

Abstract
Genomic imprinting is a common epigenetic phenomenon in mammals. Dysregulation of genomic imprinting has been implicated in a variety of human diseases. ZFP57 is a master regulator in genomic imprinting. Loss of ZFP57 causes loss of DNA methylation imprint at multiple imprinted regions in mouse embryos, as well as in embryonic stem (ES) cells. Similarly, mutations in human ZFP57 result in hypomethylation at many imprinted regions and are associated with transient neonatal diabetes and other human diseases. Mouse and human Zfp57 genes are located in the same syntenic block. However, mouse and human ZFP57 proteins only display about 50% sequence identity with different number of zinc fingers. It is not clear if they share similar mechanisms in maintaining genomic imprinting. Here we report that mouse and human ZFP57 proteins are functionally interchangeable. Expression of exogenous wild-type human ZFP57 could maintain DNA methylation imprint at three imprinted regions in mouse ES cells in the absence of endogenous mouse ZFP57. However, mutant human ZFP57 proteins containing the mutations found in human patients could not substitute for endogenous mouse ZFP57 in maintaining genomic imprinting in ES cells. Like mouse ZFP57, human ZFP57 and its mutant proteins could bind to mouse KAP1, the universal cofactor for KRAB zinc finger proteins, in mouse ES cells. Thus, we conclude that mouse and human ZFP57 are orthologs despite relatively low sequence identity and mouse ES cell system that we had established before is a valuable system for functional analyses of wild-type and mutant human ZFP57 proteins.
Keywords: ZFP57, genomic imprinting, DNA methylation imprint, loss-of-function mutation, ortholog, ES cells, Snrpn, Zac1, Dlk1-Dio3 imprinted region, bisulphite analysis
Introduction
Genomic imprinting is a kind of parental effect observed in eutherian mammals, marsupials and plants.1-5 It is essential for the survival of mammalian embryos. It is estimated that about 1% of mammalian genes may be imprinted. Based on Harwell’s website (http://www.mousebook.org/catalog.php?catalog=imprinting), roughly 150 imprinted genes have been identified so far.1 These imprinted genes display mono-allelic expression patterns so that they are preferentially expressed either from the maternal allele or from the paternal allele.2,3 Most of these imprinted genes are clustered and co-regulated by a cis-acting imprinting control region termed ICR.6 A hallmark for the ICRs is that they all contain germline-derived differentially methylated regions (DMRs) that are either methylated on the maternal chromosome or on the paternal chromosome.2-4,7 Differential methylation at the ICRs is required for maintaining genomic imprinting at the imprinted regions, i.e., parental origin-dependent mono-allelic expression of the corresponding imprinted genes.1-3,6
ZFP57 is a member of KRAB zinc finger proteins.8-10 Mouse ZFP57 binds to its cofactor mouse KAP1/Trim28 via its KRAB box.8 Similar to human ZFP57,9 mouse ZFP57 is a master regulator of genomic imprinting.8 It is required for maintaining DNA methylation imprint at a large subset of imprinted regions in mouse embryos as well as in mouse ES cells.8,11,12 We also found that the KRAB box of mouse ZFP57 is necessary for the maintenance of DNA methylation imprint at the Snrpn DMR and the IG-DMR of Dlk1-Dio3 imprinted region.11 KAP1/Trim28 is also required for maintaining genomic imprinting in mouse.11,13 ZFP57 binds to the imprinting control regions through its zinc finger domains.8,12,14 It recruits KAP1/Trim28 which in turn recruits DNA methyltransferases to maintain DNA methylation imprint at the imprinted regions.11,12
Mouse Zfp57 and its human homolog are located in the same syntenic block.8 The encoded mouse and human ZFP57 proteins only share about 50% sequence identity with different number of zinc fingers.8 Mutations in human ZFP57 cause hypomethylation at multiple imprinted regions.9,15 These mutations were found in patients with transient neonatal diabetes.9,15 With the exception of one frame-shift mutation located within the N-terminal KRAB box, these human mutations are distributed throughout the zinc finger domains of human ZFP57.9,15 Two highly conserved zinc finger domains between mouse and human ZFP57 proteins were thought to be responsible for binding to a consensus hexanucleotide sequence TGCCGC present in all imprinting control regions examined.12 Indeed, mouse and human ZFP57 peptides containing a point mutation in these two highly conserved zinc fingers had been shown to lose their DNA binding abilities to the oligonucleotides harboring the consensus hexanucleotide recognition sequence.14,16 However, it is not known how other point mutations in human ZFP57 may affect its function in genomic imprinting. By taking advantage of the mouse ES cell system that we had established before,11 we expressed wild-type human ZFP57 and three mutant human ZFP57 proteins, each containing a point mutation found in human patients, in mouse ES cells and examined their capabilities to substitute for endogenous mouse ZFP57 in maintaining genomic imprinting.
Results
Conservation between mouse and human ZFP57 proteins
In our previous study, we discovered that mouse Zfp57 gene and its human homolog are located in the same syntenic block.8 We constructed the cDNAs for three mutant human ZFP57 proteins by site-directed mutagenesis with corresponding point mutations found in human patients (Fig. 1).9 The predicted protein sequence for human ZFP57 based on the UniGene database in GenBank has an extra sequence of 20 amino acids compared with what was reported in the previous paper (Fig. 1).9 This extra sequence of 20 amino acids was confirmed by sequencing of the entire cDNA clone that we obtained (Fig. 1). Mouse and human ZFP57 proteins display about 50% identity in primary amino acid sequence alignment, with a highly conserved N-terminal KRAB box followed by different numbers of zinc fingers with variable sequence conservation.8 It is estimated that mouse ZFP57 protein (mZFP57) has at most five zinc fingers and human ZFP57 (hZFP57) may contain up to seven zinc fingers (Fig. 2A).8,9,12,14 Three point mutations (R248H, H277N, and H458D) are located, respectively, in the third (ZF3), fourth (ZF4), and seventh (ZF7) zinc fingers of human ZFP57 protein (Fig. 2A). The amino acids for these three point mutations are identical between mouse and human ZFP57 proteins (Fig. 1).9,14,16

Figure 1. Sequence alignment is shown for the 3XFLAG-tagged human ZFP57 aligned with three predicted human ZFP57 proteins in the UniGene database. We constructed a cDNA encoding human ZFP57 protein with a 3XFLAG tag (hZFP57–3XFLAG) at the carboxyl end. This hZFP57–3XFLAG fusion protein has an identical amino acid sequence with the predicted human ZFP57 protein NM_001109809 in the GenBank database until it reaches the junction of the 3XFLAG tag. The protein sequence for hZFP57–3XFLAG was aligned with three predicted human ZFP57 proteins (NM_001109809, BC157878 and BC171888) in the GenBank UniGene database. Shaded amino acids indicate three amino acids that are different in one of the three predicted proteins in the GenBank database. The amino acid positions for three mutations found in human patients are marked by an asterisk (*). Substitution mutations in bold letters are underlined and shown right above the mutated amino acids in the wild-type human ZFP57 protein. R248H, H277N, and H458D correspond to R228H, H257N, and H438D, respectively, that were reported in the original paper.9 The amino acids from D538 to K559 for the 3XFLAG tag are indicated by bold letters at the carboxyl end of hZFP57–3XFLAG.
