

Abstract
Pheochromocytoma (PCC) and abdominal paraganglioma (PGL) are neuroendocrine tumors that present with clinical symptoms related to increased catecholamine levels. About a third of the cases are associated with constitutional mutations in pre-disposing genes, of which some may also be somatically mutated in sporadic cases. However, little is known about inactivating epigenetic events through promoter methylation in these very genes. Using bisulphite pyrosequencing we assessed the methylation density of 11 PCC/PGL disease genes in 96 tumors (83 PCCs and 13 PGLs) and 34 normal adrenal references. Gene expression levels were determined by quantitative RT-PCR. Both tumors and normal adrenal samples exhibited low methylation index (MetI) in the EGLN1 (PDH2), MAX, MEN1, NF1, SDHB, SDHC, SDHD, SDHAF2 (SDH5), and TMEM127 promoters, not exceeding 10% in any of the samples investigated. Aberrant RET promoter methylation was observed in two cases only. For the VHL gene we found increased MetI in tumors as compared with normal adrenals (57% vs. 27%; P < 0.001), in malignant vs. benign tumors (63% vs. 55%; P < 0.05), and in PGL vs. PCC (66% vs. 55%; P < 0.0005). Decreased expression of the VHL gene was observed in all tumors compared with normal adrenals (P < 0.001). VHL MetI and gene expressions were inversely correlated (R = −0.359, P < 0.0001). Our results show that the VHL gene promoter has increased methylation compared with normal adrenals (MetI > 50%) in approximately 75% of PCCs and PGLs investigated, highlighting the role of VHL in the development of these tumors.
Keywords: Pheochromocytoma, paraganglioma, VHL, DNA promoter methylation, gene expression, pyrosequencing, qRT-PCR
Introduction
Pheochromocytoma (PCC) is a neuroendocrine tumor located in the adrenal medulla. Abdominal paraganglioma (PGL) is a highly related tumor occurring outside the adrenal medulla, most commonly within the sympathetic paravertebral ganglia.
The clinical profile of PCC and PGL is often characterized by increased catecholamine secretion and associated symptoms – most pronounced in tumors with epinephrine overexpression.1 The majority of patients present with common endocrine-related manifestations such as headache, palpitations, tachycardia and excessive perspiration,2 but asymptomatic tumors are also common and many are found only at autopsy.3
The last few decades have witnessed major discoveries relating to the genetic background of these tumors. Approximately one third of PCC and PGL patients carry a constitutional mutation in a predisposing gene including EGLN1 (PDH2), EPAS1 (HIF2A), KIF1Bβ, MAX, NF1, RET, SDHA, SDHAF2 (SDH5), SDHB, SDHC, SDHD, TMEM127, and VHL.4-9 Somatic mutations in some of these genes also occur, most commonly involving NF1.10,11
Studies of DNA methylation have revealed CpG hypermethylator phenotypes in PGLs carrying a SDHB,12,13 or SDHx mutation as was also demonstrated in a mouse model.14 However, possible somatic inactivation of the PCC/PGL predisposing genes by promoter methylation has so far not been fully determined. Cascon et al. reported unmethylated promoters for SDHB, SDHD, and VHL in 35 PCCs,15 and Dammann et al. described unmethylated VHL promoter in 25 PCCs.16 Later, Welander et al. reported unmethylated promoters for KIF1Bβ, MAX, NF1, RET, SDHA, SDHAF2, SDHB, SDHC, SDHD, TMEM127, and VHL in 42 sporadic PCCs.11 These three studies have employed methylation-specific PCR (MSP) for qualitative identification of hypermethylation. To unambiguously evaluate the gene promoter methylation status in these tumors, we employed quantitative Pyrosequencing in a large panel of PCCs and PGLs. Findings were subsequently compared with gene expression levels and evaluated against tumor and patient phenotypes.
Results
Increased promoter methylation density of VHL in PCC and PGL
CpG methylation density was quantified using Pyrosequencing for the promoter regions of 11 genes implicated in PCC/PGL (EGLN1 [PDH2], MAX, MEN1, NF1, RET, SDHAF2 [SDH5], SDHB, SDHC, SDHD, TMEM127, and VHL) in 96 tumors (83 PCCs, 13 PGLs) and 34 normal adrenal samples. For each sample and gene a methylation index (MetI) was defined as the mean methylation of all CpG dinucleotides covered. Overall, the methylation assessment for all genes except VHL showed no or very little methylation. With regards to EGLN1, MAX, MEN1, NF1, SDHAF2, SDHB, SDHC, SDHD, and TMEM127, MetI values below 10% were obtained for all tumors and normal adrenals (Table 1). One single CpG site (case PCC-56) within the TMEM127 gene showed an increased methylation level of 15%. For the RET gene increased methylation was recorded in two PGLs (PGL-102 and PGL-113) with MetI values of 17% and 29%, respectively, while all other tumors and normal adrenals showed MetI below 10%.
