

Abstract
The chromatin modifier EZH2 is overexpressed and associated with inferior outcome in mantle cell lymphoma (MCL). Recently, we demonstrated preferential DNA methylation of HOX genes in MCL compared with chronic lymphocytic leukemia (CLL), despite these genes not being expressed in either entity. Since EZH2 has been shown to regulate HOX gene expression, to gain further insight into its possible role in differential silencing of HOX genes in MCL vs. CLL, we performed detailed epigenetic characterization using representative cell lines and primary samples. We observed significant overexpression of EZH2 in MCL vs. CLL. Chromatin immune precipitation (ChIP) assays revealed that EZH2 catalyzed repressive H3 lysine 27 trimethylation (H3K27me3), which was sufficient to silence HOX genes in CLL, whereas in MCL H3K27me3 is accompanied by DNA methylation for a more stable repression. More importantly, hypermethylation of the HOX genes in MCL resulted from EZH2 overexpression and subsequent recruitment of the DNA methylation machinery onto HOX gene promoters. The importance of EZH2 upregulation in this process was further underscored by siRNA transfection and EZH2 inhibitor experiments. Altogether, these observations implicate EZH2 in the long-term silencing of HOX genes in MCL, and allude to its potential as a therapeutic target with clinical impact.
Keywords: mantle cell lymphoma, chronic lymphocytic leukemia, DNA methylation, EZH2, HOX genes, histone methylation
Introduction
Mantle cell lymphoma (MCL) is a clinically aggressive B-cell malignancy with a poor prognosis. The genetic hallmark of MCL is the chromosomal rearrangement caused by the t(11;14)(q13;q32), which results in overexpression of cyclin D1 and deregulated cell cycle control, an essential part of MCL pathobiology.1 Limited knowledge exists regarding the role of epigenetic modifications and their potential impact on MCL pathogenesis. In MCL, the chromatin modifier EZH2 is overexpressed in proliferating cells and associated with poor outcome,2,3 similar to other malignancies.4,5 EZH2 is a core member of polycomb repressive complex (PRC) 2, mediating repressive H3 histone K27 lysine tri methyltransferase activity (H3K27me3) of the chromatin.6 On the other hand, along with histone methyltransferase activity, EZH2 has also been reported to directly control DNA methylation through its association with and regulation of the activity of DNA methyltransferases.7
Using methylation microarrays, we recently reported differential methylation of HOX genes (n = 13) in MCL compared with the more indolent chronic lymphocytic leukemia (CLL).8 Notably, despite their differential methylation status in MCL and CLL, HOX genes were not expressed in either entity, thus indicating that both DNA methylation-dependent and independent mechanisms may operate to silence HOX genes. HOX genes are a highly conserved group of genes which encode homeodomain containing transcriptional factors that are essential during early embryonic development and regulate cell differentiation and hematopoiesis in adult cells.9,10 Inappropriate or deregulated expression of HOX genes has been implicated in the development of several cancers, including hematologic malignancies.11,12 However, HOX genes have both tumor suppressor and oncogenic activity and these contrasting actions occur in a tissue-dependent fashion.13,14
Epigenetic mechanisms, such as DNA methylation and the activity of the polycomb and trithorax group of proteins, including EZH2, have been implicated in the deregulation of HOX genes in human cancers.15,16 For instance, hypermethylation and transcriptional silencing of the complete HOXA cluster has been linked to tumor progression in breast cancer.13 Methylation changes in HOXA have also been proposed as biomarkers for grading gliomas.17 Furthermore, hypermethylation of HOXA cluster genes has been shown to correlate with disease progression in leukemia;18 however, it is not clear whether methylation is a primary determinant of gene silencing or if it occurs as a consequence of silencing mediated by other mechanisms.
Since EZH2 has been shown to regulate HOX gene expression,15,16 one possible scenario is that HOXA genes could be targets of EZH2 in MCL. To gain insight into the mechanisms involved in silencing of HOXA genes in MCL and CLL, we investigated the functional roles of repressive chromatin modifications, such as H3K27me3, as well as EZH2 in the recruitment of the DNA methylation machinery. Importantly, while HOX genes were silenced by H3K27me3 histone trimethylation in CLL, EZH2 overexpression with subsequent recruitment of methyltransferases to the HOXA promoter was critical for long-term silencing of these genes by DNA methylation in MCL. The central role for EZH2 in gene silencing was further evidenced by EZH2 siRNA experiments and by applying an EZH2 inhibitor, ultimately highlighting EZH2 as a target of potential therapeutic interest in MCL.
