

Abstract
While recent spectroscopic studies have established the presence of an interstitial carbon atom at the center of the iron-molybdenum cofactor (FeMoco) of MoFe-nitrogenase, its role is unknown. We have pursued Fe-N2 model chemistry to explore a hypothesis whereby this C-atom (previously denoted as a light X-atom) may provide a flexible trans interaction with an Fe center to expose an Fe-N2 binding site. In this context, we now report on Fe complexes of a new tris(phosphino)alkyl (CPiPr3) ligand featuring an axial carbon donor. It is established that the iron center in this scaffold binds dinitrogen trans to the Calkyl-atom anchor in three distinct and structurally characterized oxidation states. Fe-Calkyl lengthening is observed upon reduction, reflective of significant ionic character in the Fe-Calkyl interaction. The anionic (CPiPr3)FeN2- species can be functionalized by a silyl electrophile to generate (CPiPr3)Fe-N2SiR3. (CPiPr3)FeN2- also functions as a modest catalyst for the reduction of N2 to NH3 when supplied with electrons and protons at -78 °C under 1 atm N2 (4.6 equiv NH3/Fe).
INTRODUCTION
The biological reduction of atmospheric N2 to NH3 is a fascinating yet poorly understood transformation that is essential to life.1 The iron-molybdenum cofactor (FeMoco) of MoFe nitrogenase catalyzes N2 reduction and has been extensively studied.2 This cofactor has attracted the attention of inorganic and organometallic chemists for decades who have sought inspiration to explore the ability of synthetic iron and molybdenum complexes to bind and reduce dinitrogen.3,4,5,6 Advances in the past decade have included two molybdenum systems that facilitate catalytic turnover of N2 to NH3 in the presence of inorganic acid and reductant sources,7,8,9 and iron complexes that support a range of NxHy ligands relevant to nitrogen fixation,10,11,12,13 effect reductive N2 cleavage,14,15 and facilitate N2 functionalization.16,17,18
The presence of an interstitial light atom in the MoFe nitrogenase cofactor was established in 2002,19 and structural, spectroscopic, and biochemical data have more recently established its identity as a C-atom.20 The role of the C-atom is unknown. This state of affairs offers an opportunity for organometallic chemists to undertake model studies that can illuminate plausible roles for this interstitial C-atom, and hence critical aspects of the mechanism of N2 reduction catalysis. In particular, Fe-alkyl complexes that are more ionic in nature than a prototypical transition metal-alkyl may be relevant to modeling the Fe-Cinterstitial interaction of the possible N2 binding site in the cofactor (Figure 1).
Figure 1.
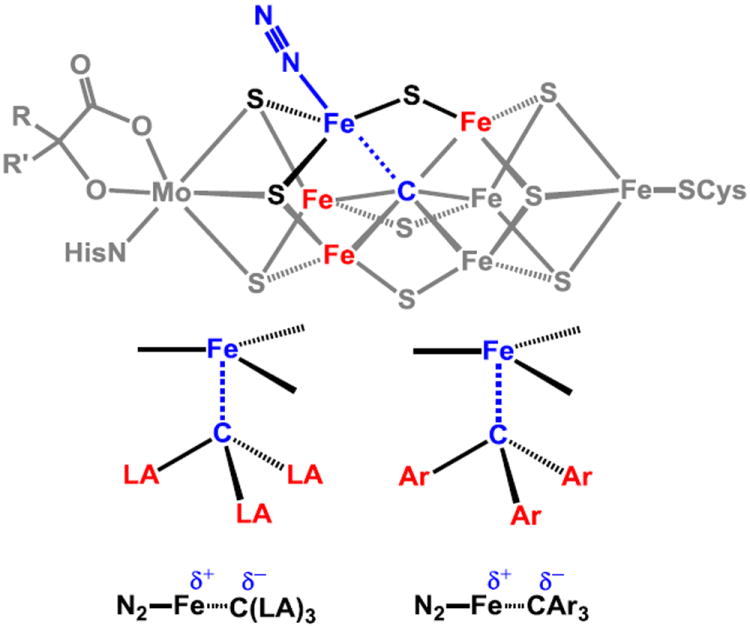
(Top) Structure of the FeMo cofactor of nitrogenase, showing a putative site for dinitrogen binding and highlighting the trigonal bipyramidal coordination environment at Fe. Possible sites of H-atoms on cofactor prior to N2 binding not shown. (Bottom) Possible role of Lewis acidic (LA) or aryl substituents in stabilizing ionic character in the N2-Fe-Calkyl interaction.
