 Suppl 3: S528–S538 10.1245/s10434-011-1991-x [DOI] [PubMed] [Google Scholar]
- 44. Wreesmann VB, Estilo C, Eisele DW, Singh B, Wang SJ (2007) Downregulation of Fanconi anemia genes in sporadic head and neck squamous cell carcinoma. ORL J Otorhinolaryngol Relat Spec 69: 218–225 10.1159/000101542 [DOI] [PubMed] [Google Scholar]
- 45. Chang C-C, Lin B-R, Chen S-T, Hsieh T-H, Li Y-J, et al. (2011) HDAC2 promotes cell migration/invasion abilities through HIF-1α stabilization in human oral squamous cell carcinoma. Journal of Oral Pathology \& Medicine: Official Publication of the International Association of Oral Pathologists and the American Academy of Oral Pathology 40: 567–575 10.1111/j.1600-0714.2011.01009.x [DOI] [PubMed] [Google Scholar]
- 46. Chang H-H, Chiang C-P, Hung H-C, Lin C-Y, Deng Y-T, et al. (2009) Histone deacetylase 2 expression predicts poorer prognosis in oral cancer patients. Oral Oncol 45: 610–614 10.1016/j.oraloncology.2008.08.011 [DOI] [PubMed] [Google Scholar]
- 47. Huebbers CU, Preuss SF, Kolligs J, Vent J, Stenner M, et al. (2013) Integration of HPV6 and downregulation of AKR1C3 expression mark malignant transformation in a patient with juvenile-onset laryngeal papillomatosis. PLoS ONE 8: e57207 10.1371/journal.pone.0057207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chaiwongkot A, Vinokurova S, Pientong C, Ekalaksananan T, Kongyingyoes B, et al. (2013) Differential methylation of E2 binding sites in episomal and integrated HPV 16 genomes in preinvasive and invasive cervical lesions. Int J Cancer 132: 2087–2094 10.1002/ijc.27906 [DOI] [PubMed] [Google Scholar]
- 49. Park I-S, Chang X, Loyo M, Wu G, Chuang A, et al. (2011) Characterization of the methylation patterns in human papillomavirus type 16 viral DNA in head and neck cancers. Cancer Prev Res (Phila) 4: 207–217 10.1158/1940-6207.CAPR-10-0147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Balderas Loaeza A, Anaya Saavedra G, Ramirez Amador VA, Guido Jimenez MC, Kalantari M, et al. (2007) Human papillomavirus-16 DNA methylation patterns support a causal association of the virus with oral squamous cell carcinomas. Int J Cancer 120: 2165–2169 10.1002/ijc.22563 [DOI] [PubMed] [Google Scholar]
- 51. Peter M, Stransky N, Couturier J, Hupé P, Barillot E, et al. (2010) Frequent genomic structural alterations at HPV insertion sites in cervical carcinoma. J Pathol 221: 320–330 10.1002/path.2713 [DOI] [PubMed] [Google Scholar]
- 52. Gallego MI, Schoenmakers EF, Van de Ven WJ, Lazo PA (1997) Complex genomic rearrangement within the 12q15 multiple aberration region induced by integrated human papillomavirus 18 in a cervical carcinoma cell line. Mol Carcinog 19: 114–121 doi:10.1002/(SICI)1098-2744(199707)19:2<114::AID-MC6>3.0.CO;2-F [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(TIF)
(TIF)
(TIF)
(DOCX)