Table 1. Results of methylation analyses in the tumors and normal adrenals.
| Tumors | Adrenals | Tumors vs. Adrenals | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Gene | MetI | MetI | MetI | MetI | |||||
| symbol | Gene name | n | mean % | range % | n | mean % | range % | P value | |
| EGLN1 | Egl nine homolog 1 | 95 | 1.6 | 0 - 5.6 | 12 | 1.2 | 0 - 2.4 | n.s. | |
| MAX | Myc associated factor X | 96 | 1.0 | 0 - 4.7 | 6 | 0.9 | 0 - 3.2 | n.s. | |
| MEN1 | Multiple endocrine neoplasia type 1 | 95 | 1.6 | 0 - 5.7 | 11 | 1.6 | 0.9 - 4.6 | n.s. | |
| NF1 | Neurofibromin 1 | 96 | 1.0 | 0 - 3.8 | 4 | 0.6 | 0 - 1.5 | n.s. | |
| RET | Rearranged during transfection proto-oncogene | 96 | 2.8 | 0 - 28.8 | 12 | 2.9 | 1.0 - 3.8 | n.s. | |
| SDHAF2 | Succinate dehydrogenase complex assembly factor 2 | 96 | 0.8 | 0 - 4.0 | 12 | 0.8 | 0 - 2.5 | n.s. | |
| SDHB | Succinate dehydrogenase complex, subunit B | 93 | 1.1 | 0 - 1.7 | 15 | 1.5 | 0.6 - 2.0 | n.s. | |
| SDHC | Succinate dehydrogenase complex, subunit C | 95 | 0.3 | 0 - 1.5 | 18 | 0.6 | 0 - 2.3 | n.s. | |
| SDHD | Succinate dehydrogenase complex, subunit D | 96 | 1.5 | 0 - 3.5 | 15 | 0.7 | 0 - 1.5 | n.s. | |
| TMEM127 | Transmembrane protein 127 | 96 | 1.5 | 0 - 9.0 | 11 | 2.3 | 0.9 - 5.0 | n.s. | |
| VHL | Von Hippel-Lindau tumor suppressor | 96 | 56.5 | 19.1 - 88.6 | 34 | 27.0 | 14.4 - 43.8 | p < 0.0001 | |
n.s., not statistically significant; MetI, methylation index
By contrast, increased methylation was frequently observed for the VHL promoter (Fig. S1; Table 2). Comparing VHL methylation between sample groups (Fig. 1) revealed increased MetI in tumors as compared with normal adrenals (mean MetI 57% vs. 27%; P < 0.0001). DNA from 4 normal adrenal medullary samples (NAM-1–4) also showed low methylation levels (mean MetI = 31%) comparable to the normal adrenals. Altogether 72/96 (75%) of the tumors had a MetI > 50% while the MetI of the normal adrenals did not exceed 44% in any single case (Table 1). We further observed a difference in methylation level between specific CpG sites within the analyzed VHL promoter sequence (Table 2; Fig. 2; Fig. S1). This was particularly pronounced at CpG 1 and 2 (Chr3: 10 183 196 and Chr3: 10 183 198 respectively). Specifically, we observed a statistically significant (P < 0.0001, Table 2) higher methylation level in tumors as compared with normal adrenals at CpG 1 (mean methylation 80% vs. 37%), CpG 2 (72% vs. 36%), CpG 3 (45% vs. 19%), and CpG 4 (30% vs. 15%) (Fig. 2).
Table 2. Results and correlations of methylation analyses of the VHL promotes.
| Mean methylation | |||||
|---|---|---|---|---|---|
| Parameter | CpG 1 | CpG 2 | CpG 3 | CpG 4 | MetI |
| Tumors vs. Adrenals | |||||
| Tumors | 79.9 | 72.1 | 44.2 | 30.0 | 56.6 |
| Adrenals | 37.3 | 36.4 | 18.9 | 15.3 | 27.0 |
| P value | < 0.0001* | < 0.0001* | < 0.0001* | < 0.0001* | < 0.0001* |
| PGLs vs. PCCs | |||||
|---|---|---|---|---|---|
| PGLs | 88.7 | 81.7 | 48.9 | 45.7 | 66.3 |
| PCCs | 78.6 | 70.6 | 43.5 | 27.5 | 55.0 |
| P value | 0.002* | 0.003* | 0.293 | 0.001* | 0.001* |
| Malignant vs. Benign | |||||
|---|---|---|---|---|---|
| Malignant | 85.6 | 79.3 | 46.7 | 38.9 | 62.7 |
| Benign | 78.6 | 70.6 | 43.7 | 27.9 | 55.2 |
| P value | 0.097 | 0.011* | 0.482 | 0.004* | 0.009* |
| Methylation vs. gene expression | |||||
|---|---|---|---|---|---|
| VHL (assay 1) R = | -0.35 | -0.31 | -0.24 | -0.26 | -0.36 |
| P value | < 0.0001* | 0.0003* | 0.0071* | 0.0027* | < 0.0001* |
| Methylation vs. gene expression | |||||
|---|---|---|---|---|---|
| VHL (assay 2) R = | -0.23 | -0.33 | -0.10 | -0.24 | -0.27 |
| P value | 0.009* | 0.0001* | 0.252 | 0.006* | 0.002* |
Statistically significant (P < 0.05); R, Pearson correlation coefficient.