We have suggested that a possible role played by the interstitial C-atom is to provide a flexible Fe-Cinterstitial interaction that exposes an Fe-N2 binding site on a belt iron atom trans to the Fe-C linkage (Figure 1).3,15,21,22,23 Subsequent modulation of the Fe-C interaction and hence the local Fe geometry as a function of the N2 reduction state might enable the Fe center to stabilize the various NxHy intermediates sampled along a pathway to NH3.
To test the chemical feasibility of this hypothesis for Fe-mediated N2 reduction, our group has previously employed phosphine-supported Fe complexes in approximately trigonal geometries (pseudotetrahedral, trigonal pyramidal, or trigonal bipyramidal) to bind and functionalize dinitrogen. Tripodal trisphosphine ligands featuring an axial donor (X = N, Si, B) and aryl backbones have been used to canvass the ability of low-valent iron in such geometries to bind and activate dinitrogen (Figure 2).23,24,25 The (TPiPrB)Fe-system (TPRB = tris(o-phosphinoaryl)borane) has proven rich in this context, and has most recently been shown to be a modestly effective catalyst for the reduction of N2 to NH3 in the presence of proton and electron sources at low temperature and 1 atm N2.21 An important feature of the (TPiPrB)Fe-system is the presence of a flexible Fe-B interaction. 15,25 This flexibility may facilitate the formation of intermediates featuring Fe-Nx π-bonding (e.g., Fe=NNH2, Fe≡N, Fe=NH) during catalysis. Whether the aforementioned hypothesis concerning a hemi-labile role for the interstitial C-atom of FeMoco is correct or not, these inorganic model studies lend credibility to the idea so far as the principles of coordination chemistry are concerned.
Figure 2.
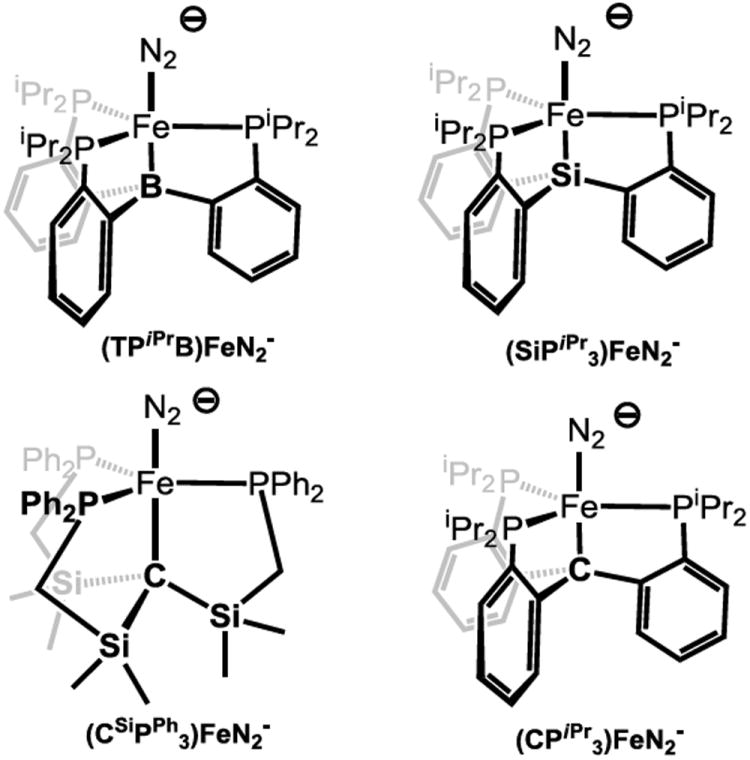
Select trigonal bipyramidal scaffolds previously studied by our lab, and the present (CPiPr3)FeN2- system.
To extend our studies to systems that place a C-atom in a position trans to an Fe-N2 binding site we have sought related ligand scaffolds that feature a C-atom anchor. In designing these scaffolds we have hypothesized that the proposed flexibility of the Fe-C linkage in the FeMo cofactor may be facilitated by the ability of the environment around the interstitial carbide—five additional electropositive Fe atoms—to stabilize developing negative charge on the carbon. With this in mind we have previously reported iron complexes of a tris(phosphino)alkyl ligand whose axial carbon binding site is flanked by three electropositive silyl groups (Figure 2) which may play a role in stabilizing the substantial ionic character of this Fe-Calkyl bond (Figure 1).22
Herein we report a new tris(phosphino)alkyl ligand, (CPiPr3), featuring aryl linkers bound to the axial carbon. We reasoned that possible delocalization of negative or positive charge buildup into the aryl π-system would allow for increased flexibility in the Fe-C bond; this flexibility is expected to facilitate possible catalytic N2 functionalization and reduction, as discussed above. Additionally, as this ligand is closely structurally related to the SiP3, TPB, and NP3 ligands whose iron coordination chemistry we have extensively explored, Fe complexes of CP3iPr are of obvious comparative interest and would be particularly beneficial with regard to considering the role an Fe-Cinterstitial interaction might play in facilitating N2 binding and reduction within the cofactor. To this end, we embarked on the synthesis of the new ligand (CPiPr3)H and the development of its Fe-N2 chemistry.