Results and Discussion
Overexpression of EZH2 in MCL compared with CLL
In line with previous studies in MCL,2,3 we observed a significantly higher EZH2 expression level in MCL (n = 20) compared with CLL (n = 116) using RQ-PCR (fold difference (FD) 1.97 and P < 0.0001). When EZH2 expression levels were compared separately with favorable-prognostic IGHV-mutated CLL samples (n = 61), the FD was even more pronounced (FD 2.77, P < 0.0001), whereas the comparison against poor-prognostic IGHV-unmutated CLL samples (n = 55) rendered a slightly lower FD (FD 1.35, P < 0.02). These results are also in agreement with previous reports in diffuse large B cell lymphoma (DLBCL) and follicular lymphoma, thus indicating that EZH2 overexpression in B-cell lymphomas might be considered as a sign of clinical aggressiveness.4
The entire HOXA gene cluster is differentially methylated in MCL vs. CLL
An important finding in our previous genome-wide methylation array study of MCL and CLL concerned the identification of 13 differentially methylated HOX genes. While these genes were hypermethylated in MCL and predominantly hypomethylated in CLL, none were expressed in either entity.8 In order to gain insight into the epigenetic mechanisms involved in HOX gene silencing in each disease, we selected for further analysis genes from the HOXA cluster located on chromosome 7, i.e., HOXA2, HOXA9, and HOXA13 (Fig. 1).8 Similar to our recent finding that the methylation status of the HOXA13 gene differed between MCL and CLL,8 by analyzing 9 MCL and 12 CLL samples using pyrosequencing we also observed differential methylation between MCL vs. CLL for the HOXA9 gene (Fig. 2). Hence, using a more quantitative methodology such as pyrosequencing, significantly higher methylation levels of HOXA genes were validated in MCL vs. CLL.
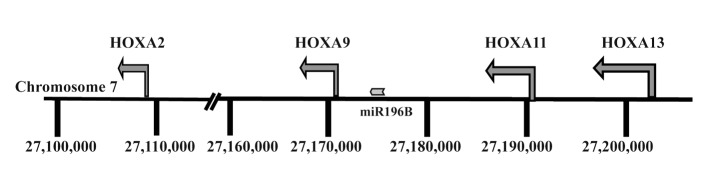
Figure 1. Physical map showing the location of the HOXA and miR196B genes on chromosome 7.
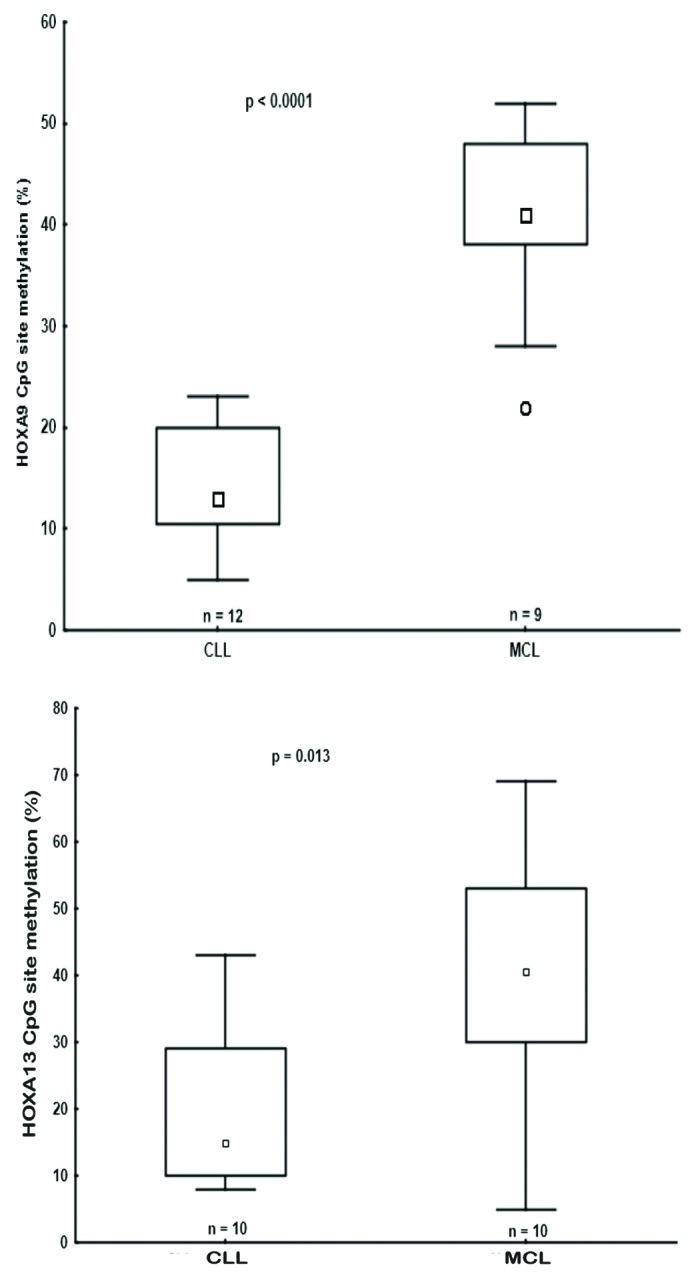
Figure 2.HOXA9 and HOXA13 genes were differentially methylated in CLL and MCL primary samples. Box plots showing DNA methylation levels of the HOXA9/HOXA13 genes in CLL and MCL primary samples, as quantified by pyrosequencing.