Figure 1. Comparison of VHL promoter methylation in normal adrenals and benign or malignant tumors. (A) Box plot showing Methylation index (MetI) for VHL in the different sample groups. (B) Column chart showing individual MetI for each sample. *WHO malignant cases.

Figure 2. Methylation at each of the 4 individual CpGs of the VHL promoter in normal adrenals and tumors. The scatterplot shows Pyrosequencing values for each CpG and case analyzed together with the Methylation index (MetI). Normal adrenals, benign and malignant tumors are indicated with colors according to legend.
Increased VHL promoter methylation in malignant tumors and PGL
The results from VHL methylation analysis were compared with tumor and patient characteristics (Table S1). This revealed higher MetI values in malignant as compared with benign tumors (mean 63% vs. 55%; P = 0.009). Furthermore, we found higher MetI in PGLs vs. PCCs (mean 66% vs. 55%; P = 0.001). No significant correlations were found between MetI values and gender (P = 0.5), age at surgery (P = 0.65), tumor weight or size (P = 0.4), epinephrine levels (P = 0.14), norepinephrine levels (P = 0.53), dopamine levels (P = 0.7), constitutional SDHB mutations (P = 0.1) or clinical MEN 2A syndrome (P = 0.31). CpG 4 (Chr3: 10,183,220) showed the largest difference in methylation levels between PGLs and PCCs (mean methylation level 46% vs. 28%, P < 0.001) and between benign and malignant tumors (39% vs. 28%, P = 0.004) (Table 2).
Suppression of VHL gene expression
We studied the VHL gene expression in 74 tumors together with 13 normal adrenals using two individual VHL assays with qRT-PCR. To identify endogenous controls, we assessed 32 candidate housekeeping genes. We found 5 genes to be stably expressed among PCC, PGL and normal adrenal references. These include: B2M, CASC3, MRPL19, PSMC4, and UBC (Fig. S2). Significant changes were found in tumors as compared with normal adrenals. In general, the VHL gene expression was lower in tumors than in normal adrenals (P < 0.0001 for assay 1 and P = 0.002 for assay 2, Fig. 3). Furthermore, we found significantly lower VHL gene expression on PGLs as compared with PCCs for assay 2 (P = 0.047) but not for assay 1 (P = 0.72). Regarding tumor weight, we found significantly lower VHL expression in smaller tumors (Pearson correlation test R = 0.24, P = 0.007 with assay 2, R = 0.11, P = 0.2 with assay 1). Elevated norepinephrine levels were found significantly correlated with higher VHL gene expression (P = 0.02 for assay 1, P = 0.15 for assay 2). No significant correlations were found between VHL gene expression and gender (P = 0.8 for assay 1, P = 0.5 for assay 2), age at surgery (P = 0.58 for assay 1, P = 0.73 for assay 2), epinephrine levels (P = 0.5 for both assay 1 and 2), dopamine levels (P = 0.9 for assay 1, P = 0.3 for assay 2), constitutional SDHB mutations (P = 0.1), or clinical MEN 2A syndrome (P = 0.8 for assay 1, P = 0.6 for assay 2). Furthermore, no significant correlation was found between VHL gene expression and malignancy (P = 0.97 for assay 1, P = 0.63 for assay 2).

Figure 3. Comparison of promoter methylation density and gene expression for VHL. (A) Boxplots showing relative VHL mRNA expression (y-axis) in the sample groups (x-axis) determined by qRT-PCR using two different assays. (B) Scatterplot including trend line showing MetI values (x-axis) and relative mRNA expression levels determined by assay 1 (y-axis).
Inverse correlation between VHL methylation status and mRNA expression
To further investigate the effect of VHL promoter methylation on its gene expression we compared MetIs for the promoter with VHL mRNA expression levels as determined by each of the two qRT-PCR assays. Pearson correlation analysis showed significant correlation (R = 0.796, P < 0.0001) between the two mRNA expression assays supporting that no methodological errors are present since the same gene was analyzed with dissimilar assays. Furthermore, MetI values were inversely correlated with VHL expression (Pearson correlation test R = -0.359, P < 0.0001 for assay 1 and R = -0.232, P = 0.0077 for assay 2). With regard to the individual CpGs, all sites showed inverse correlation between methylation level and VHL expression for assay 1 (Table 2).