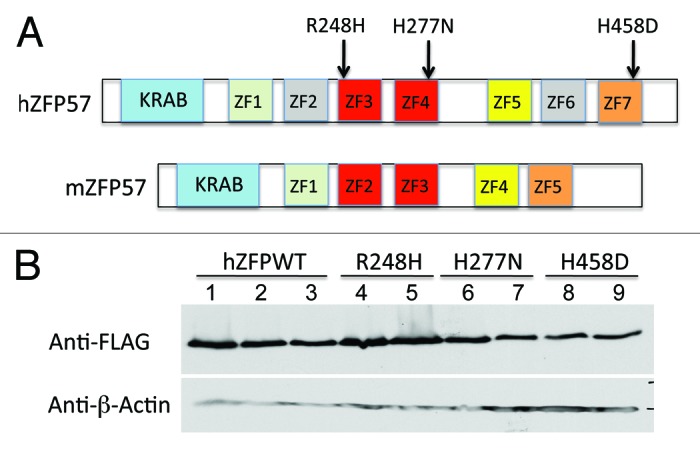
Figure 2. Wild-type and mutant human ZFP57 proteins were expressed in mouse ES cells lacking endogenous mouse ZFP57. (A) Schematic diagrams are shown for the wild-type human ZFP57 (hZFP57) and mouse ZFP57 (mZFP57) proteins. Same colors indicate the conserved domains with high amino acid sequence identities. Arrows with corresponding amino acid substitutions above them indicate the positions of three point mutations found in human patients. Both hZFP57 and mZFP57 contain a highly conserved KRAB box (in blue) at the N-terminus. The full-length hZFP57 is predicted to have seven zinc finger (ZF) domains. R248H and H277N are located in ZF3 and ZF4 of hZFP57 (both in red), respectively. H458D is located in the last zinc finger ZF7 of hZFP57 (in orange). By contrast, mouse ZFP57 (mZFP57) protein only has five zinc finger domains (ZF1-ZF5). ZF3 and ZF4 of hZFP57 are highly homologous to ZF2 and ZF3 of mZFP57. ZF1, ZF5, and ZF7 of hZFP57 share high sequence similarities to the corresponding ZF1, ZF4 and ZF5 of mZFP57. (B) Western blot analysis of the whole-cell lysate isolated from the Zfp57-null mouse ES clones expressing the wild-type human ZFP57 (hZFPWT) or one of three mutant human ZFP57 proteins (R248H, H277N and H458D) with a 3XFLAG tag at the C-terminus. Lanes 1–3, three independent Zfp57-null mouse ES clones expressing the wild-type human ZFP57 (hZFPWT). Lanes 4–5, two independent Zfp57-null mouse ES clones expressing the R248H mutant human ZFP57 protein. Lanes 6–7, two independent Zfp57-null mouse ES clones expressing the H277N mutant human ZFP57 protein. Lanes 8–9, two independent Zfp57-null mouse ES clones expressing the H458D mutant human ZFP57 protein. Top panel, western blot probed with mouse monoclonal anti-FLAG antibody. Bottom panel, western blot probed with mouse monoclonal antibody against β-actin.
The most highly conserved regions between mouse and human ZFP57 proteins are their N-terminal KRAB boxes and two zinc fingers in the middle.8 These two highly conserved zinc fingers correspond to the second (ZF2) and third (ZF3) zinc fingers of mouse ZFP57 protein and the third (ZF3) and fourth (ZF4) zinc fingers of human ZFP57 protein, respectively (Fig. 2A).8,12,14 Indeed, these two zinc fingers were thought to form the DNA binding domain of ZFP57 proteins that recognized the consensus hexanucleotide sequence TGCCGC.12,14,16 Two of three point mutations in this study are located in these two highly conserved zinc fingers (R248H in ZF3 and H277N in ZF4 of hZFP57) (Fig. 2A). The third point mutation (H458D) is located in the seventh zinc finger (ZF7) of hZFP57 that is homologous to ZF5 of mZFP57 (Fig. 2A). ZF1 and ZF5 of hZFP57 share high sequence similarities to ZF1 and ZF4 of mZFP57, respectively (Fig. 2A). However, ZF2 and ZF6 of hZFP57 are not conserved in mZFP57 (Fig. 2A).
Expression of human ZFP57 in mouse ES cells
Through PCR amplification of the cDNAs encoding the wild-type human ZFP57 and the 3XFLAG tag, we inserted a tag of the 3XFLAG epitope at the carboxyl end of human ZFP57 (Fig. 1). Similarly, the 3XFLAG tag was fused in-frame with three mutant human ZFP57 proteins at the carboxyl end carrying a point mutation found in human patients. The wild-type human ZFP57 and three mutant human ZFP57 proteins were expressed in the mouse ES cell system that we had constructed previously for testing the abilities of the exogenous proteins to substitute for the endogenous mouse ZFP57 in maintaining DNA methylation imprint.11 In this mouse ES cell system, both alleles of the endogenous mouse Zfp57 gene were flanked by two LoxP sites and were converted into two deleted null alleles of Zfp57 upon Cre recombinase-mediated excision (Fig. 3D).11 Also upon Cre recombinase-mediated integration, the plasmid vector carrying an exogenous transgene was integrated into the hprt locus on the X chromosome and the exogenous protein product was constitutively expressed from the transgene driven by a chick β-actin and CMV hybrid promoter.11 Multiple independent ES clones were obtained for expressing the wild-type or one of three mutant human ZFP57 proteins. We confirmed that both alleles of endogenous mouse Zfp57 gene for these mouse ES clones were deleted after Cre recombinase-mediated excision by PCR-based genotyping assays (Fig. 3D). Whole-cell lysate was obtained from these ES clones and subjected to western blot analysis with mouse monoclonal antibody against the FLAG epitope to detect the tagged human ZFP57–3XFLAG proteins (Fig. 2B). Mouse ES clones expressed the wild-type and mutant human ZFP57–3XFLAG proteins at similar levels when the ES cells for these ES clones were directly lysed in the whole-cell lysis buffer for western blot analysis (Fig. 2B).
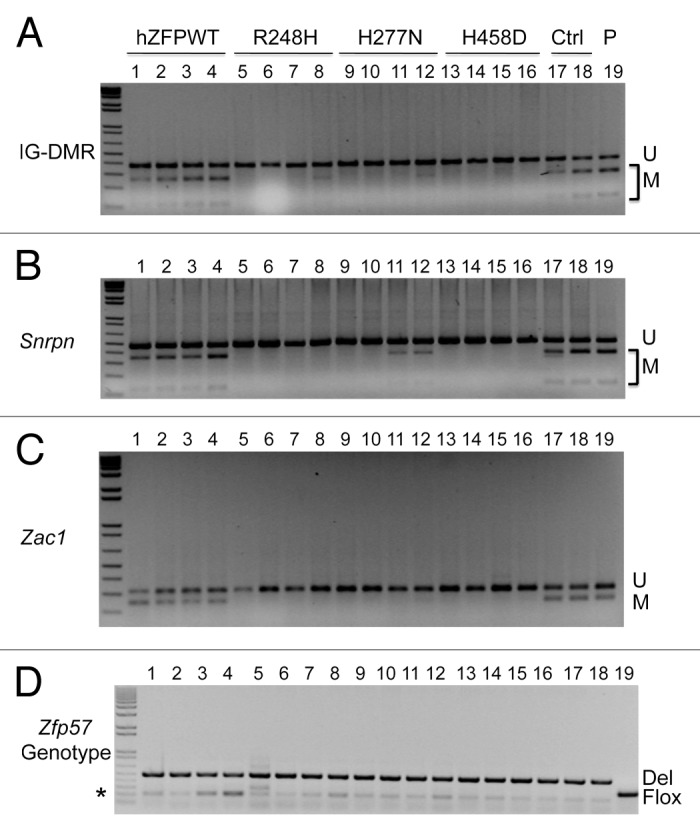
Figure 3. COBRA analysis indicates that wild-type but not mutant human ZFP57 can substitute for endogenous mouse ZFP57 in maintaining DNA methylation imprint at three imprinted regions in mouse ES cells. The mouse ES cell system we constructed before was used to drive expression of wild-type or mutant human ZFP57–3XFLAG proteins in the Zfp57-null mouse ES cells.11 Exogenous human ZFP57–3XFLAG protein was constitutively expressed from an integrated transgene at the hprt locus after Cre recombinase-mediated recombination.11 Simultaneously, expression of the endogenous mouse ZFP57 was turned off after excision of both floxed alleles of the mouse Zfp57 gene. Genomic DNA was isolated from these ES clones grown on gelatin-coated plates before being subjected to COBRA. Lanes 1–4, four independent Zfp57-null mouse ES clones expressing the wild-type human ZFP57–3XFLAG protein (hZFPWT). Lanes 5–8, four independent Zfp57-null mouse ES clones expressing the R248H mutant human ZFP57–3XFLAG protein. Lanes 9–12, four independent Zfp57-null mouse ES clones expressing the H277N mutant human ZFP57–3XFLAG protein. Lanes 13–16, four independent Zfp57-null mouse ES clones expressing the H458D mutant human ZFP57–3XFLAG protein. Lanes 17–18, two independent control (Ctrl) Zfp57-null mouse ES clones expressing mouse ZFP57 tagged with a Myc epitope and six histidines at the C-terminal end. Lane 19, parental (P) mouse ES cells with two floxed alleles of Zfp57. U, unmethylated bisulphite PCR product after restriction enzyme digestion. M, methylated bisulphite PCR product after restriction enzyme digestion. (A) COBRA analysis of the IG-DMR region at the Dlk1-Dio3 imprinted domain with TaqI digestion. (B) COBRA analysis of the Snrpn imprinted region with HhaI digestion. (C) COBRA analysis of the Zac1 imprinted region with TaqI digestion. (D) PCR genotyping of the endogenous mouse Zfp57 alleles in the ES clones.8 Flox, the floxed allele of Zfp57 with two LoxP sites flanking two exons of Zfp57.8 As expected, parental cells in Lane 19 contain two floxed alleles. One ES clone expressing R248H mutant human ZFP57 in Lane 5 also has some ES cells containing a floxed allele of mouse Zfp57. Del, the deleted allele of Zfp57 after Cre recombinase-mediated excision of the floxed allele of Zfp57.8 Asterisk (*), the wild-type allele of mouse Zfp57. It appears that a small fraction of the cells were the feeder cells carried over with the ES cells when the ES clones were plated on gelatin-coated plates for genomic DNA preparation.