In addition to HOX genes, a CpG island flanking the miR196B is located in the HOXA cluster between the HOXA9 and HOXA10 genes (Fig. 1). Previous studies have indicated that methylation of CpG islands regulates not only the expression of HOX genes but also the nearby microRNA miR196b promoter.19 Using pyrosequencing to measure the methylation level at the miR196B promoter site, as for the HOXA genes, a significant difference in the level of methylation was observed between disease entities, i.e., hypermethylation in MCL (n = 8) and hypomethylation in CLL (n = 10) (P = 0.0004, Fig. S1A), in line with earlier studies in other malignancies.19,20 However, similar to HOXA genes, the expression levels of miR196b were extremely low in both MCL and CLL samples/cell lines as compared with normal human fibroblasts (~100 fold lower expression; Fig. S1B).
Therefore, we conclude that the entire HOXA cluster, including miR196B, is silenced in both MCL and CLL, however this is accomplished via different mechanisms, with DNA methylation likely playing a more prominent role in MCL. That said, since the methylation level of HOXA cluster genes was < 50% in MCL samples (Fig. 2), we cannot exclude that this could either be due to differential methylation of the HOXA alleles or due to other cell types present in the samples (although all MCL samples contained >70% tumor cells).
Differential enrichment of H3K27me3, EZH2, and DMNT1/3b at HOXA promoters in MCL vs. CLL
Since HOX genes are regulated by EZH2, we wanted to investigate if HOXA genes are targets for silencing in MCL and CLL. First, we investigated if the HOXA2, HOXA9, and HOXA13 genes carry other epigenetic modifications in their upstream regulatory regions. For this reason, we performed ChIP assays on MCL and CLL cell lines (Granta 519 and HG3) using antibodies for the repressive histone mark H3K27me3 and EZH2, respectively. Normal human fibroblast cells were used as a negative control for histone methylation, since HOXA13 is highly expressed in these cells.8 Overall, results from the ChIP assays indicate clear differences in H3K27me3 modification between MCL vs. CLL cell lines, with the Granta 519 cell line showing several-fold higher enrichment of H3K27me3 for all three HOXA genes (i.e., HOXA2, HOXA9, and HOXA13) compared with the HG3 cell line (Fig. 3). Furthermore, the degree of H3K27me3 enrichment correlated well with EZH2 binding, i.e., high in MCL and low in CLL (Fig. 3).

Figure 3. Differential EZH2 levels correlated with DNMT1 recruitment in CLL vs. MCL cell lines. Enrichment of H3K27me3, EZH2, DNMT1, and DNMT3b levels at the HOXA gene promoters in the Granta 519 and HG3 cell lines was measured using ChIP with the respective antibody. The data represents values from triplicates plotted over IgG. A human fibroblast cell line was used as a positive control.
Next, we investigated if the differential methylation status of HOXA genes could be related to differences in recruitment of DNA methyltransferases (DNMTs) to the HOXA promoters. To this end, we performed ChIP assays with DNMT1 and DNMT3b antibodies and showed that both DNMT1 and DNMT3b binding levels at the HOX gene promoters were associated with the DNA methylation levels, i.e., higher in the MCL cell line compared with the CLL cell line (Fig. 3). Nevertheless, the HOX genes also showed some background levels of H3K27me3, EZH2, and DNMTs in the control human fibroblast cell line. When extending these ChIP analyses to primary patient material (3 MCL and 3 CLL samples), similar results were obtained for DNMT1, DNMT3b, and EZH2, although the differences in fold enrichment were less pronounced as compared with the more homogeneous cell line data (Fig. S2).
Hence, a higher EZH2 occupancy at the HOXA promoters in MCL may lead to increased enrichment of H3K27me3 and recruitment of DNMT1/3b that in turn execute DNA methylation. Yet, what could be the reasons behind higher EZH2 binding in MCL?
Regulation of EZH2 expression by the cyclin D1/p16-pRB-E2F pathway in MCL
The frequent loss of the p16INK4 tumor suppressor gene (which is an inhibitor of cyclin D1) by genomic deletion or promoter hypermethylation has been shown to be associated with high proliferation and shorter survival in MCL.21,22 Interestingly, in a human mammary breast cancer cell line, it was demonstrated that downregulation of the p16INK4 gene resulted in overexpression of the E2F transcriptional factor and polycomb PcG complex proteins, such as EZH2, leading to DNA hypermethylation of the HOXA9 gene promoter.23 In order to investigate the p16INK4 expression levels, we analyzed 12 MCL and 12 CLL patient samples by RQ-PCR and observed considerably higher expression levels in CLL compared with MCL (P = 0.002) (Fig. 4A). Furthermore, we analyzed protein levels of p16, E2F, and EZH2 in the CLL and MCL cell lines using western blotting. p16 protein expression was low and E2F and EZH2 proteins were high in the MCL (Granta 519) cell line as compared with the CLL cell line (HG3) (Fig. 4B). The lower expression of p16 in the Granta MCL cell line was expected since this cell line has been shown to carry a homozygous deletion of p16.24 Also, due to the presence of both a mutation in ATM and del(17p), this cell line is more likely to represent aggressive MCL. Hence, reduced p16 expression may lead to increased EZH2 expression in MCL.