Discussion
Using a quantitative approach we assessed the methylation status of the promoters of 11 PCC/PGL susceptibility genes in a large cohort of PCCs, PGLs and adrenal reference samples. We found significant hypermethylation of the VHL gene promoter in the tumor samples compared with normal adrenal references. VHL gene expression correlated inversely with the epigenetic status of the tumors, showing lower expression in tumors and higher expression in adrenal references. Given that gene silencing by promoter CpG methylation is an important epigenetic mechanism in tumorigenesis,17 our results indicate that VHL promoter hypermethylation could play a role in the development of PCC and PGL. Welander et al. have shown lower expression of the VHL gene in tumors with loss of one gene copy.11 Other mechanisms for VHL inactivation could encompass involvement of microRNAs,18 mutations or, as our results indicate, promoter hypermethylation. Taken together our findings suggest that epigenetic VHL silencing is a frequently occurring and potentially disease-related event in PCC and PGL development.
In this study we observed very low methylation levels for all tested susceptibility genes except VHL. This is essentially in agreement with previous, non-quantitative publications discussing methylation analysis of PCC/PGL susceptibility genes.11,15,16 However, our observations contrast previously published conclusions concerning the VHL gene. This discrepancy may have several plausible explanations. Most important are the methodological differences; the previous studies that report unmethylated VHL gene promoters employed MSP11,15,16 and analyzed sequences located 127 nucleotides (6 CpG sites) downstream of the sequence analyzed in this study (Fig. 4). We found that the methylation density varies within the sequence of 4 CpG dinucleotides covered by our VHL assay (Table 2; Fig. S1). It is therefore possible that the previously assessed CpG sites, located downstream of our assay, are indeed unmethylated—as surmised by Welander et al. and Dammann et al. However, a methylated VHL promoter has previously been found by Moore et al. in renal cell carcinomas employing primers targeting a sequence 302 nucleotides (38 CpG sites) downstream of the sequence analyzed in our study (Fig. 4).19 Previously, a genome-wide methylation analysis (Illumina Infinium HM27 DNA methylation assay) was undertaken for 145 PCCs and PGLs14 which did not report hypermethylation in the VHL promoter. This discrepancy with our data most likely stems from the fact that the CpG sites covered by the Illumina 27K chip are dissimilar to those analyzed in this study. The nearest location for any individual CpG site covered by the Illumina assay is found ~1 kb downstream of the sequence assessed herein.
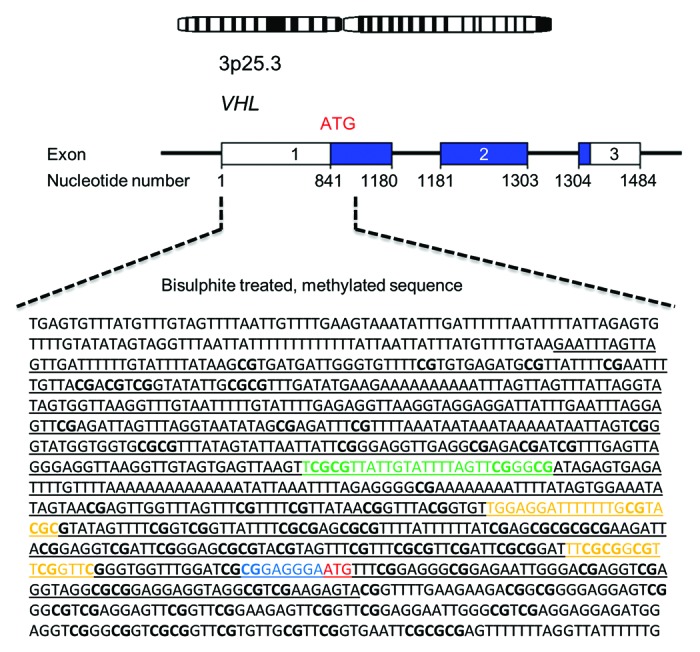
Figure 4. Schematic overview of the VHL promoter region. Underlined letters in black represent the annotated promoter sequence (GenBank accession No AF010238), letters in green denote the sequence analyzed in this study, letters in yellow denote the primer sites analyzed by Dammann et al., letters in blue represent the sequence analyzed by Moore et al., bold letters represent CpG islands and letters in red mark the translation start site.
The increased VHL MetI levels in the tumors compared with normal adrenal references suggest that his is an important event in tumor development. A microarray study comparing rat PCCs with normal adrenal medullas revealed a number of differentially expressed genes, but did not identify VHL as one of them, indicating a difference between rat and human PCCs.20 We also observed significantly increased MetI levels for VHL in malignant as compared with benign cases and the VHL gene expression was inversely correlated with MetI values. However, the VHL gene expression did not show any statistically significant difference between malignant and benign cases. Likely, the variations in VHL gene expression may be present but can be too small to detect. Previously, gene expression profiling studies in human have not identified VHL as a differentially expressed gene between malignant and benign PCCs,21-24 which is in agreement with our observations.