RESULTS AND DISCUSSION
Ligand Synthesis
Whereas the ligands (SiPiPr3)H and TPiPrB are straightforward to synthesize by the addition of lithiated o-phosphinophenyl precursors to HSiCl3 and BCl3,24,26 the preparation of (CPiPr3)H via an analogous method by addition of phosphinoaryllithium moieties to a C1 source (e.g., triple addition to dimethylcarbonate followed by deoxygenation of the resultant triarylmethanol product) has proven ineffective in our hands. However, an orthogonal synthetic approach based on elaboration of an initially formed triarylmethane scaffold afforded a viable approach to the preparation of (CPiPr3)H on a multigram scale and in reasonable yields. This synthesis of (CPiPr3)H follows an approach inspired by a previously reported synthesis of Ph2P(o-C6H4CH2C6H4-o)PPh2,27 and hinges on the sequential formation and cleavage of two diaryliodonium ions to give the tris(2-halophenyl)methane precursor (5) (Scheme 1).
Scheme 1.
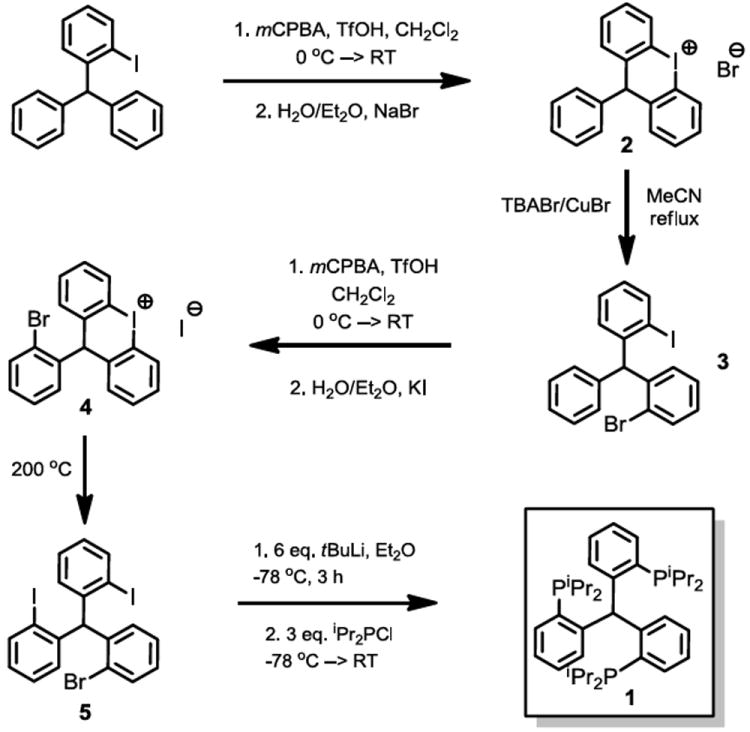
Synthesis of (CPiPr3)H (1).
The synthesis of o-iodotriphenylmethane has been previously reported28 and is readily effected in three steps from commercially available 2-nitrobenzaldehyde on a 20-gram scale. Cyclization of this species to the diaryliodonium bromide salt (2) is accomplished by a previously reported technique.29 Slow but clean ring-opening of 2 by CuBr and [TBA][Br] in acetonitrile gives 2-bromo-2’-iodotriphenylmethane (3). The 2-bromo-2’-iodotriphenylmethane species was targeted rather than 2,2’-diiodotriphenylmethane in order to mitigate the possibility of complications from excessive oxidation in the next step.
Formation of a second diaryliodonium cation as its iodide salt follows via an analogous procedure to regioselectively generate 4, which can be straightforwardly decomposed to 2-bromo-2’,2”-diiodotriphenylmethane (5) by heating to 200 °C for 15 minutes under an inert atmosphere. Each step in the synthesis of 5 from o-iodotriphenylmethane can be accomplished in 75% yield or more (overall yield: 38% over five steps).