Figure 4. Differential expression of p16 in CLL and MCL. (A) Box plots showing relative p16 expression levels in CLL and MCL primary samples quantified using RQ-PCR. (B) Western blot analysis of p16, EZH2, E2F, and β actin in the HG3 and Granta 519 cell line.
The HOXA DNA methylation status is determined by the EZH2 levels
Since EZH2 can physically interact with DNMT1 and DNMT3b and recruit them to the gene promoters to silence HOX genes,7,16,25 this may be the case for MCL. To corroborate the direct role of EZH2 in orchestrating DNA methylation of HOX genes, EZH2 expression was first abrogated using RNA interference. In this experiment, siRNA knockdown of EZH2 mRNA revealed a significant decrease in EZH2 protein levels in the MCL cell line as determined by western blot analysis. EZH2 knockdown also resulted in an expected decrease in the levels of H3K27me3, and a corresponding increase in the levels of the HOXA9, HOXA2, and HOXA13 proteins (Fig. 5A). These results were also validated at the mRNA level using RQ-PCR (Fig. S3A).

Figure 5. Downregulation of EZH2 using siRNA results in loss of DNA methylation and re-expression of HOXA genes in MCL. (A) Western blot analysis of EZH2, all three HOXA proteins (HOXA2, HOXA9, and HOXA13) and H3K27me3 expression after treatment with control siRNA or EZH2 siRNA in the Granta 519 MCL cell line. Histone H3 and GAPDH were used as internal loading controls. (B) Bisulfite sequencing analysis of the HOXA2 and HOXA9 genes in the Grant 519 cell line after treatment with EZH2 siRNA and control siRNA. The methylation status of 10 clones is presented for each sample; each circle represents one CpG site indicating either cytosine (open circles), methyl cytosine (filled circles) or non-CpG site (missing circles). (C) Enrichment of H3K27me3, EZH2, DNMT1, and DNMT3b levels at the HOXA gene promoters in the Granta519 MCL cell line using control siRNA and EZH2 siRNA treated. The data represents values from triplicates plotted over IgG.
To investigate the impact of decreased EZH2 levels on the DNA methylation status of the HOXA genes, bisulfite sequencing was also performed, verifying the loss of DNA methylation at most CpG sites analyzed on both the HOXA2 and HOXA9 genes (Fig. 5B). Since the downregulation of EZH2 resulted in re-expression of HOXA genes, we also measured the degree of histone methylation and binding of DNMTs to HOXA gene promoters upon EZH2 depletion using ChIP. As expected, there was a significant loss of H3K27me3 as well as DNMT1 and DNMT3B at the HOXA promoters in response to EZH2 depletion (Fig. 5C).
To further investigate the role of EZH2 in the recruitment of DNMTs to HOXA gene promoters, EZH2 was depleted by treating the MCL cell line with the H3K27me3 histone methyl inhibitor 3-Deazaneplanocin A (DZNep), which is a cyclopentenyl analog of 3-deazaadenosine. Previously, this drug has been shown to deplete EZH2 levels and to inhibit H3K27me3 in acute myeloid leukemia5 and multiple myeloma cells in a dose- and time-dependent manner.26 When the MCL cell line was treated with increasing concentrations of DZNep, ranging from 0 to 10µM, a significant reduction in EZH2 as well as H3K27me3 levels was detected, followed by activation of the HOXA9 gene (Fig. 6).
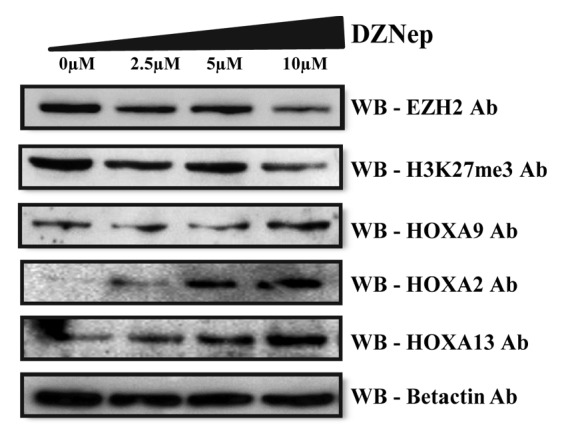
Figure 6. Re-expression of HOXA proteins after DZNep treatment. Western blot analysis of EZH2, H3K27me3, and HOXA proteins (HOXA2, HOXA9, and HOX13) levels in the Granta 519 cell line treated with increasing concentrations of DZNep ranging from 0–10 µM for 4 d. Beta actin levels are used as internal loading control.
EZH2: a potential novel target for epigenetic therapy?