A CpG hypermethylator phenotype has been described for the PGL and SDHB12,13 as well as SDHx14 mutated entity. In this context it is of interest to note that the two cases with RET hypermethylation were malignant PGL harboring SDHB mutation (PGL-102 and PGL-113). Neither VHL promoter methylation nor VHL gene expression correlated with SDHB mutations and accompanying CpG island methylator phenotype (CIMP).13 The possible association to clinical VHL syndrome could not be evaluated as this was only observed in one patient.
In summary, our results show that the VHL gene promoter is hypermethylated in pheochromocytomas and paragangliomas, accompanied by stunted gene expression, a finding that highlights the importance of VHL inactivation in the etiology of these tumors.
Materials and Methods
Tumor samples and controls
Fresh frozen tissue material was obtained from the Biobank of the Karolinska University Hospital, Stockholm, Sweden. These samples had been collected with written or oral informed consent, as documented in patients’ files, and sampling was approved by the local ethical committee. All tissues included were dissected and handled in accordance with established biobank protocols. The cohort included 96 tumors (83 PCCs, 13 PGLs; representative for the ratio of PCCs vs. abdominal PGLs operated at the Karolinska University Hospital) (Table S1). Thirty samples of non-tumorous adrenal tissues were used as normal adrenal references (Table S2). These samples were obtained in conjunction with renal or adrenal surgery and histopathologically verified as non-tumorous by two expert pathologists independently. In addition, we included 4 samples of commercially available DNA from normal adrenal medullas (NAM-1–4, Table S2) obtained from autopsy of patients deceased from unrelated causes (Clinomics Biosciencies Inc.).
Cases were characterized for gender, age of onset, hormonal status, existence of multifocal tumors, occurrence of multiple or bilateral tumors, tumor weight and tumor size, recurrence, duration of follow-up, patient outcome and time for metastasis. In addition, occurrence of constitutional SDHB mutations and clinical VHL, MEN 2A, and NF1 syndrome is given in Table S1. In the current paper malignant cases were identified based on metastasis and/or extensive local invasion according to the Armed Forces Institute of Pathology (AFIP) guidelines,25 due to higher sensitivity in detecting potentially deleterious tumors (herein referred to as malignant). Additionally, we have given the classification according to the World Health Organization (WHO) guidelines1 (referred to as WHO malignant), since these are also widely accepted for the classification of these tumors. The WHO criteria base malignancy solely on the presence of distant metastases.1 A small representative piece was cut from 115/130 (88%) of all analyzed samples. The piece was thawed and fixed in formaldehyde solution, embedded in paraffin, sectioned and stained with Haematoxylin and Eosin. Tumor or normal cell content was subsequently determined by two expert pathologists, independently revealing representative tumor or normal cell content of more than 70% in 92% of the included cases (data not shown). Cases that revealed tumor or normal cell content <50% were excluded from the study.
Preparation of DNA and RNA
Extraction of genomic DNA was performed using DNeasy Blood and Tissue DNA isolation kit (QIAGEN AB). Quantification and initial quality check of the extracted DNA was performed using a NanoDrop A100 (Thermo Scientific). EpiTect Bisulphite conversion kit was used for bisulphite conversion of 500 ng of genomic DNA from all tumors and normal samples (QIAGEN AB). The method is described in detail in a preceding publication.26
Extraction of total RNA was performed using the mirVana miRNA Isolation Kit (Ambion, Life Technologies, Applied Biosystems) with no changes to the original protocol. RNA quality was measured using an Agilent 2100 Bioanalyzer (Agilent) with the RNA Nano 6000 chips and reagents (Agilent) (Tables S1 and S2).
Pyrosequencing and data analysis
Pyrosequencing was performed to assess the methylation level of PCC and PGL susceptibility genes in 96 tumors (83 PCCs, 13 PGLs) and 34 adrenal/adrenal medullary reference samples. The assays for EGLN1, MAX, MEN1, NF1, RET, SDHAF2, TMEM127, and VHL are commercially available (PyroMark Assay Database, Geneglobe, QIAGEN). The SDHB, SDHC and SDHD assays were ad hoc-designed using PyroMark Assay Design software version 2.0 (QIAGEN). Both newly designed and commercially available assays were extensively tested for assay bias as previously described13 (results not shown). Assay specifications and sequences are summarized in Table S3. We also made several attempts at designing primers for SDHA, and for the VHL region corresponding to the target sites in an article by Dammann et al.,16 however these assays did not produce reliable results and were therefore excluded.
Pyrosequencing has been described in detail in a previous publication.27 In short, pre-made master mix—including DNA polymerase from QIAGEN—was used to amplify 1 µl of bisulphite–treated DNA in a PCR reaction, using the following conditions: 95 °C for 15 min, followed by 45 cycles of: (95 °C 30 s, 56 °C [all assays except SDHB, SDHC, and SDHD; 53 °C] 30 s, 72 °C 30 s), and 72 °C for 10 min. The PCR product was then run in a PyroMark Q96 ID pyrosequencer, employing unique run files for each separate assay (available for download through QIAGEN). Quantification of methylation density was done using the PyroMark Q96 software. Negative controls (water sample, no primer sample, no DNA sample and 0% methylated commercially available DNA [QIAGEN]) and positive controls (50% and 100% methylated commercially available DNA [QIAGEN]) were included in all experiments.