Interaction of human ZFP57 with mouse KAP1
ZFP57 is a KRAB zinc finger protein. Previously, we demonstrated that mouse ZFP57 binds to its cofactor KAP1 via its KRAB box.8,11 KAP1 is an obligate cofactor for a few hundred KRAB zinc finger proteins in mouse and human genomes.10,17 KAP1 is likely an abundant protein in mouse ES cells.18 We found that endogenous KAP1 protein level remained constant in the mouse ES clones expressing wild-type or mutant human ZFP57 proteins (Fig. 4A). To determine if human ZFP57 can also bind to mouse KAP1 in mouse ES cells lacking endogenous mouse ZFP57, we subjected these mouse ES clones to co-immunoprecipitation (co-IP) interaction assay when the ES cells were lysed in RIPA buffer (see Materials and Methods). Mouse monoclonal anti-FLAG M2 affinity gel (Sigma catalog #A2220) was used for co-IP to pull down other proteins associated with 3XFLAG-tagged human ZFP57 proteins in mouse ES cells containing two deleted alleles of mouse Zfp57 (Fig. 3D). When the immunoprecipitate was probed with the affinity-purified rabbit polyclonal antibodies against mouse KAP1, a specific band corresponding to mouse KAP1 was present in the co-IP samples derived from the Zfp57-null mouse ES clones expressing wild-type human ZFP57 (Lane 2) or expressing one of three mutant human ZFP57 proteins (R248H in Lane 3, H277N in Lane 4, H458D in Lane 5) (Fig. 4A). As expected, no KAP1 was present in the immunoprecipitate derived from the Zfp57-null ES clone 2B6 when precipitated with mouse monoclonal anti-FLAG M2 affinity gel (Lane 1) (Fig. 4A). Since KAP1 is ubiquitously expressed and is an abundant protein in mouse ES cells,18 we reason that KAP1 was not a limiting factor in the co-IP assay. We estimated the relative binding affinities of mouse KAP1 for wild-type or mutant human ZFP57 proteins based on the ratio of KAP1 vs. 3XFLAG-tagged human ZFP57 proteins present in the immunoprecipitate (Fig. 4B). Compared with the wild-type human ZFP57 in Lane 2, we observed slightly more KAP1 present in Lane 4 and Lane 5 for the human ZFP57 point mutants, H277N and H458D, respectively, and slightly less KAP1 in Lane 3 for the human ZFP57 mutant R248H (Fig. 4A and B). We repeated the co-IP at least three times for the wild-type and mutant human ZFP57 proteins (data not shown). It appears that wild-type and mutant human ZFP57 proteins had comparable binding affinities for mouse KAP1 in mouse ES cells based on the co-IP interaction assay.
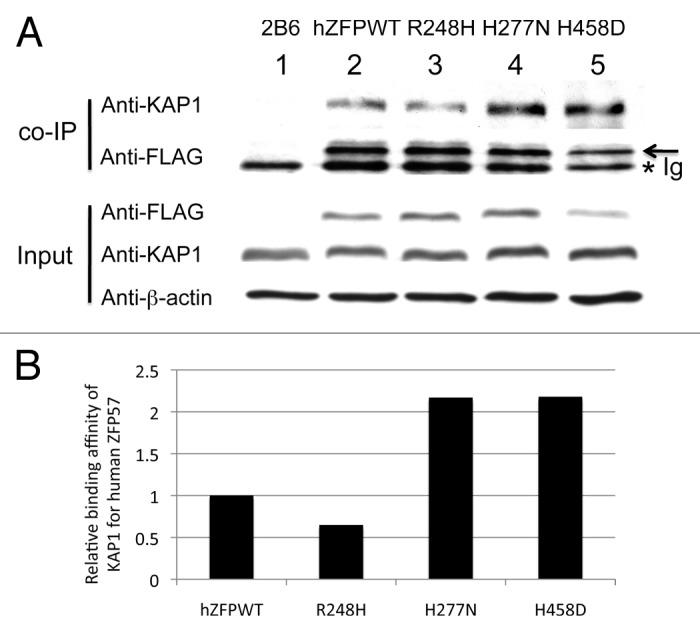
Figure 4. Wild-type and mutant human ZFP57 proteins bound to mouse KAP1 in mouse ES cells. (A) Co-immunoprecipitation (co-IP) was performed for endogenous mouse KAP1 and constitutively expressed exogenous human wild-type or one of the mutant ZFP57 proteins in mouse ES cells. ES cells were lysed in RIPA buffer for co-IP. Mouse monoclonal anti-FLAG M2 affinity gel (Sigma catalog #A2220) was used for immunoprecipitation (IP) to pull down other proteins associated with constitutively expressed exogenous wild-type or mutant human ZFP57 proteins in mouse ES cells. The immunoprecipitate was subjected to western blot (WB) analysis with affinity-purified rabbit polyclonal antibodies against mouse KAP1.20 The immunoprecipitate was also probed with mouse anti-FLAG monoclonal antibody (Sigma catalog #F1804). An asterisk indicates the position of immunoglobulin (Ig). An arrow marks the position of the wild-type or mutant human ZFP57 proteins with the 3XFLAG tag. The input samples were probed with these two antibodies as well as the mouse monoclonal anti-β-actin antibody (Sigma catalog #A1978). Lane 1, a Zfp57-null ES clone 2B6.8,11 Lane 2, an ES clone expressing wild-type human ZFP57 (hZFPWT). Lane 3, an ES clone expressing R248H mutant human ZFP57–3XFLAG protein. Lane 4, an ES clone expressing H277N mutant human ZFP57–3XFLAG protein. Lane 5, an ES clone expressing H458D mutant human ZFP57–3XFLAG protein. (B) Wild-type and mutant human ZFP57 proteins exhibited similar binding affinities for KAP1 in mouse ES cells. The intensities of each band in the western blot were measured by Image J. Relative binding affinities were calculated based on the ratios of the band intensity of anti-KAP1 over the band intensity of anti-FLAG for each ES sample in the co-IP western blot in A, with the binding affinity of the wild-type human ZFP57 for mouse KAP1 set at 1. We reason that mouse KAP1 is much more abundant than human ZFP57 in mouse ES cells and KAP1 was not a limiting factor in the co-IP assay. Thus, we did not take the concentration of KAP1 in various ES clones into account in estimating the relative binding affinities between mouse KAP1 and human ZFP57 proteins.
Human ZFP57 could substitute for mouse ZFP57 in maintaining genomic imprinting at three imprinted regions
Mouse and human ZFP57 proteins appear to play similar roles in maintaining genomic imprinting. Mouse Zfp57 is a maternal-zygotic effect gene.8 Loss of just the zygotic function of ZFP57 led to partial loss of DNA methylation imprint, whereas elimination of both maternal and zygotic ZFP57 resulted in complete loss of methylation at a large subset of imprinted regions.8 Similarly, mutations in human ZFP57 had been shown to be associated with hypomethylation at multiple imprinted regions as well.9 Mouse Zfp57 gene and its human counterpart are located in the same syntenic block.8 However, mouse and human ZFP57 proteins only share about 50% sequence identity with different number of zinc fingers.8 We wonder if mouse and human ZFP57 are functionally interchangeable. Thus, we tested if exogenous human ZFP57 could substitute for endogenous mouse ZFP57 in maintaining genomic imprinting in the mouse ES cell system that we established before.11 Based on PCR genotyping, both floxed (Flox) alleles of Zfp57 present in the parental ES cells (Lane 19) were excised by transiently expressed Cre recombinase and converted into two deleted (Del) alleles of Zfp57 in almost all ES clones analyzed in this study except that one ES clone expressing the human ZFP57 point mutant, R248H (Lane 5), still retained a floxed allele of Zfp57 in a small fraction of ES cells (Fig. 3D). We isolated genomic DNA samples from these ES clones grown on gelatin-coated plates and then subjected these individual mouse ES clones to DNA methylation analysis to determine if constitutive expression of wild-type human ZFP57 protein or any of three mutant human ZFP57 proteins can substitute for endogenous mouse ZFP57 in maintaining genomic imprinting in mouse ES cells (Fig. 3).