In conclusion, this study for the first time highlights the epigenetic silencing mechanisms underlying differential HOX gene regulation in MCL vs. CLL. In the case of CLL, HOX genes appear to be silenced primarily by H3K27me3 histone methylation, which is catalyzed by EZH2, while in MCL EZH2 also binds to DNMTs thereby recruiting the DNA methylation machinery for efficient silencing of the HOXA genes. More specifically, HOXA gene promoters exhibited greater enrichment with EZH2 in MCL as compared with CLL, which in turn correlated with increased DNMT1 recruitment and CpG methylation, thus indicating that the level of EZH2 indeed determines the level of DNA methylation through DNMTs. This is in contrast to normal B cells which expressed HOXA genes at higher levels comparable to the expression levels in normal fibroblast cells (Fig. S3).8
Given that DNA methylation is a more stable epigenetic mark compared with histone modifications, EZH2-mediated specific methylation of the HOXA promoters could be a critical step in conferring an aggressive phenotype to MCL. This important function of EZH2 has also been reflected in B-cell activation and lymphomagenesis, where EZH2 acts as an epigenetic switch in promoting H3K27me3 toward DNA hypermethylation.27 This transition toward DNA methylation reduces epigenetic plasticity and locks the target genes in a stable repressive state, and hence prevents any major transcriptional changes irrespective of external cues. Accordingly, our current data implies that EZH2-mediated DNA methylation ensures the stable repression of HOXA genes, a step that may be critical in the aggressive phenotype of MCL. Our novel observations also advocate that EZH2 plays a crucial role in HOX gene silencing in MCL, which was also confirmed by our siRNA and EZH2 inhibitor experiments, and that DNA methylation occurs as a secondary event following EZH2 recruitment to the HOX promoter. This is further supported by the fact that most MCL patient samples did not show complete methylation of HOXA cluster genes.
The key role for EZH2 in MCL lymphomagenesis is also underscored by the recent finding that the MYC oncogene upregulates EZH2 in MCL, eventually leading to decreased expression of the tumor suppressor miRNA, miR29.28-30 Furthermore, EZH2 inhibition was very recently pointed out as a potential treatment strategy for the germinal center subtype of DLBCL.4,5,31-33 For all these reasons, as well as the well-established notion that EZH2 overexpression is associated with tumor invasion, tumor progression and poor prognosis in many different cancer types,34-36 our novel observations highlight EZH2 as a potential novel target for epigenetic-based therapy in MCL. Nevertheless, systematic investigations of EZH2 target genes are required before this can be attempted in a clinical setting.
Methods
Patient material
In this present study, we included tumor samples from MCL and CLL patients collected from the biobanks at Uppsala University Hospital and Karolinska University Hospital, Sweden, and G. Papanicolaou Hospital, Thessaloniki, Greece. All MCL cases fulfilled the diagnostic criteria of the World Health Organization Classification.8 All CLL samples (poor-prognostic subset #1 and favorable prognostic subset #4) were diagnosed according to recently revised criteria showing a typical CLL immunophenotype.37 Clinical and molecular data are summarized in Table S1. Sorted CD19+ B cells from healthy control was obtained from 3H Biomedical, Uppsala, Sweden.
Cell lines and cell culture conditions
Two EBV transformed cell lines, one CLL (HG3)38 and one MCL (Granta 519)39 were used for ChIP assays and siRNA transfection assays. The cell lines were cultured in RPMI 1640 (Invitrogen) supplemented with glutamine (4 mM glutamine for Granta 519 and 2 mM Glutamine for HG3), 10% fetal bovine serum (FBS; Invitrogen), and 1× penicillin/streptomycin (Invitrogen, Carlsbad, CA, USA).
Chromatin immunoprecipitation assay
Chromatin immune precipitation was performed using the Shearing module kit and OneDay ChIP Kit™ (Diagenode) according to the manufacturer’s instructions. Antibodies used were: EZH2 polyclonal antibody (Diagenode, pAB-039-050), H3K27me3 polyclonal antibody (Diagenode, pAB-069-050), Dnmt1 monoclonal antibody (Imgenex, IMG-261A), Dnmt3b polyclonal antibody (Diagenode, pAB-076-005), and IgG (negative control, OneDay ChIP Kit™Diagenode). The detailed protocol is listed in the Supplemental Material.
Gene and protein expression analysis
Total RNA was prepared using the TRIzol method (Invitrogen Life Technologies) or the AllPrep DNA/RNA kit (Qiagen) according to the manufacturer’s instructions. The reverse transcription reaction was performed using MMLV-RT or Superscript II reverse transcriptase (Invitrogen) and random hexamers (Fermentas) according to the manufacturer’s protocol. RQ-PCR analysis for the determination of the p16 and EZH2 mRNA levels was performed as described in the Supplementary Material. Results for EZH2 mRNA expression in CLL that were used for comparison to MCL have been reported previously.40
Western blot analysis was performed using total cell lysates and the detailed protocol has been provided in the Supplementary Material.