For each assay and sample, a methylation index (MetI) was calculated as the mean methylation of all CpG dinucleotides covered in the assay. Subsets of cases were re-analyzed with repeated PCRs and subsequent Pyrosequencing to validate the initial results. These tests had similar end results (±2%) for all CpG dinucleotides in all cases (data not shown). Subsets of cases (including both PCCs and PGLs) were bisulphite treated a second time and subsequently analyzed using PCR and Pyrosequencing, with analogous end results (data not shown). DNA from multiple cases was also re-extracted for the same experiments and Pyrosequencing showed the same end results (±2%) for all CpGs in all cases tested (data not shown), indicating the robustness and reproducibility of the methods employed.
Quantitative reverse transcription real-time PCR (qRT-PCR)
qRT-PCR analysis was performed for 74 of the tumors, together with 13 of the non-tumorous adrenal reference samples where RNA was available. The 4 normal medulla samples were not included. cDNA was synthesized from 100 ng total RNA using High Capacity cDNA Reverse Transcription Kit, following manufacturer’s protocol (Applied Biosystems). For the qRT-PCR, 1 µl of cDNA template was used together with PCR reaction mix and the particular TaqMan gene expression assay: VHL mRNA assay 1 (Hs00184451_m1, Applied Biosystems) and VHL mRNA assay 2 (Hs01650959_m1) targeting total VHL transcripts.
As recommended by the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines,28 we tested 32 different endogenous control genes in triplicates for 6 different PCC, PGL, and normal adrenal tissue samples. These genes are included in the TaqMan Array Human Endogenous control plate (Applied Biosystems). Results from the testing of the endogenous control run are summarized in Fig. S2, and their specifications are summarized in Table S3. The 5 most stable endogenous controls chosen for further experiments were selected from the ones displaying the smallest variation among the samples i.e., B2M, CASC3, MRPL19, PSMC4, and UBC. The geometrical mean of the expression of these genes were used to normalize all expression data. Triplicates of PCC, PGL, and normal adrenal samples were run together with endogenous controls and non-template controls in 384-well plates using standard quantitative method of amplification in a 7900HT Real-Time PCR System (Applied Biosystems). Relative expression levels for the VHL gene were reported as 2−∆CT.
Statistics
Pearson correlation test was used to compare methylation density and gene expression for methylated genes and to compare age at surgery with methylation density and gene expression. Unpaired student t-test was performed to analyze differences in the methylation and gene expression levels between the tumor groups and normal adrenals, respectively. Unpaired student t-test was also used to compare gender with methylation status and gene expression. Comparisons between tumor subgroups were always done employing the AFIP criteria for malignancy. When relevant, WHO malignant cases are separately specified.
A P value of < 0.05 was regarded as statistically significant. The statistical analyses were performed using STATISTICA v10 (STATSOFT) and Figures 2 and 3B were generated with Think Cell (think-cell Software GmBH)
Supplementary Material
Acknowledgments
The authors wish to thank Ms Susanne Bergström for valuable Think-Cell guidance and Ms Lisa Ånfalk at the Department of Oncology-Pathology, Karolinska University Hospital Solna, for excellent sample collection and handling.