Lithiation of 5 with six equiv of tert-butyllithium at -78 °C followed by treatment with three equiv of diisopropylphosphine chloride gives the desired tris(o-diisopropylphosphinophenyl) methane, (CPiPr3)H (1) in 67% yield (Scheme 1). The protonated form of the ligand, 1, is characterized by a single peak in its phosphorus NMR spectrum at -9.1 ppm. The 1H NMR spectrum, while indicative of three-fold symmetry, also shows features suggestive of a rigid ligand scaffold where rotation about the phosphine-carbon bonds is hindered; in particular, four magnetically inequivalent sets of resonances are observed for the isopropyl methyl hydrogens. Additionally, the central C-H methine proton is shifted markedly downfield (8.15 ppm) and manifests as a quartet due to through-space coupling to the three phosphorus atoms. Similar NMR properties were observed for the central methine proton in a related trisphosphine ligand based on a tris(indolyl)methane scaffold.30
Metallation at iron and precursor complexes
We initially hoped to effect metallation of 1 by first deprotonating it to give an alkali metal complex followed by transmetallation with an iron (II) halide or other transition metal precursor. To our frustration, 1 proved unexpectedly difficult to deprotonate even with very strong bases such as benzyl potassium and Schlosser’s base,31 perhaps due in part to the steric protection of the methine proton; additionally, the acidity of this proton is likely not as high as for bare triphenylmethane since the ligand bulk limits the extent to which the aryl rings can approach a coplanar configuration to afford resonance stabilization of a resulting carbanion.32 Furthermore, the strategy used for metallation of the (SiPiPr3)H ligand on iron—using methyl Grignard with FeCl2 to generate a methyl iron complex which then eliminates methane with concomitant formation of the iron-silicon bond24—was not effective for (CPiPr3)H. It appeared to instead result in reduction of iron without the formation of the desired iron-carbon bond. Thus, it was necessary to develop a different protocol for the formation of a (CPiPr3)Fe-complex featuring an iron-carbon bond.
Combining 1 and iron(II) iodide in toluene cleanly affords the tetracoordinate, κ2-bisphosphine diiodide highspin iron(II) complex (6) as a yellow powder (Scheme 2). Its solid-state structure (Figure 3) shows a tetrahedral environment at the iron center and a bidentate binding mode for the ligand. One-electron reduction of 6 in benzene or toluene using a range of reagents including sodium amalgam, potassium graphite, or alkylmagnesium/lithium reagents, results in the formation of the deep brick-red four-coordinate iron(I) complex {(CPiPr3)H}FeI (7). The bromide congener, {(CPiPr3)H}FeBr (8), is analogously prepared and has been crystallographically characterized (Figure 3); its most notable feature is the endo orientation of the unactivated methine C-H. This proton is located within the ligand cage pointed nearly linearly towards the iron center. Both 7 and 8 are unstable with respect to disproportionation to Fe(0), (CPiPr3)H, and {(CPiPr3)H}FeX2 (X = I, Br), especially in coordinating solvents. However, if appropriate conditions are employed, 7 is sufficiently longlived to be generated and used without further purification for subsequent reactions.
Scheme 2.
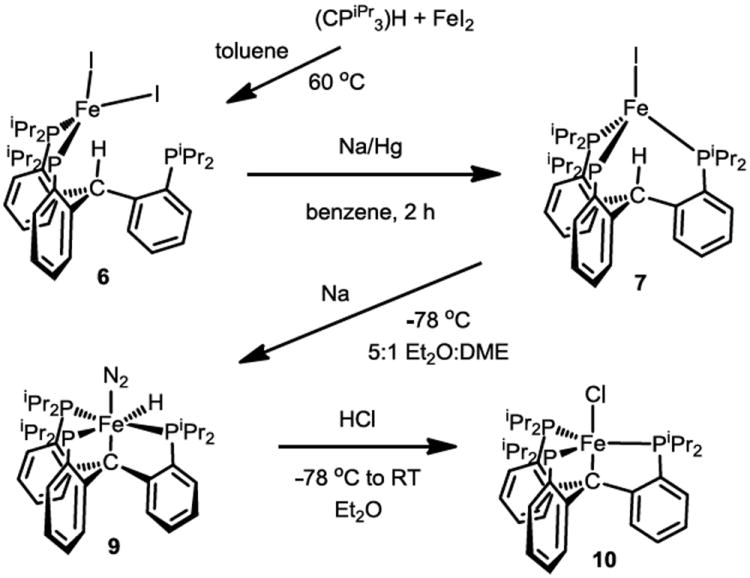
Synthesis of iron complexes of (CPiPr3)H.
Figure 3.