Pyrosequencing and bisulfite sequencing
Both pyrosequencing and bisulfite sequencing were performed as in our previous publications8,41 and detailed protocols are provided in the Supplementary Material.
siRNA transfections and DZNep treatment assay
Granta 519 cells were transfected with predesigned siRNA against EZH2 using Stealth RNAi siRNA, containing a mixture of three oligos (HSS103462; HSS176652, and HSS176653) in equal concentrations (Invitrogen). The siRNA negative control (Invitrogen) was used as control siRNA. Transient transfection was performed on an Amaxa Nucleofection Device (Lonza Cologne AG) according to the manufacturer's instruction. In brief, Granta 519 cells were split at a density of 5 × 105/ml in the medium 48 h before transfection. Thereafter, 4 × 106 cells were collected and resuspended in 100 µl human cell line nucleofector solution C with 100 pmol of EZH2 siRNA mix or control siRNA using the X-01 electroporation program.
The Granta 519 cells were treated with the EZH2 inhibitor, 3-Deazaneplanocin A (DZNep) (Cayman chemicals) for three days using different concentrations ranging from 0‒10 µM.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Authorship
Kanduri M designed and performed the research, analyzed the data and wrote the paper. Papakonstantinou N, Ntoufa S, and Sutton L performed research. Sander B, Stamatopoulos K, and Kanduri C analyzed data and wrote the paper. Rosenquist R supervised the research and wrote the paper.
Supplemental Materials
Supplemental materials may be found here: http://www.landesbioscience.com/journals/epigenetics/article/26546/
Footnotes
Previously published online: www.landesbioscience.com/journals/epigenetics/article/26546
References
- 1.Rosenwald A, Wright G, Wiestner A, Chan WC, Connors JM, Campo E, Gascoyne RD, Grogan TM, Muller-Hermelink HK, Smeland EB, et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell. 2003;3:185–97. doi: 10.1016/S1535-6108(03)00028-X. [DOI] [PubMed] [Google Scholar]
- 2.van Kemenade FJ, Raaphorst FM, Blokzijl T, Fieret E, Hamer KM, Satijn DP, Otte AP, Meijer CJ. Coexpression of BMI-1 and EZH2 polycomb-group proteins is associated with cycling cells and degree of malignancy in B-cell non-Hodgkin lymphoma. Blood. 2001;97:3896–901. doi: 10.1182/blood.V97.12.3896. [DOI] [PubMed] [Google Scholar]
- 3.Visser HP, Gunster MJ, Kluin-Nelemans HC, Manders EM, Raaphorst FM, Meijer CJ, Willemze R, Otte AP. The Polycomb group protein EZH2 is upregulated in proliferating, cultured human mantle cell lymphoma. Br J Haematol. 2001;112:950–8. doi: 10.1046/j.1365-2141.2001.02641.x. [DOI] [PubMed] [Google Scholar]
- 4.McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A, 3rd, Diaz E, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108–12. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- 5.Fiskus W, Wang Y, Sreekumar A, Buckley KM, Shi H, Jillella A, Ustun C, Rao R, Fernandez P, Chen J, et al. Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood. 2009;114:2733–43. doi: 10.1182/blood-2009-03-213496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simon JA, Lange CA. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat Res. 2008;647:21–9. doi: 10.1016/j.mrfmmm.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 7.Viré E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden JM, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439:871–4. doi: 10.1038/nature04431. [DOI] [PubMed] [Google Scholar]
- 8.Halldórsdóttir AM, Kanduri M, Marincevic M, Mansouri L, Isaksson A, Göransson H, Axelsson T, Agarwal P, Jernberg-Wiklund H, Stamatopoulos K, et al. Mantle cell lymphoma displays a homogenous methylation profile: a comparative analysis with chronic lymphocytic leukemia. Am J Hematol. 2012;87:361–7. doi: 10.1002/ajh.23115. [DOI] [PubMed] [Google Scholar]
- 9.Sauvageau G, Lansdorp PM, Eaves CJ, Hogge DE, Dragowska WH, Reid DS, Largman C, Lawrence HJ, Humphries RK. Differential expression of homeobox genes in functionally distinct CD34+ subpopulations of human bone marrow cells. Proc Natl Acad Sci U S A. 1994;91:12223–7. doi: 10.1073/pnas.91.25.12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akam M. Hox and HOM: homologous gene clusters in insects and vertebrates. Cell. 1989;57:347–9. doi: 10.1016/0092-8674(89)90909-4. [DOI] [PubMed] [Google Scholar]
- 11.Argiropoulos B, Humphries RK. Hox genes in hematopoiesis and leukemogenesis. Oncogene. 2007;26:6766–76. doi: 10.1038/sj.onc.1210760. [DOI] [PubMed] [Google Scholar]