Glossary
Abbreviations:
- AFIP
armed forces institute of pathology
- bp
base pair
- CIMP
CpG island methylator phenotype
- EGLN1 (PDH2)
egl nine homolog 1 gene
- EPAS (HIF2A)
endothelial PAS domain 1 gene
- KIF1Bβ
kinesin family member 1B beta gene
- MAX
myc associated factor X gene
- MEN 2A
multiple endocrine neoplasia type 2A
- MetI
methylation index
- NF1
neurofibromatosis type 1 syndrome
- NF1
neurofibromin 1 gene
- PCC
pheochromocytoma
- PGL
abdominal paraganglioma
- RET
rearranged during transfection proto-oncogene
- SDHA
succinate dehydrogenase complex, subunit A gene
- SDHAF2 (SDH5)
succinate dehydrogenase complex assembly factor 2 gene
- SDHB succinate dehydrogenase complex
subunit B gene
- SDHC succinate dehydrogenase complex
subunit C gene
- SDHD succinate dehydrogenase complex
subunit D gene
- TMEM127
transmembrane protein 127 gene
- VHL
von Hippel-Lindau syndrome
- VHL
von Hippel-Lindau tumor suppressor gene
- WHO
world health organization
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
The study was financially supported by Karolinska Institutet, StratCan (Strategic Research Program in Cancer at Karolinska Institutet), the Swedish Research Council, The Cancer Society in Stockholm, Gustav V Jubilee Foundation, the Swedish Cancer Society, as well as the Regional Agreement on Medical Training and Clinical Research (ALF) between Stockholm County Council and Karolinska Institutet.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/epigenetics/article/26686
Footnotes
Previously published online: www.landesbioscience.com/journals/epigenetics/article/26686
References
- 1.DeLellis R. Pathology and genetics of tumours of endocrine organs. Lyon: IARC Press; 2004. 136–166 p. [Google Scholar]
- 2.Bravo EL, Gifford RW., Jr. Current concepts. Pheochromocytoma: diagnosis, localization and management. N Engl J Med. 1984;311:1298–303. doi: 10.1056/NEJM198411153112007. [DOI] [PubMed] [Google Scholar]
- 3.Sutton MG, Sheps SG, Lie JT. Prevalence of clinically unsuspected pheochromocytoma. Review of a 50-year autopsy series. Mayo Clin Proc. 1981;56:354–60. [PubMed] [Google Scholar]
- 4.Neumann HPH, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, Schipper J, Klisch J, Altehoefer C, Zerres K, et al. Freiburg-Warsaw-Columbus Pheochromocytoma Study Group Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002;346:1459–66. doi: 10.1056/NEJMoa020152. [DOI] [PubMed] [Google Scholar]
- 5.Jafri M, Maher ER. The genetics of phaeochromocytoma: using clinical features to guide genetic testing. Eur J Endocrinol. 2012;166:151–8. doi: 10.1530/EJE-11-0497. [DOI] [PubMed] [Google Scholar]
- 6.Yeh I-T, Lenci RE, Qin Y, Buddavarapu K, Ligon AH, Leteurtre E, Do Cao C, Cardot-Bauters C, Pigny P, Dahia PLM. A germline mutation of the KIF1B beta gene on 1p36 in a family with neural and nonneural tumors. Hum Genet. 2008;124:279–85. doi: 10.1007/s00439-008-0553-1. [DOI] [PubMed] [Google Scholar]
- 7.Bayley J-P, Kunst HPM, Cascon A, Sampietro ML, Gaal J, Korpershoek E, Hinojar-Gutierrez A, Timmers HJLM, Hoefsloot LH, Hermsen MA, et al. SDHAF2 mutations in familial and sporadic paraganglioma and phaeochromocytoma. Lancet Oncol. 2010;11:366–72. doi: 10.1016/S1470-2045(10)70007-3. [DOI] [PubMed] [Google Scholar]
- 8.Astuti D, Ricketts CJ, Chowdhury R, McDonough MA, Gentle D, Kirby G, Schlisio S, Kenchappa RS, Carter BD, Kaelin WG, Jr., et al. Mutation analysis of HIF prolyl hydroxylases (PHD/EGLN) in individuals with features of phaeochromocytoma and renal cell carcinoma susceptibility. Endocr Relat Cancer. 2011;18:73–83. doi: 10.1677/ERC-10-0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Comino-Méndez I, de Cubas AA, Bernal C, Álvarez-Escolá C, Sánchez-Malo C, Ramírez-Tortosa CL, Pedrinaci S, Rapizzi E, Ercolino T, Bernini G, et al. Tumoral EPAS1 (HIF2A) mutations explain sporadic pheochromocytoma and paraganglioma in the absence of erythrocytosis. Hum Mol Genet. 2013;22:2169–76. doi: 10.1093/hmg/ddt069. [DOI] [PubMed] [Google Scholar]
- 10.Burnichon N, Buffet A, Parfait B, Letouzé E, Laurendeau I, Loriot C, Pasmant E, Abermil N, Valeyrie-Allanore L, Bertherat J, et al. Somatic NF1 inactivation is a frequent event in sporadic pheochromocytoma. Hum Mol Genet. 2012;21:5397–405. doi: 10.1093/hmg/dds374. [DOI] [PubMed] [Google Scholar]
- 11.Welander J, Larsson C, Bäckdahl M, Hareni N, Sivlér T, Brauckhoff M, Söderkvist P, Gimm O. Integrative genomics reveals frequent somatic NF1 mutations in sporadic pheochromocytomas. Hum Mol Genet. 2012;21:5406–16. doi: 10.1093/hmg/dds402. [DOI] [PubMed] [Google Scholar]