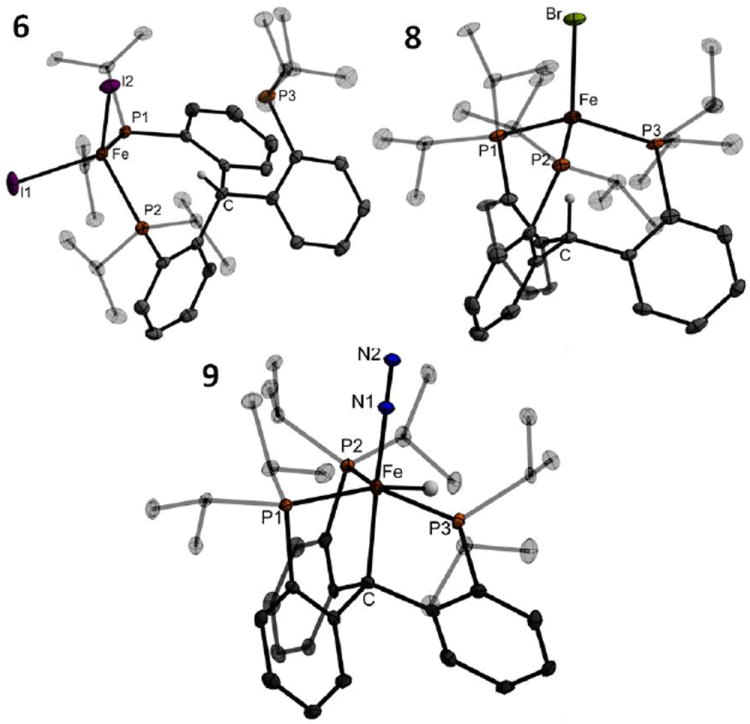
Crystal structures of {(CPiPr3)H}FeI2 (6, top left), {(CPiPr3)H}FeBr (8, top right), and (CPiPr3)Fe(H)(N2) (9, bottom). Ellipsoids shown at 50% probability; hydrogen atoms (except the triarylmethine C-H and Fe-H hydride) and solvent molecules omitted for clarity.
Further reduction of 7 with sodium metal in a 5:1 mixture of Et2O and DME at -78 °C causes formal insertion of the Fe center into the C-H bond of the (CPiPr3)H ligand and uptake of atmospheric N2 to give yellow, diamagnetic (CPiPr3)Fe(H)(N2) (9). The position of the iron hydride is identifiable in the XRD difference map of 9, as is the presence of an Fe-C bond at 2.155(2) Å (Figure 3). IR data for 9 show a strong N-N vibration at 2046 cm-1 and an Fe-H vibration at 1920 cm-1., The properties of 9 can be compared to the isostructural (SiPiPr3)Fe(H)(N2) and [(NPiPr3)Fe(H)(N2)]+ complexes23,33 and other closely related species such as {[P(CH2CH2PiPr2)3]Fe(H)(N2)}+;34 the vibrational and metrical properties of the N2 ligand suggest a more activated dinitrogen moiety in 9 relative to its congeners.
Deprotonation of 9 to afford (CPiPr3)FeN2- was canvassed but proved unsuccessful. A more circuitous but ultimately effective route to (CPiPr3)FeN2- proceeded via treatment of 9 with anhydrous HCl in Et2O to afford dark red-orange (CPiPr3)FeCl (10) in good yield (Scheme 2). The crystal structure of 10 was not reliably determined due to its propensity to crystallize in a cubic space group with extensive whole molecule disorder. Complex 10 is paramagnetic and its room temperature solution magnetic moment of 4.9 μB is suggestive of a high-spin, S = 2 ground state. A lower spin state might have been reasonably anticipated to arise from a presumably strong-field ligand set comprised of three diisopropylarylphosphines and an alkyl group. For comparison, (SiPiPr)3FeCl exhibits an intermediate S = 1 ground state.24 The Calkyl anchor in 10 thereby appears to be a weaker-field donor than the silyl anchor in (SiPiPr)3FeCl.
Synthesis and characterization of the {(CPiPr3)FeN2}n (n = 0, -1, +1) series
Reduction of the chloride precursor 10 affords entry into the desired series of trigonal bipyramidal iron dinitrogen complexes. Stirring 10 over sodium metal in THF produces the neutral low-spin Fe(I) complex (CPiPr3)FeN2 (11) (υ(NN) = 1992 cm-1) (Scheme 3). Complex 11 is low-spin and paramagnetic (S = 1/2); it has been crystallographically characterized (Figure 4) and shows a distortion from trigonal symmetry with one widened P-Fe-P angle (132.5°), as expected due to the Jahn-Teller active ground state. The N2 vibrational frequency and N-N bond length (1.134(4) Å) show that the dinitrogen ligand in this complex is somewhat more activated than that in the isoelectronic (SiPiPr3)FeN2 complex (υ(NN) = 2003 cm-1, N-N = 1.1245(2) Å) or in the neutral Fe(0) complex (TPiPrB)FeN2 (υ(NN) = 2011 cm-1).17,25 These differences are relatively small and as such are difficult to reliably interpret. But given the fact that (CPiPr3) appears to have a weaker-field donor set than (SiPiPr3) according to the observed ground spin states of (CPiPr3)FeCl (S = 2) and (SiPiPr3)FeCl (S = 1), one might have reasonably anticipated (SiPiPr3)FeN2 to have a lower υ(NN) than (CPiPr3)FeN2.