- 12.Eklund EA. The role of HOX genes in myeloid leukemogenesis. Curr Opin Hematol. 2006;13:67–73. doi: 10.1097/01.moh.0000208467.63861.d6. [DOI] [PubMed] [Google Scholar]
- 13.Novak P, Jensen T, Oshiro MM, Wozniak RJ, Nouzova M, Watts GS, Klimecki WT, Kim C, Futscher BW. Epigenetic inactivation of the HOXA gene cluster in breast cancer. Cancer Res. 2006;66:10664–70. doi: 10.1158/0008-5472.CAN-06-2761. [DOI] [PubMed] [Google Scholar]
- 14.Strathdee G, Sim A, Soutar R, Holyoake TL, Brown R. HOXA5 is targeted by cell-type-specific CpG island methylation in normal cells and during the development of acute myeloid leukaemia. Carcinogenesis. 2007;28:299–309. doi: 10.1093/carcin/bgl133. [DOI] [PubMed] [Google Scholar]
- 15.Nagel S, Venturini L, Marquez VE, Meyer C, Kaufmann M, Scherr M, MacLeod RA, Drexler HG. Polycomb repressor complex 2 regulates HOXA9 and HOXA10, activating ID2 in NK/T-cell lines. Mol Cancer. 2010;9:151. doi: 10.1186/1476-4598-9-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu X, Gong Y, Yue J, Qiang B, Yuan J, Peng X. Cooperation between EZH2, NSPc1-mediated histone H2A ubiquitination and Dnmt1 in HOX gene silencing. Nucleic Acids Res. 2008;36:3590–9. doi: 10.1093/nar/gkn243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Di Vinci A, Casciano I, Marasco E, Banelli B, Ravetti GL, Borzì L, Brigati C, Forlani A, Dorcaratto A, Allemanni G, et al. Quantitative methylation analysis of HOXA3, 7, 9, and 10 genes in glioma: association with tumor WHO grade and clinical outcome. J Cancer Res Clin Oncol. 2012;138:35–47. doi: 10.1007/s00432-011-1070-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strathdee G, Holyoake TL, Sim A, Parker A, Oscier DG, Melo JV, Meyer S, Eden T, Dickinson AM, Mountford JC, et al. Inactivation of HOXA genes by hypermethylation in myeloid and lymphoid malignancy is frequent and associated with poor prognosis. Clin Cancer Res. 2007;13:5048–55. doi: 10.1158/1078-0432.CCR-07-0919. [DOI] [PubMed] [Google Scholar]
- 19.Tsai KW, Hu LY, Wu CW, Li SC, Lai CH, Kao HW, Fang WL, Lin WC. Epigenetic regulation of miR-196b expression in gastric cancer. Genes Chromosomes Cancer. 2010;49:969–80. doi: 10.1002/gcc.20804. [DOI] [PubMed] [Google Scholar]
- 20.Schotte D, Lange-Turenhout EA, Stumpel DJ, Stam RW, Buijs-Gladdines JG, Meijerink JP, Pieters R, Den Boer ML. Expression of miR-196b is not exclusively MLL-driven but is especially linked to activation of HOXA genes in pediatric acute lymphoblastic leukemia. Haematologica. 2010;95:1675–82. doi: 10.3324/haematol.2010.023481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hutter G, Scheubner M, Zimmermann Y, Kalla J, Katzenberger T, Hübler K, Roth S, Hiddemann W, Ott G, Dreyling M. Differential effect of epigenetic alterations and genomic deletions of CDK inhibitors [p16(INK4a), p15(INK4b), p14(ARF)] in mantle cell lymphoma. Genes Chromosomes Cancer. 2006;45:203–10. doi: 10.1002/gcc.20277. [DOI] [PubMed] [Google Scholar]
- 22.Pinyol M, Hernandez L, Cazorla M, Balbín M, Jares P, Fernandez PL, Montserrat E, Cardesa A, Lopez-Otín C, Campo E. Deletions and loss of expression of p16INK4a and p21Waf1 genes are associated with aggressive variants of mantle cell lymphomas. Blood. 1997;89:272–80. [PubMed] [Google Scholar]
- 23.Reynolds PA, Sigaroudinia M, Zardo G, Wilson MB, Benton GM, Miller CJ, Hong C, Fridlyand J, Costello JF, Tlsty TD. Tumor suppressor p16INK4A regulates polycomb-mediated DNA hypermethylation in human mammary epithelial cells. J Biol Chem. 2006;281:24790–802. doi: 10.1074/jbc.M604175200. [DOI] [PubMed] [Google Scholar]
- 24.Jadayel DM, Lukas J, Nacheva E, Bartkova J, Stranks G, De Schouwer PJ, et al. Potential role for concurrent abnormalities of the cyclin D1, p16CDKN2 and p15CDKN2B genes in certain B cell non-Hodgkin's lymphomas. Functional studies in a cell line (Granta 519). Leukemia: official journal of the Leukemia Society of America. Leukemia Research Fund, UK. 1997;11:64–72. doi: 10.1038/sj.leu.2400555. [DOI] [PubMed] [Google Scholar]