COBRA analysis was performed on the genomic DNA samples isolated from four independent mouse ES clones that constitutively express wild-type human ZFP57 protein (Lanes 1–4 of Fig. 3). We also performed COBRA analysis on genomic DNA harvested from four independent mouse ES clones for each mutant human ZFP57 protein (Lanes 5–16 of Fig. 3). As positive controls for COBRA, we utilized genomic DNA samples from the parental (P) ES clone expressing endogenous mouse ZFP57 (Lane 19 of Fig. 3), as well as the ES clones expressing the exogenous mouse ZFP57 with a tag of the Myc epitope and six histidines at the carboxyl end in the absence of endogenous mouse ZFP57 (Lane 17 and Lane 18 of Fig. 3).
As shown in Figure 3A, significant DNA methylation was observed at the IG-DMR of Dlk1-Dio3 imprinted region in all ES clones expressing the wild-type human ZFP57 (Lanes 1–4 of Fig. 3A) as well as in all the positive control samples (Lanes 17–19 of Fig. 3A). By contrast, DNA methylation imprint was almost completely missing in the ES clones expressing the mutant human ZFP57 proteins (Lanes 5–16 of Fig. 3A). These results demonstrate that wild-type human ZFP57 but not mutant human ZFP57 proteins can substitute for endogenous mouse ZFP57 in maintaining DNA methylation imprint at the IG-DMR of Dlk1-Dio3 imprinted region.
We also performed COBRA analysis for the Snrpn DMR region of these ES clones (Fig. 3B). Similar to the IG-DMR region, significant DNA methylation was detected at the Snrpn DMR in the ES clones expressing the wild-type human ZFP57 protein (Lanes 1–4 of Fig. 3B). DNA methylation was also found at the Snrpn DMR in the positive control ES clones expressing mouse ZFP57 (Lanes 17–18) as well as in the parental ES cells (Lane 19) (Fig. 3B). Faint methylated (M) COBRA product was observed in two out of four ES clones expressing H277N mutant human ZFP57 protein (Lanes 11–12 of Fig. 3B). However, the Snrpn DMR was completely unmethylated in other ES clones expressing the mutant human ZFP57 proteins (Lanes 5–10 and Lanes 13–16 of Fig. 3B). These data prove that wild-type human ZFP57 but not mutant human ZFP57 proteins can substitute for endogenous mouse ZFP57 in maintaining DNA methylation imprint at the Snrpn DMR.
In addition, we performed COBRA analysis of the Zac1 DMR region for these ES clones (Fig. 3C). Significant DNA methylation was detected at the Zac1 DMR in the ES clones expressing the wild-type human ZFP57 protein (Lanes 1–4) that was comparable to the methylation level observed in the control ES clones expressing mouse ZFP57 (Lanes 17–18) or in the parental ES cells (Lane 19) (Fig. 3C). By contrast, Zac1 DMR was completely unmethylated in the ES clones expressing any of three mutant human ZFP57 proteins (Lanes 5–16 of Fig. 3C). These data suggest that wild-type human ZFP57 but not mutant human ZFP57 proteins can substitute for endogenous mouse ZFP57 in maintaining DNA methylation imprint at the Zac1 DMR as well.
Besides these three imprinted regions, we also examined DNA methylation imprint at some other imprinted domains. However, DNA methylation imprint was already variably lost in the parental ES cells at the Peg1, Peg3, Peg5, Peg10, and Rasgrf1 DMR regions (C Ray, F Bell and X Li, data not shown). Thus, we could not use this mouse ES cell system to test if human ZFP57 proteins can replace endogenous mouse ZFP57 protein in maintaining DNA methylation imprint at these five other imprinted regions.
Maintenance of DNA methylation imprint by human ZFP57 was confirmed at three imprinted regions by bisulphite sequencing
To further confirm if human ZFP57 could indeed substitute for the endogenous mouse ZFP57 in maintaining genomic imprinting in mouse ES cells, we subjected the bisulphite mutagenized genomic DNA samples from these ES clones to bacterial colony bisulphite sequencing. Compared with the parental cells in Clone #19 (Fig. 5E), DNA methylation imprint was largely maintained at the IG-DMR of the Dlk1-Dio3 imprinted region in 1 out of 2 positive control ES clones expressing exogenous mouse ZFP57 (Clone #18 in Fig. 5E) and 2 out of 4 ES clones expressing the wild-type human ZFP57 (Clone #3–4 in Fig. 5A). Medium-level of DNA methylation was present at the IG-DMR of Clone #2 expressing the wild-type human ZFP57 (Fig. 5A). Low levels of DNA methylation imprint was also observed in Clone #17 (Fig. 5E) and Clone #1 (Fig. 5A) expressing exogenous mouse ZFP57 and human ZFP57, respectively. Except for Clone #8 expressing R248H mutant protein with some sporadic methylation at the CpG sites of the IG-DMR, all other 11 ES clones expressing one of three mutant human ZFP57 proteins had completely lost DNA methylation imprint at the IG-DMR region (Fig. 5B–D). Thus, bisulphite sequencing results at the IG-DMR were consistent with what we had observed with the COBRA analysis of these ES clones (compare Fig. 5 with Fig. 3A). These data indicate that wild-type human ZFP57 but not the mutant human ZFP57 proteins can functionally replace the endogenous mouse ZFP57 protein in the maintenance of DNA methylation imprint at the IG-DMR of the Dlk1-Dio3 imprinted region.
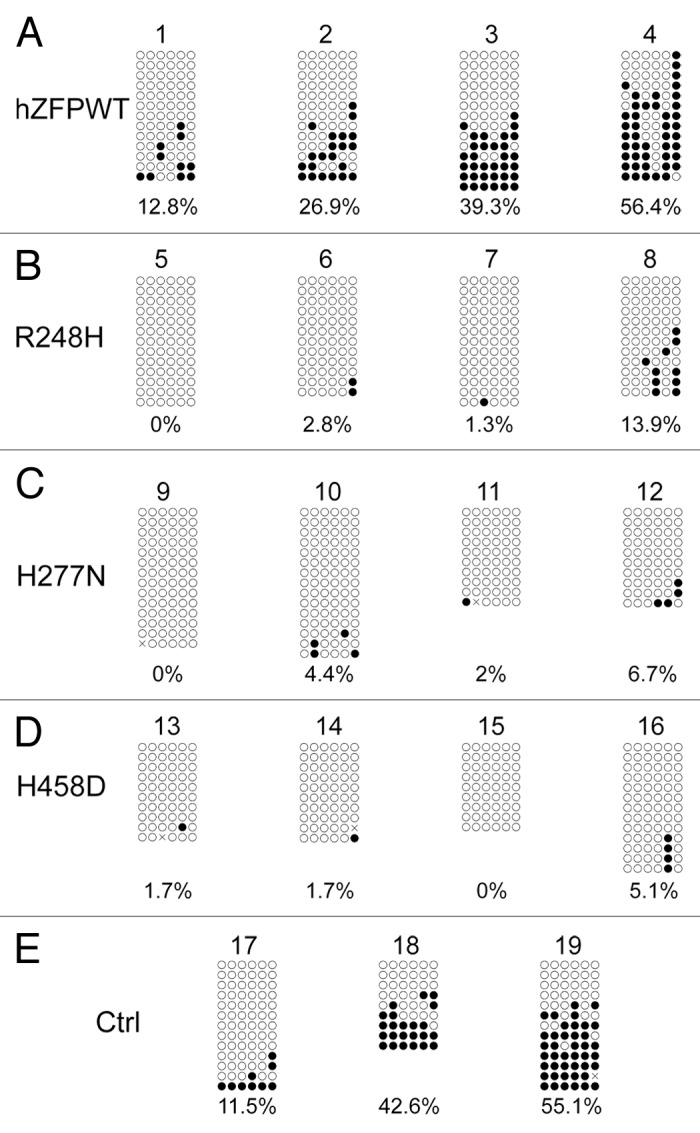
Figure 5. Bisulphite sequencing result confirms that wild-type human ZFP57 can substitute for endogenous mouse ZFP57 in maintaining DNA methylation imprint at the IG-DMR of the Dlk1-Dio3 imprinted region in mouse ES cells. Genomic DNA samples from the ES clones plated on the gelatin-coated plates were subjected to bisulphite mutagenesis, PCR amplification and bacterial colony sequencing. Filled circle, a methylated CpG. Unfilled circle, an unmethylated CpG. Cross (X), a CpG site that its methylation status cannot be determined. Each row stands for a template DNA molecule directly sequenced from a single bacterial colony containing the bisulphite PCR product. Numbers 1 to 19 indicate the same ES clones that were subjected to COBRA analysis in Figure 3. Percentage of methylated CpG sites in all sequenced bacterial colonies for each ES clone is listed directly below the diagram illustrating methylated and unmethylated CpG sites. (A) Four independent Zfp57-null mouse ES clones expressing the wild-type human ZFP57–3XFLAG protein (hZFPWT). (B) Four independent Zfp57-null mouse ES clones expressing the R248H mutant human ZFP57–3XFLAG protein. (C) Four independent Zfp57-null mouse ES clones expressing the H277N mutant human ZFP57–3XFLAG protein. (D) Four independent Zfp57-null mouse ES clones expressing the H458D mutant human ZFP57–3XFLAG protein. (E) control (Ctrl) ES clones. 17 and 18, two independent Zfp57-null mouse ES clones expressing mouse ZFP57 tagged with a Myc epitope and six histidines at the C-terminal end. 19, parental (P) mouse ES cells with two floxed alleles of Zfp57.