- 12.Geli J, Kiss N, Karimi M, Lee J-J, Bäckdahl M, Ekström TJ, Larsson C. Global and regional CpG methylation in pheochromocytomas and abdominal paragangliomas: association to malignant behavior. Clin Cancer Res. 2008;14:2551–9. doi: 10.1158/1078-0432.CCR-07-1867. [DOI] [PubMed] [Google Scholar]
- 13.Kiss NB, Muth A, Andreasson A, Juhlin CC, Geli J, Bäckdahl M, Höög A, Wängberg B, Nilsson O, Ahlman H, et al. Acquired hypermethylation of the P16INK4A promoter in abdominal paraganglioma: relation to adverse tumor phenotype and predisposing mutation. Endocr Relat Cancer. 2013;20:65–78. doi: 10.1530/ERC-12-0267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Letouzé E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, Janin M, Menara M, Nguyen AT, Benit P, et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 2013;23:739–52. doi: 10.1016/j.ccr.2013.04.018. [DOI] [PubMed] [Google Scholar]
- 15.Cascon A, Ruiz-Llorente S, Fraga MF, Leton R, Telleria D, Sastre J, Diez JJ, Martinez Diaz-Guerra G, Diaz Perez JA, Benitez J, et al. Genetic and epigenetic profile of sporadic pheochromocytomas. J Med Genet. 2004;41:e30. doi: 10.1136/jmg.2003.012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dammann R, Schagdarsurengin U, Seidel C, Trümpler C, Hoang-Vu C, Gimm O, Dralle H, Pfeifer GP, Brauckhoff M. Frequent promoter methylation of tumor-related genes in sporadic and men2-associated pheochromocytomas. Exp Clin Endocrinol Diabetes. 2005;113:1–7. doi: 10.1055/s-2004-830522. [DOI] [PubMed] [Google Scholar]
- 17.Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301:89–92. doi: 10.1038/301089a0. [DOI] [PubMed] [Google Scholar]
- 18.Valera VA, Walter BA, Linehan WM, Merino MJ. Regulatory Effects of microRNA-92 (miR-92) on VHL Gene Expression and the Hypoxic Activation of miR-210 in Clear Cell Renal Cell Carcinoma. J Cancer. 2011;2:515–26. doi: 10.7150/jca.2.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moore LE, Nickerson ML, Brennan P, Toro JR, Jaeger E, Rinsky J, Han SS, Zaridze D, Matveev V, Janout V, et al. Von Hippel-Lindau (VHL) inactivation in sporadic clear cell renal cancer: associations with germline VHL polymorphisms and etiologic risk factors. PLoS Genet. 2011;7:e1002312. doi: 10.1371/journal.pgen.1002312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elkahloun AG, Powers JF, Nyska A, Eisenhofer G, Tischler AS. Gene expression profiling of rat pheochromocytoma. Ann N Y Acad Sci. 2006;1073:290–9. doi: 10.1196/annals.1353.033. [DOI] [PubMed] [Google Scholar]
- 21.Waldmann J, Fendrich V, Holler J, Buchholz M, Heinmöller E, Langer P, Ramaswamy A, Samans B, Walz MK, Rothmund M, et al. Microarray analysis reveals differential expression of benign and malignant pheochromocytoma. Endocr Relat Cancer. 2010;17:743–56. doi: 10.1677/ERC-09-0118. [DOI] [PubMed] [Google Scholar]
- 22.Eisenhofer G, Huynh T-T, Pacak K, Brouwers FM, Walther MM, Linehan WM, Munson PJ, Mannelli M, Goldstein DS, Elkahloun AG. Distinct gene expression profiles in norepinephrine- and epinephrine-producing hereditary and sporadic pheochromocytomas: activation of hypoxia-driven angiogenic pathways in von Hippel-Lindau syndrome. Endocr Relat Cancer. 2004;11:897–911. doi: 10.1677/erc.1.00838. [DOI] [PubMed] [Google Scholar]
- 23.Dahia PLM, Ross KN, Wright ME, Hayashida CY, Santagata S, Barontini M, Kung AL, Sanso G, Powers JF, Tischler AS, et al. A HIF1alpha regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS Genet. 2005;1:72–80. doi: 10.1371/journal.pgen.0010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thouënnon E, Elkahloun AG, Guillemot J, Gimenez-Roqueplo A-P, Bertherat J, Pierre A, Ghzili H, Grumolato L, Muresan M, Klein M, et al. Identification of potential gene markers and insights into the pathophysiology of pheochromocytoma malignancy. J Clin Endocrinol Metab. 2007;92:4865–72. doi: 10.1210/jc.2007-1253. [DOI] [PubMed] [Google Scholar]
- 25.Lack EE. Tumors of the adrenal glands and extraadrenal paraganglia. Washington DC, USA: American Registry of Pathology in collaboration with the Armed Forces Institute of Pathology. 2007. 274–276 p. [Google Scholar]
- 26.Sulaiman L, Juhlin CC, Nilsson I-L, Fotouhi O, Larsson C, Hashemi J. Global and gene-specific promoter methylation analysis in primary hyperparathyroidism. Epigenetics. 2013;8:646–55. doi: 10.4161/epi.24823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Juhlin CC, Kiss NB, Villablanca A, Haglund F, Nordenström J, Höög A, Larsson C. Frequent promoter hypermethylation of the APC and RASSF1A tumour suppressors in parathyroid tumours. PLoS One. 2010;5:e9472. doi: 10.1371/journal.pone.0009472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55:611–22. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.