- 25.Acharyya S, Sharma SM, Cheng AS, Ladner KJ, He W, Kline W, Wang H, Ostrowski MC, Huang TH, Guttridge DC. TNF inhibits Notch-1 in skeletal muscle cells by Ezh2 and DNA methylation mediated repression: implications in duchenne muscular dystrophy. PLoS One. 2010;5:e12479. doi: 10.1371/journal.pone.0012479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kalushkova A, Fryknäs M, Lemaire M, Fristedt C, Agarwal P, Eriksson M, Deleu S, Atadja P, Osterborg A, Nilsson K, et al. Polycomb target genes are silenced in multiple myeloma. PLoS One. 2010;5:e11483. doi: 10.1371/journal.pone.0011483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Béguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, Shen H, Yang SN, Wang L, Ezponda T, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell. 2013;23:677–92. doi: 10.1016/j.ccr.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang X, Zhao X, Fiskus W, Lin J, Lwin T, Rao R, Zhang Y, Chan JC, Fu K, Marquez VE, et al. Coordinated silencing of MYC-mediated miR-29 by HDAC3 and EZH2 as a therapeutic target of histone modification in aggressive B-Cell lymphomas. Cancer Cell. 2012;22:506–23. doi: 10.1016/j.ccr.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Hartmann E, Fernàndez V, Moreno V, Valls J, Hernández L, Bosch F, Abrisqueta P, Klapper W, Dreyling M, Hoster E, et al. Five-gene model to predict survival in mantle-cell lymphoma using frozen or formalin-fixed, paraffin-embedded tissue. J Clin Oncol. 2008;26:4966–72. doi: 10.1200/JCO.2007.12.0410. [DOI] [PubMed] [Google Scholar]
- 30.Zhao JJ, Lin J, Lwin T, Yang H, Guo J, Kong W, Dessureault S, Moscinski LC, Rezania D, Dalton WS, et al. microRNA expression profile and identification of miR-29 as a prognostic marker and pathogenetic factor by targeting CDK6 in mantle cell lymphoma. Blood. 2010;115:2630–9. doi: 10.1182/blood-2009-09-243147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiao Y. Enhancer of zeste homolog 2: A potential target for tumor therapy. Int J Biochem Cell Biol. 2011;43:474–7. doi: 10.1016/j.biocel.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 32.Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, Sacks JD, Raimondi A, Majer CR, Song J, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012;8:890–6. doi: 10.1038/nchembio.1084. [DOI] [PubMed] [Google Scholar]
- 33.Fiskus W, Rao R, Balusu R, Ganguly S, Tao J, Sotomayor E, Mudunuru U, Smith JE, Hembruff SL, Atadja P, et al. Superior efficacy of a combined epigenetic therapy against human mantle cell lymphoma cells. Clin Cancer Res. 2012;18:6227–38. doi: 10.1158/1078-0432.CCR-12-0873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu J, Yu J, Rhodes DR, Tomlins SA, Cao X, Chen G, Mehra R, Wang X, Ghosh D, Shah RB, et al. A polycomb repression signature in metastatic prostate cancer predicts cancer outcome. Cancer Res. 2007;67:10657–63. doi: 10.1158/0008-5472.CAN-07-2498. [DOI] [PubMed] [Google Scholar]
- 35.Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003;22:5323–35. doi: 10.1093/emboj/cdg542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RG, Otte AP, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–9. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- 37.Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Döhner H, Hillmen P, Keating MJ, Montserrat E, Rai KR, et al. International Workshop on Chronic Lymphocytic Leukemia Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111:5446–56. doi: 10.1182/blood-2007-06-093906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosén A, Bergh AC, Gogok P, Evaldsson C, Myhrinder AL, Hellqvist E, Rasul A, Björkholm M, Jansson M, Mansouri L, et al. Lymphoblastoid cell line with B1 cell characteristics established from a chronic lymphocytic leukemia clone by in vitro EBV infection. Oncoimmunology. 2012;1:18–27. doi: 10.4161/onci.1.1.18400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amin HM, McDonnell TJ, Medeiros LJ, Rassidakis GZ, Leventaki V, O’Connor SL, Keating MJ, Lai R. Characterization of 4 mantle cell lymphoma cell lines. Arch Pathol Lab Med. 2003;127:424–31. doi: 10.5858/2003-127-0424-COMCLC. [DOI] [PubMed] [Google Scholar]
- 40.Papakonstantinou NNS, Papadopoulos G, Hatzigeorgiou A, Anagnostopoulos A, Chlichlia A, Ghia P, Muzio M, Belessi C, Stamatopoulos K. Distinct profiles of miRNAs modulating immune signalling pathways in different subsets of chronic lymphocytic leukemia. 17th Congress of the European Haematology Association. Amsterdam, 2012. [Google Scholar]
- 41.Kanduri M, Cahill N, Göransson H, Enström C, Ryan F, Isaksson A, Rosenquist R. Differential genome-wide array-based methylation profiles in prognostic subsets of chronic lymphocytic leukemia. Blood. 2010;115:296–305. doi: 10.1182/blood-2009-07-232868. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.