We performed similar bisulphite sequencing analysis for Snrpn DMR in these ES clones. As shown in Figure 6, DNA methylation imprint was maintained at the Snrpn DMR in the parental ES cells (Clone #19 in Fig. 6E), two positive control ES clones expressing exogenous mouse ZFP57 (Clone #17–18 in Fig. 6E) and four ES clones expressing wild-type human ZFP57 (Clone #1–4 in Fig. 6A). Similar to the COBRA analysis (Fig. 3B), a low level of DNA methylation was observed for two ES clones expressing the human ZFP57 point mutant, H277N, (Clone #11–12 in Fig. 6C). However, none of the other ES clones expressing the mutant human ZFP57 proteins exhibited any significant methylation at the Snrpn DMR (Fig. 6B–D). Together with the COBRA data (Fig. 3B), we conclude that human ZFP57 protein can substitute for mouse ZFP57 in maintaining genomic imprinting at the Snrpn imprinted region in mouse ES cells.

Figure 6. Bisulphite sequencing result confirms that wild-type human ZFP57 can substitute for endogenous mouse ZFP57 in maintaining DNA methylation imprint at the Snrpn DMR region in mouse ES cells. Genomic DNA samples from the ES clones plated on the gelatin-coated plates were subjected to bisulphite mutagenesis, PCR amplification and bacterial colony sequencing. Filled circle, a methylated CpG. Unfilled circle, an unmethylated CpG. Cross (X), a CpG site with unknown methylation status. Each row stands for a template DNA molecule directly sequenced from a single bacterial colony containing the bisulphite PCR product. Numbers 1 to 19 indicate the same ES clones that were subjected to COBRA analysis in Figure 3. Percentage of methylated CpG sites in all sequenced bacterial colonies for each ES clone is listed directly below the diagram illustrating methylated and unmethylated CpG sites. (A) Four independent Zfp57-null mouse ES clones expressing the wild-type human ZFP57–3XFLAG protein (hZFPWT). (B) Four independent Zfp57-null mouse ES clones expressing the R248H mutant human ZFP57–3XFLAG protein. (C) Four independent Zfp57-null mouse ES clones expressing the H277N mutant human ZFP57–3XFLAG protein. (D) Four independent Zfp57-null mouse ES clones expressing the H458D mutant human ZFP57–3XFLAG protein. (E) Control (Ctrl) ES clones. Numbers 17 and 18, two independent Zfp57-null mouse ES clones expressing mouse ZFP57 tagged with a Myc epitope and six histidines at the C-terminal end. 19, parental (P) mouse ES cells with two floxed alleles of Zfp57.
We also examined DNA methylation imprint at the Zac1 imprinted region in these ES clones by bisulphite sequencing (Fig. 7). We observed similar levels of DNA methylation imprint at the Zac1 imprinted region in the parental ES cells (Clone #19 in Fig. 7E), two positive control ES clones expressing exogenous mouse ZFP57 (Clone #17–18 in Fig. 7E) and four ES clones expressing wild-type human ZFP57 (Clone #1–4 in Fig. 7A). Very little methylation was present in the ES clones expressing one of three mutant human ZFP57 proteins (Fig. 7B–D). Bisulphite sequencing results of these ES clones are consistent with the COBRA analysis at the Zac1 imprinted region (compare Fig. 7 with Fig. 3C). These data demonstrate that human ZFP57 can maintain DNA methylation imprint at the Zac1 imprinted region in mouse ES cells in the absence of endogenous mouse ZFP57.

Figure 7. Bisulphite sequencing result confirms that wild-type human ZFP57 can substitute for endogenous mouse ZFP57 in maintaining DNA methylation imprint at the Zac1 DMR region in mouse ES cells. Genomic DNA samples from the ES clones plated on the gelatin-coated plates were subjected to bisulphite mutagenesis, PCR amplification and bacterial colony sequencing. Filled circle, a methylated CpG. Unfilled circle, an unmethylated CpG. Cross (X), a CpG site with unknown methylation status. Each row stands for a template DNA molecule directly sequenced from a single bacterial colony containing the bisulphite PCR product. Numbers 1 to 19 indicate the same ES clones that were subjected to COBRA analysis in Figure 3. Percentage of methylated CpG sites in all sequenced bacterial colonies for each ES clone is listed directly below the diagram illustrating methylated and unmethylated CpG sites. (A) Four independent Zfp57-null mouse ES clones expressing the wild-type human ZFP57–3XFLAG protein (hZFPWT). (B) Four independent Zfp57-null mouse ES clones expressing the R248H mutant human ZFP57–3XFLAG protein. (C) Four independent Zfp57-null mouse ES clones expressing the H277N mutant human ZFP57–3XFLAG protein. (D) Four independent Zfp57-null mouse ES clones expressing the H458D mutant human ZFP57–3XFLAG protein. (E) Control (Ctrl) ES clones. Numbers 17 and 18, two independent Zfp57-null mouse ES clones expressing mouse ZFP57 tagged with a Myc epitope and six histidines at the C-terminal end. Number 19, parental (P) mouse ES cells with two floxed alleles of Zfp57.
It appears that many of the sequenced bacterial clones had exact same CpG methylation patterns at these three imprinted regions (Figs. 5–7). This was particularly the case for the ES clones expressing one of the three mutant human ZFP57 proteins because most sequenced bacterial clones were completely unmethylated. Besides methylation patterns at the CpG sites, it is known that unconverted non-CpG sites in the sequenced imprinted regions due to incomplete bisulphite mutagenesis can also be used to identify unique clones that were amplified from a unique starting template DNA molecule. We applied this DNA methylation analysis method to the same bisulphite sequencing data we obtained through random bacterial colony sequencing of bisulphite PCR product. Indeed, we found that there were some changes in percentage of DNA methylation for each sequenced sample when only unique clones were used for quantification of DNA methylation level at these three imprinted regions (Figs. S1–S3). For example, percentage of DNA methylation was somewhat increased at the IG-DMR or Snrpn DMR in five ES clones expressing the wild-type human or mouse ZFP57 protein (see Clone #1–3, Clone #17 and Clone #19, compare Fig. 5 with Fig. S1 and compare Fig. 6 with Fig. S2). It remained the same for some ES samples (e.g., Clone #4 and Clone #18 in Fig. 5 and Fig. S1). Percentage of DNA methylation was somewhat decreased in other ES samples (e.g., Clone #18 in Fig. 6 and Fig. S2, Clone #1 in Fig. 7 and Fig. S3). Nevertheless, we found that DNA methylation imprint was generally maintained in the ES clones expressing wild-type human or mouse ZFP57 protein but not in the ES clones expressing one of three mutant human ZFP57 proteins (Figs. S1–S3). As explained in Discussion, both DNA methylation analysis methods have some advantages and some disadvantages. Therefore, the sequence analysis results with both methods were presented as two separate sets of figures (Figs. 5–7; Figs. S1–S3). In general, they are in agreement with each other.