Scheme 3.
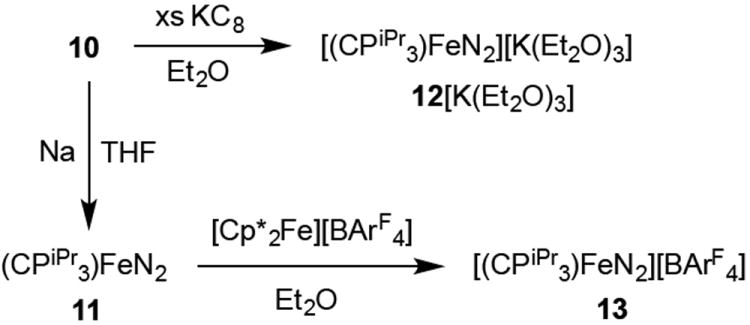
Synthesis of the dinitrogen adduct series (CPiPr3)FeN2 (11), (CPiPr3)FeN2- (12), and (CPiPr3)FeN2+ (13).
Figure 4.
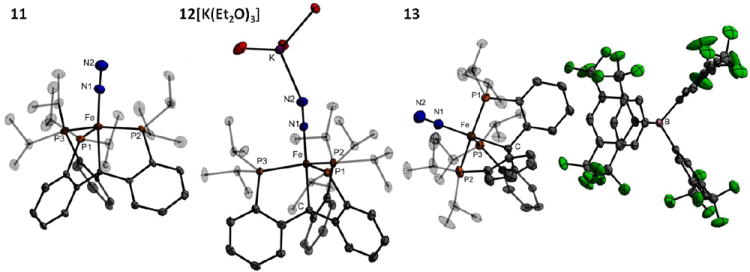
Crystal structures of (CPiPr3)FeN2 (11, left), (CPiPr3)FeN2- (12[K(Et2O)3], center, ethyl groups of coordinated Et2O molecules omitted), and (CPiPr3)FeN2+ (13, right). Ellipsoids are shown at 50% probability and hydrogen atoms are omitted for clarity.
Both a one-electron oxidation and a one-electron reduction of 11 are accessible (Figure 5). The Fe(II/I) couple appears at -1.20 V (vs Fc/Fc+) and is quasi-reversible; the current in the cathodic wave is diminished and an irreversible reduction wave appears at -1.65 V. This is very similar electrochemical behavior to what has been documented for (SiPiPr3)FeN2 and suggests that the same phenomenon is responsible for the observations in this system17—that is, N2 coordinates reversibly to the {(CPiPr3)Fe}+ complex; partial loss of N2 upon oxidation of (CPiPr3)FeN2 is likely responsible for the quasi-reversibility of the (II/I) couple, and the reduction at -1.65 V is most reasonably attributed to the cationic species {(CPiPr3)Fe(L)}+ (where L may be THF, or may be a vacant site), which then takes up N2 upon reduction. The Fe(I/0) couple is fully reversible, consistent with the formation of a stable (CPiPr3)FeN2- anion. This reduction occurs at an unusually negative potential (-2.55 V vs Fc/Fc+). For comparison, the reduction of (SiPiPr3)FeN2 to (SiPiPr3)FeN2- occurs at -2.2 V.17
Figure 5.
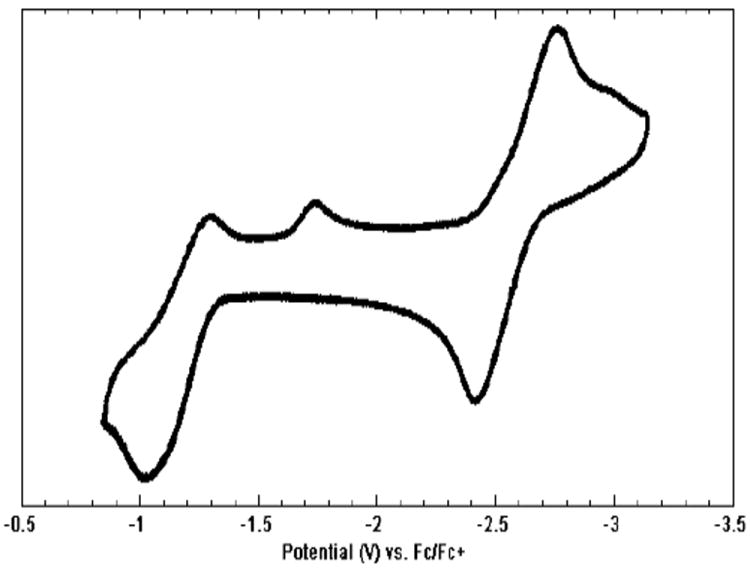
Cyclic voltammogram of 11; scan rate 0.5 V/s.