Taken together, both COBRA and bisulphite sequencing analyses showed that wild-type human ZFP57 but not mutant human ZFP57 proteins could functionally substitute for mouse ZFP57 in maintaining genomic imprinting at the IG-DMR of the Dlk1-Dio3 imprinted region, the Snrpn imprinted region and the Zac1 imprinted region.
Discussion
Despite that they are located in the same syntenic block regions, mouse and human ZFP57 proteins only share about 50% sequence identity based on amino acid sequence alignment. They even have different numbers of zinc fingers. Two zinc fingers in the middle of mouse and human ZFP57 proteins have relatively high sequence conservation and have been proposed to be responsible for recognition of the hexanucleotide consensus target sequence present in all imprinting control regions.12,14,16 The other most highly conserved region is the N-terminal KRAB box that mediates the interaction between KRAB zinc finger proteins and their universal cofactor KAP1/Trim28. Indeed, our co-IP results in this study indicate that human ZFP57 proteins can bind to mouse KAP1/Trim28 in mouse ES cells. These findings have prompted us to hypothesize that human ZFP57 may be able to form similar functional complexes in mouse ES cells and substitute for endogenous mouse ZFP57 in maintaining DNA methylation imprint. These predictions were borne out. Based on COBRA and bisulphite sequencing analyses, three imprinted control regions remained methylated in mouse ES cells expressing the wild-type human ZFP57 when the endogenous mouse Zfp57 gene product was ablated by Cre recombinase. These results demonstrate that mouse and human ZFP57 proteins are orthologs despite relatively low sequence identity and they are functionally interchangeable in maintaining genomic imprinting. It will be interesting to test if ZFP57 homologous proteins from other species will also be able to functionally substitute for mouse ZFP57 in genomic imprinting. This may provide insights into the evolution of ZFP57 family proteins as well as evolution of genomic imprinting. This will be the subject of a future study.
Mutations in human ZFP57 cause hypomethylation at multiple imprinted regions, similar to what had been observed in the Zfp57 knockout mutant mouse. These point mutations are located mainly in or around the zinc fingers of human ZFP57 protein.9,15 Interestingly, the peptides containing one of two point mutations in the most highly conserved two zinc fingers lost their abilities to bind to the oligonucleotides containing the consensus target sequence TGCCGC for ZFP57.14,16However, it is unclear whether the full-length human ZFP57 containing any of these two point mutations may also have lost their DNA-binding abilities for the target sequence. It is still unknown if these are strong loss-of-function mutations either. Thus, we had constructed three full-length mutant human ZFP57 proteins that contain a point mutation found in human patients (Fig. 1). The amino acids for these three point mutations are identical between mouse and human ZFP57 proteins. Two of them are the same substitution mutations that had been shown to affect DNA-binding ability of the peptides containing two conserved zinc fingers in previous studies. One mutation is located in the last zinc finger of human ZFP57 protein and is also identical between mouse and human ZFP57 proteins. As expected, all three mutant human ZFP57 proteins failed to maintain DNA methylation imprint in mouse ES cells in the absence of endogenous mouse ZFP57 protein (Figs. 3 and 5–7). Therefore, our results have confirmed that two point mutations in two highly conserved zinc finger regions are strong loss-of-function mutations. Furthermore, point mutations in other regions may also act as strong loss-of-function mutations like H458D in the last zinc finger of human ZFP57. This means that other zinc fingers of human ZFP57 may also be involved in binding to target DNA sequences, together with two highly conserved zinc fingers. Alternatively, other zinc fingers may mediate the interactions of human ZFP57 with other proteins or other components of the complex required for the maintenance of DNA methylation imprint. All three mutant human ZFP57 proteins in this study could bind to the universal cofactor KAP1 presumably through an intact N-terminal KRAB box with relative binding affinities comparable to the wild-type human ZFP57. However, they could not substitute for endogenous mouse ZFP57 in maintaining genomic imprinting. Thus, we hypothesize that some unknown components that interact with ZFP57 may be needed for maintaining genomic imprinting besides KAP1.
Transient transfection of Cre recombinase was sufficient to cause complete excision of both floxed alleles of Zfp57 in almost all ES clones except for ES Clone #5 expressing human point mutant ZFP57 protein, R248H (Fig. 3D). This suggests that any DNA methylation imprint present in the genomic DNA samples expressing the wild-type or a mutant human ZFP57 protein was not due to residual parental ES cells carrying an intact Zfp57 floxed allele that could still express endogenous mouse ZFP57. Indeed, no methylation was observed at three imprinted regions even in ES Clone #5 despite that it still had some parental ES cells with an incompletely excised floxed allele of Zfp57 (Fig. 43D).
We cultured ES clones on gelatin-coated plates before genomic DNA preparation in order to reduce the percentage of feeder cells present in the ES cell population for each ES clone. However, a small fraction of feeder cells were carried over with ES cells when these ES clones were plated onto gelatin-coated plates. To minimize feeder cell contamination in the ES cell populations, we usually diluted ES cells before plating them onto gelatin-coated plates, and would not harvest them for genomic DNA preparation until the ES cell culture became almost confluent. Indeed, only a faint PCR product corresponding to the wild-type allele of mouse Zfp57 was present in most ES genomic DNA samples based on PCR genotyping (Fig. 3D). Nevertheless, a relatively strong PCR product derived from the wild-type allele of mouse Zfp57 was observed in ES Clone #4 expressing the wild-type human ZFP57 (Fig. 3D). We reason that presence of small fractions of feeder cells did not create a significant problem for our COBRA and bisulphite sequencing analyses of most ES genomic DNA samples because high level of DNA methylation was only observed in the ES clones expressing the wild-type mouse or human ZFP57 protein but not in the ES clones expressing any of three mutant human ZFP57 proteins (Fig. 3D). Furthermore, the PCR product for the wild-type allele of Zfp57 may be preferentially amplified in three-primer PCR reactions for genotyping because of its much smaller size than that of the deleted allele of Zfp57 (see Materials and Methods). Therefore, the actual percentage of feeder cells present in these ES clones may be less than it appeared in the gel shown in Figure 3D. If a large portion of genomic DNA for ES Clone #4 did come from the carryover feeder cells, then some DNA molecules presented in COBRA and bisulphite sequencing results for this ES genomic DNA sample may have reflected the DNA methylation status of three imprinted regions in the feeder cells instead (Figs. 3 and 5–7). This may partially explain relatively high DNA methylation levels observed at the IG-DMR and Snrpn DMR for this ES genomic DNA sample (Figs. 3, 5, and 6), although it is not clear why DNA methylation level at the Zac1 DMR was shown to be lower in this ES clone compared with most other ES clones expressing the wild-type mouse or human ZFP57 (Fig. 7).
It is also notable that two ES clones (Clone #1 and Clone #17) expressing the wild-type human or mouse ZFP57 did not retain much DNA methylation imprint at the IG-DMR region (Fig. 5; Fig. S1). By contrast, we observed substantial amount of DNA methylation at the Snrpn DMR in these two ES clones (Fig. 6; Fig. S2). DNA methylation level was actually higher at the Zac1 DMR in these two ES clones compared with most other ES clones expressing the wild-type mouse or human ZFP57 protein (Fig. 7; Fig. S3). Thus, it is likely that there are some variations in the DNA methylation levels at different imprinted regions in these ES clones. Interestingly, there were a lot of variations in percentage of methylation among six CpG sites of the sequenced IG-DMR region within each ES clone expressing wild-type human or mouse ZFP57 (Fig. 5; Fig. S1). By contrast, methylation level was relatively more constant at each CpG site of the sequenced Snrpn DMR or Zac1 DMR region within each ES clone (Figs. 6 and 7; Figs. S2–S3). This may imply that DNA methylatioin imprint at the IG-DMR may not be stably maintained as it is in the other two imprinted regions.