The Fe-N2 adduct triad {(CPiPr3)FeN2}n (n = 0 (11), -1 (12), +1 (13)) proved synthetically accessible. Treatment of 10 with an excess of potassium graphite (KC8) in Et2O results in immediate reduction to the very dark brown-blue CPiPr3FeN2- anion (12). The IR spectrum of a thin film deposited from diethyl ether solution shows a υ(NN) vibration at 1870 cm-1, suggestive of a close ion pair with the potassium ion capping the N2 moiety. Accordingly, treatment of the potassium complex with two equivalents of 12-crown-4 results in the formation of [(CPiPr3)FeN2][K(12-crown-4)2] (12[K(12-crown-4)2]) with a shift of the υ(NN) vibration to 1905 cm-1. The anion has been crystallographically characterized (Figure 4) as its K(Et2O)3 salt, [(CPiPr3)FeN2][K(Et2O)3] (12[K(Et2O)3]; the bulk material after drying is solvated by 0.5 molecules of Et2O per anion, 12[K(Et2O)0.5]).
Oxidation of 11 with one equivalent of [Cp*2Fe][BArF4] (ArF = 3,5-trifluoromethylphenyl; Cp* = pentamethylcyclopentadienide) in Et2O gives rise to [(CPiPr3)FeN2][BArF4] (13) as an orange crystalline solid, which has also been structurally characterized (Figure 4). The dinitrogen ligand in 13 (υ(NN) = 2128 cm-1), is labile and in solution under an N2 atmosphere appears to be in equilibrium with a solvated or vacant cation [(CPiPr3)Fe(L)]+; in addition to the electrochemical properties discussed above, evidence from UV-Vis spectroscopy is consistent with the loss of coordinated N2 under vacuum (see Supporting Information).
Whereas a related series was accessible for the silylanchored {(SiPiPr3)FeN2}n system (n = 0, +1, -1),17 only the anion (CSiPPh3)FeN2- proved accessible for the previously reported Calkyl-anchored system.22 Hence, the present {(CPiPr3)FeN2}n series allows for a direct comparison of how the anchoring atom (Si vs C) responds across three redox states when positioned trans to an N2 ligand of an isostructural trigonal bipyramidal framework.
In the case of the {(SiPiPr3)FeN2}n series, the Fe-Si bond distance decreases upon reduction from 2.298(7) Å in the (SiPiPr3)FeN2+ cation to 2.2526(9) Å in the (SiPiPr3)FeN2- anion. In direct contrast, the Fe-C bond distance in {(CPiPr3)FeN2}n increases upon reduction, from 2.081(3) Å in 13 to 2.152(3) Å in 11 to 2.1646(17) Å in 12. The different responses manifest in these two systems may be due to the electropositive silicon atom binding more strongly to the more electron-rich iron, whereas the more electronegative Calkyl binds more strongly to the higher-valent, more electron-deficient iron center.
Notably, the overall change in the bond length is greater in the CPiPr3 case (0.084 Å from 13 to 12) than for the more covalent SiPiPr3 system, where the overall change is only 0.045 Å despite the longer total bond length. This suggests a greater degree of flexibility in the Fe-Calkyl interaction. A similar conclusion was drawn for the {(CSiPPh3)Fe(CO)}n (n = +1, 0, -1) series, where an even more pronounced Fe-C lengthening was observed upon reduction.22
In the case of the (TPiPrB)Fe system, a highly flexible Fe-B interaction has been observed as a function of the ligand positioned trans to the B-atom that may be important to its success in activating N2 in both stoichiometric and catalytic reactions.15,21,35 However, an analogous series of N2 complexes has not been characterized to allow for direct comparison. Whereas the anion [(TPiPrB)]FeN2]- has been studied by X-ray crystallography (Fe-B = 2.311(2) Å), the [(TPiPrB)Fe]+ cation does not coordinate N2 at atmospheric pressure, and attempts to obtain the crystal structure of neutral (TPiPrB)FeN2 have been unsuccessful.25,35 Nonetheless, our chemical intuition is that the Fe-B linkage in (TPiPrB)Fe will be appreciably more flexible than the Fe-C linkage in (CPiPr3)Fe.