After our detailed methylation analysis, we found that most sequenced bacterial clones exhibited different methylation patterns at the CpG sites or contained unique unconverted non-CpG sites. Thus, they were unique clones. However, some bacterial clones did appear to have the same CpG methylation patterns without unique unconverted non-CpG sites. Therefore, these were non-unique clones. Due to high conversion efficiencies of the Zymo Research’s bisulphite mutagenesis kit, sometimes it was hard to find any unconverted non-CpG sites. While unique clones were derived from different template DNA molecules, it does not necessarily mean that non-unique clones were amplified from the same original template DNA molecules. We reason that at least some of the non-unique clones shown here were probably amplified from different template DNA molecules because sufficient amount of template DNA was used for PCR amplification after bisulphite mutagenesis to ensure that there was enough diversity for the starting template DNA molecules. In addition, we constantly achieved good PCR amplification efficiency for all DNA samples (Fig. 3). Thus, we believe that it was unlikely that non-unique clones with the same CpG methylation pattern but without unique unconverted non-CpG sites were all amplified from the same original template DNA molecule.
If we measure DNA methylation level solely based on unique clones, we could possibly over-estimate the actual DNA methylation level at these three imprinted regions (e.g., compare Clone # 17 in Fig. 6 and Fig. S2, Clone #19 in Fig. 6 and Fig. S2). Alternatively, we could under-estimate the actual DNA methylation level (e.g., compare Clone # 18 in Fig. 6 and Fig. S2). For the reasons stated above, it seems to us that DNA methylation analysis with just unique clones is not always advantageous and sometimes it may not be accurate either. Thus, we included the data from both analyses with one set of figures covering every sequenced bacterial clones (Figs. 5–7) and the other set of figures listing just unique clones (Figs. S1–S3). No matter which methylation analysis method was used, our findings remained the same: ES clones expressing the wild-type mouse or human ZFP57 protein retained significant DNA methylation imprint at these three imprinted regions whereas ES clones expressing a mutant human ZFP57 protein could not maintain DNA methylation imprint.
Our analyses of three mutant human ZFP57 proteins are reminiscent of what have been observed with mutations in other human proteins with DNA-binding abilities such as human p53.19 Indeed, mutations in the DNA-binding domain of p53 result in three different phenotypes: (1) defective in direct contact with DNA base pairs but with normal protein stability; (2) disrupted local structure of protein; (3) denatured protein.19 In this case, two human ZFP57 point mutations (R248H and H277N) may belong to the first class mutation with defect in DNA contact. The point mutation H458D could be a mutation in the first class with defect in DNA contact if the last zinc finger (ZF7) of human ZFP57 is also required for DNA binding. Alternatively, H458D could be a second-class mutation with disrupted local structure of protein that affects its interaction with other components in the ZFP57 complex. Since all three mutant proteins could bind to mouse KAP1 in co-IP with comparable binding affinities similar to the wild-type human ZFP57, it is unlikely that any of these three mutations belongs to the third class of mutation that results in denatured proteins. More mutations in human ZFP57 may need to be examined to test this hypothesis.
In summary, the results obtained so far with our ES cell system have proven that it is a valuable system for structure/function analysis of wild-type and mutant human ZFP57 proteins. Previously, we had successfully demonstrated that KAP1 facilitated the interactions between ZFP57 and DNA methyltransferases with this mouse ES cell system.11 We will continue to take advantage of this ES cell system to dissect the molecular mechanisms of ZFP57-associated complexes in genomic imprinting and human diseases.
Materials and Methods
Construction of cDNAs encoding mutant human ZFP57
Site-directed mutagenesis approach was employed to introduce the point mutations into the cDNA encoding human ZFP57. The QuikChange kit from Stratagene was used for this mutagenesis experiment.
Tagging human ZFP57 with the 3XFLAG epitope
PCR was used to amplify and fuse the cDNA fragments for human ZFP57 and the 3XFLAG epitope. The cDNA fragment for the 3XFLAG epitope was amplified by PCR from the plasmid p3XFLAG-CMV (Sigma). Then it was digested with SalI and EcoRV before being ligated into the SalI and EcoRV sites of the pBluescript II KS(-) vector containing the cDNA for human ZFP57. After this ligation, the 3XFLAG epitope was fused in-frame with the C-terminal end of human ZFP57 to generate the hybrid cDNA. Similarly, three mutant human proteins were tagged with the 3XFLAG epitope at the C-terminal end.
Expression of human ZFP57 proteins in mouse ES cells
The mouse ES cell system that we had constructed previously was employed to express wild-type and mutant human ZFP57 proteins.11 Specifically, the cDNA fragments encoding the wild-type and mutant human ZFP57–3XFLAG proteins were digested with KpnI and EcoRV before being inserted into the KpnI and SmaI sites of the p2Lox vector containing the LoxP and LoxM sites for Cre recombinase-mediated integration.11 The resultant plasmids were individually transfected into the mouse ES cells by electroporation, together with the pCAGGS-Cre plasmid that expresses Cre recombinase under the control of chicken β-actin and CMV hybrid promoter. Transient expression of Cre recombinase resulted in integration of the cDNA fragment into the hprt locus on the X chromosome of mouse ES cells.11 This integration event also restored expression of the puromycin-resistant gene product at the hprt locus of this mouse ES cell system.11 One to two days after electroporation, mouse ES cells were subjected to selection with 1.5 μg of puromycin in the ES cell medium until puromycin-resistant ES cell colonies appeared in about 10 days. ES colonies were picked individually and expanded on the irradiated feeder cells. They were plated on gelatin-coated plates for one generation to remove most of the feeder cells before being harvested for genomic DNA preparation. These genomic DNA samples that were depleted of feeder cells were used for PCR-based genotyping and DNA methylation analysis.
Tagged wild-type or mutant human ZFP57–3XFLAG proteins were constitutively expressed from the integrated transgene at the hprt locus driven by the chicken β-actin and CMV hybrid promoter placed immediately in front of the human ZFP57 cDNA fragment. In the meantime, both floxed alleles of the endogenous mouse Zfp57 gene were deleted upon Cre recombinase-mediated excision. Deletion of endogenous mouse Zfp57 gene was confirmed by PCR-based genotyping (Fig. 3D).8,11
Harvesting whole-cell lysate for western blot analysis
Expression of wild-type and mutant human ZFP57 proteins was examined by western blot analysis with whole-cell lysate samples directly harvested from the ES clones. ES clones were grown on gelatin-coated 6-well plates to deplete the feeder cells. Once the ES cells reached confluency, 200 μl of whole-cell lysis buffer comprising of 50 mM Tris, 10 mM EDTA, 1% SDS and 20 mM DTT was added to each well after removing the medium from the ES cells. The cells were scrapped off the plates in the whole-cell lysis buffer and the lysate samples were heated at 95 °C for 10 min to denature proteins before being loaded for western blot analysis. Mouse monoclonal anti-FLAG antibody (Sigma, catalog # F1804) was used for western blot analysis to detect 3XFLAG-tagged human ZFP57 proteins.
Co-immunoprecipitation (co-IP) analysis of human ZFP57 and mouse KAP1
RIPA buffer was used for isolation of protein samples for co-IP analysis. This RIPA buffer was made of 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 2 mM EDTA, 1% NP-40, 0.1% SDS. PMSF was freshly added to the RIPA buffer before use at a final concentration of 1 mM, together with the complete cocktail protease inhibitors from Roche (catalog # 11697498001). ES clones were grown on gelatin-coated 6-well plates to remove most of the feeder cells. Once the ES cells reached confluency, 0.5 ml of RIPA buffer with PMSF and cocktail protease inhibitors was added to each well to lyse the ES cells. Anti-c-Myc agarose affinity gel (Sigma catalog # A7470) was used for pre-clearing of the lysate samples for co-IP. Mouse monoclonal anti-FLAG M2 affinity gel (Sigma catalog #A2220) was used for immunoprecipitation. Affinity-purified rabbit polyclonal antibodies against mouse KAP1 were used to probe the immunoprecipitate and input samples.8,11,20 Mouse monoclonal anti-FLAG antibody (Sigma, catalog # F1804) was used for western blot analysis to detect 3XFLAG-tagged human ZFP57 proteins in the immunoprecipitate and input samples. The input samples were also probed with the mouse monoclonal anti-β-actin antibody (Sigma catalog #A1978).
PCR-based genotyping of the endogenous Zfp57 alleles in mouse ES cells
Similar to our previous study,8 we used three primers for genotyping the wild-type, floxed (Flox) and deleted (Del) alleles of Zfp57 in mouse ES cells by PCR amplification. The size for the PCR product of the wild-type allele is 226 bp. The PCR products amplified from the floxed allele and deleted allele are 307 bp and 512 bp in size, respectively.
Combined bisulphite restriction analysis (COBRA)
ES clones for genomic DNA preparation were grown on gelatin-coated 6-well plates to deplete most of the feeder cells. Once the ES cells reached confluency on 6-well plates, they were harvested for genomic DNA preparation. Roughly 500 ng of genomic DNA per sample was subjected to bisulphite mutagenesis with EZ DNA Methylation-GoldTM Kit (Zymo Research). COBRA analysis was performed afterwards as it was in the previous study.11
Bacterial colony bisulphite sequencing
Bisulphite mutagenized genomic DNA samples were amplified by PCR and ligated into the pGEM-T vector system (Promega catalog #A3610). Individual bacterial colonies were picked and directly sequenced by Macrogen USA. DNA methylation sequence analysis was performed with a web-based program called QUMA (http://quma.cdb.riken.jp/).
PCR primers for imprinted regions
After bisulphite mutageneis, a portion of genomic DNA derived from these ES clones was used for two rounds of PCR amplification for COBRA and bacterial colony bisulphite sequencing. The first-round outside PCR primers for the Snrpn DMR are SN-bis-OF 5′-AGTATTTGAT AATTTGGTTG GGTTTTATG and SN-bis-OR 5′-TCAAAAATCT TAATAAACCC AAATC. The second-round nested PCR primers for the Snrpn DMR are SN-bis-IF 5′-ATGTAATATG ATATAGTTTA GAAATTAG and SN-bis-IR 5′-CCACAAACCC AACTAACCTT CCTC. For the IG-DMR of the Dlk1-Dio3 imprinted region, the first-round outside PCR primers are IG-nF1 5′-TGTGGATCCT AGAGATGTTT TTGTTGA and IG-nR1 5′-TTCGGATCCC TACAACTTAA AAATTTCTCC AACC. The second-round nested PCR primers for the IG-DMR are IG-F2416 5′-TTTTAGTTTT TTGGGTTTTA GAGAA and IG-A2799 5′-TTTTAGTTTT TTGGGTTTTA GAGAA. For the Zac1 DMR, the first-round outside PCR primers are Zac1 DMR-Bis-OF1 5′-GGTTAGGGTA GGTAAGTAGT G and Zac1 DMR-Bis-OR1 5′-CAAAACCAAA ACCCTTACTA AC. The second-round nested PCR primers for the Zac1 DMR are Zac1 DMR-Bis-OF2 5′-GAGGTGATAA ATTTTGAATT TGGGTG and Zac1 DMR-Bis-OR2 5′-CTCCCAAAAA TTCTTAAAAA TCCAAC.
Supplementary Material
Acknowledgments
The work in the author’s laboratory was supported by the grants from NIH (GM093335), New York State (NYSTEM Contract #C026434) and American Heart Association (09SDG2400151). Li X conceived the study. Li X and Ray C wrote the manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: http://www.landesbioscience.com/journals/epigenetics/article/26544/
Footnotes
Previously published online: www.landesbioscience.com/journals/epigenetics/article/26544
References
- 1.Li X. Genomic imprinting is a parental effect established in mammalian germ cells. Curr Top Dev Biol. 2013;102:35–59. doi: 10.1016/B978-0-12-416024-8.00002-7. [DOI] [PubMed] [Google Scholar]
- 2.Bartolomei MS, Ferguson-Smith AC. Mammalian genomic imprinting. Cold Spring Harb Perspect Biol. 2011;3:a002592. doi: 10.1101/cshperspect.a002592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barlow DP. Genomic imprinting: a mammalian epigenetic discovery model. Annu Rev Genet. 2011;45:379–403. doi: 10.1146/annurev-genet-110410-132459. [DOI] [PubMed] [Google Scholar]
- 4.Bartolomei MS. Genomic imprinting: employing and avoiding epigenetic processes. Genes Dev. 2009;23:2124–33. doi: 10.1101/gad.1841409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Edwards CA, Ferguson-Smith AC. Mechanisms regulating imprinted genes in clusters. Curr Opin Cell Biol. 2007;19:281–9. doi: 10.1016/j.ceb.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 6.Ben-Porath I, Cedar H. Imprinting: focusing on the center. Curr Opin Genet Dev. 2000;10:550–4. doi: 10.1016/S0959-437X(00)00126-X. [DOI] [PubMed] [Google Scholar]
- 7.Ferguson-Smith AC. Genomic imprinting: the emergence of an epigenetic paradigm. Nat Rev Genet. 2011;12:565–75. doi: 10.1038/nrg3032. [DOI] [PubMed] [Google Scholar]
- 8.Li X, Ito M, Zhou F, Youngson N, Zuo X, Leder P, Ferguson-Smith AC. A maternal-zygotic effect gene, Zfp57, maintains both maternal and paternal imprints. Dev Cell. 2008;15:547–57. doi: 10.1016/j.devcel.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mackay DJ, Callaway JL, Marks SM, White HE, Acerini CL, Boonen SE, Dayanikli P, Firth HV, Goodship JA, Haemers AP, et al. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat Genet. 2008;40:949–51. doi: 10.1038/ng.187. [DOI] [PubMed] [Google Scholar]
- 10.Corsinotti A, Kapopoulou A, Gubelmann C, Imbeault M, Santoni de Sio FR, Rowe HM, Mouscaz Y, Deplancke B, Trono D. Global and stage specific patterns of Krüppel-associated-box zinc finger protein gene expression in murine early embryonic cells. PLoS One. 2013;8:e56721. doi: 10.1371/journal.pone.0056721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zuo X, Sheng J, Lau HT, McDonald CM, Andrade M, Cullen DE, Bell FT, Iacovino M, Kyba M, Xu G, et al. Zinc finger protein ZFP57 requires its co-factor to recruit DNA methyltransferases and maintains DNA methylation imprint in embryonic stem cells via its transcriptional repression domain. J Biol Chem. 2012;287:2107–18. doi: 10.1074/jbc.M111.322644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quenneville S, Verde G, Corsinotti A, Kapopoulou A, Jakobsson J, Offner S, Baglivo I, Pedone PV, Grimaldi G, Riccio A, et al. In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide to affect chromatin and DNA methylation of imprinting control regions. Mol Cell. 2011;44:361–72. doi: 10.1016/j.molcel.2011.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Messerschmidt DM, de Vries W, Ito M, Solter D, Ferguson-Smith A, Knowles BB. Trim28 is required for epigenetic stability during mouse oocyte to embryo transition. Science. 2012;335:1499–502. doi: 10.1126/science.1216154. [DOI] [PubMed] [Google Scholar]
- 14.Liu Y, Toh H, Sasaki H, Zhang X, Cheng X. An atomic model of Zfp57 recognition of CpG methylation within a specific DNA sequence. Genes Dev. 2012;26:2374–9. doi: 10.1101/gad.202200.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boonen SE, Mackay DJ, Hahnemann JM, Docherty L, Grønskov K, Lehmann A, Larsen LG, Haemers AP, Kockaerts Y, Dooms L, et al. Transient neonatal diabetes, ZFP57, and hypomethylation of multiple imprinted loci: a detailed follow-up. Diabetes Care. 2013;36:505–12. doi: 10.2337/dc12-0700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baglivo I, Esposito S, De Cesare L, Sparago A, Anvar Z, Riso V, Cammisa M, Fattorusso R, Grimaldi G, Riccio A, et al. Genetic and epigenetic mutations affect the DNA binding capability of human ZFP57 in transient neonatal diabetes type 1. FEBS Lett. 2013;587:1474–81. doi: 10.1016/j.febslet.2013.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Emerson RO, Thomas JH. Adaptive evolution in zinc finger transcription factors. PLoS Genet. 2009;5:e1000325. doi: 10.1371/journal.pgen.1000325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolf D, Goff SP. Embryonic stem cells use ZFP809 to silence retroviral DNAs. Nature. 2009;458:1201–4. doi: 10.1038/nature07844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bullock AN, Fersht AR. Rescuing the function of mutant p53. Nat Rev Cancer. 2001;1:68–76. doi: 10.1038/35094077. [DOI] [PubMed] [Google Scholar]
- 20.Schultz DC, Friedman JR, Rauscher FJ., 3rd Targeting histone deacetylase complexes via KRAB-zinc finger proteins: the PHD and bromodomains of KAP-1 form a cooperative unit that recruits a novel isoform of the Mi-2alpha subunit of NuRD. Genes Dev. 2001;15:428–43. doi: 10.1101/gad.869501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.