The Calkyl-Fe interactions in both (CPiPr3)FeN2- (12) and (CSiPPh3)FeN2- reflect a higher degree of ionic character than in a prototypical Fe-Calkyl bond, with (CSiPPh3)FeN2- being most striking in this context.22 Comparative DFT studies of (CSiPPh3)FeN2- and (CPiPr3)FeN2- including NBO analyses, support this view,22,36 predicting strong polarization of the σ-bond pair towards the C-atom (23% Fe/ 77% C in (CSiPPh3)FeN2-; 27% Fe/ 73% C in (CPiPr3)FeN2-) (Figure 6). As expected, the Fe-C bond in 12 is slightly more covalent than that in (CSiPPh3)FeN2-, where the axial carbon is flanked by electropositive silicon atoms. Comparative NBO analyses for (CSiPPh3)FeN2-, (SiPiPr3)FeN2-, and simplified model systems were discussed at greater length in a previous report.22
Figure 6.
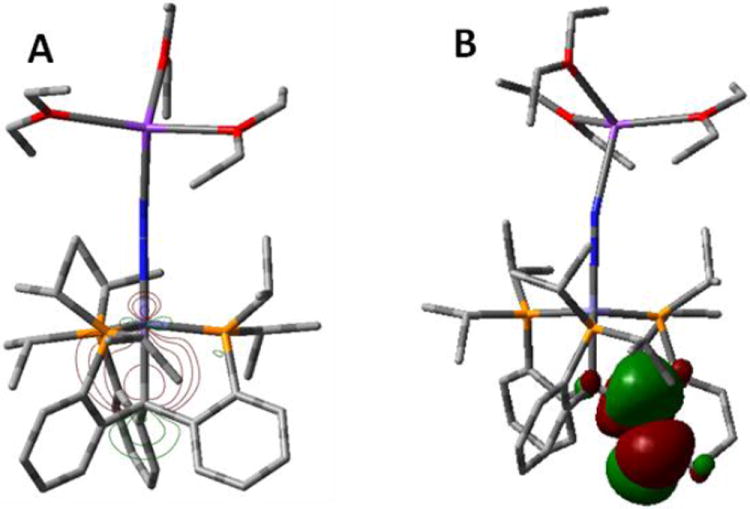
(A) Isocontour plot of the Fe-Calkyl σ bond of 12[K(Et2O)3] located from NBO analyses. (B) Contour plot of one of the Caryl π* orbitals which accepts delocalized electron density from the Fe-Calkyl σ bond.
Second-order perturbation analysis from an NBO calculation indicates the presence of stabilizing donoracceptor interactions between filled and virtual orbitals, representing deviations from a simple Lewis structure description due to electronic delocalization.36 In the case of 12, significant interactions between the filled Fe-Calkyl σ bond and π* orbitals of the aryl rings (Cipso-Cortho) are evident (Figure 6). Three primary donor-acceptor interactions (one to each ring) are located, representing stabilizations of 6.70 kcal/mol, 5.99 kcal/mol, and 5.95 kcal/mol. This result suggests that stabilization of the negative charge on carbon by delocalization onto the aryl rings is at least partially responsible for the observed ionic character of the Fe-C bond, and hence for its increased flexibility. We suggest that a similar stabilization of ionic character at an N2-Fe-Cinterstitial site of the cofactor may facilitate N2 binding.
Reactivity studies
To compare the reactivity of (CPiPr3)FeN2- at the bound N2 ligand with (SiPiPr3)FeN2-, (CSiPPh3)FeN2-, and (TPiPrB)FeN2-, treatment of 12 with TMSCl at -78 °C was examined and afforded the diamagnetic diazenido complex (CPiPr3)FeN2SiMe3 (14) (υ(NN) = 1736 cm-1). This product, though it has not been structurally characterized, is spectroscopically similar to those obtained for the structurally related Si- and B-anchored systems.15,17
More interesting is the comparative behavior of (CPiPr3)FeN2- on treatment with proton/electron equivalents at low temperature. Numerous studies have explored the possibility of Fe-N2 protonation/reduction to release ammonia,3,4,5,6,37 which in all but one case21 afforded low chemical yields of NH3 (ca. ≤ 10% per Fe in one step; 35% per Fe overall in two independent synthetic steps14). The previously described C-anchored system (CSiPPh3)FeN2- (Figure 2) follows a similar trend, affording negligible NH3 on treatment at low temperature with [H(Et2O)2][BArF4] and KC8. The Si-anchored system (SiPiPr3)FeN2- also affords sub-stoichiometric NH3 yields (35% per Fe) when similarly treated, and instead produces some N2H4 (~45% per Fe) when H(Et2O)BF4 and CrCl2 are employed.24
By contrast, cooling a solution of 12[K(Et2O)0.5] in Et2O at -78 °C followed by the addition of 40 equiv KC8 and then 38 equiv [H(Et2O)2][BArF4] leads to the formation of 4.6 ± 0.8 equiv NH3 (230% per Fe; average of 8 runs; Eq. 1), a yield that establishes a modest degree of N2 reduction catalysis at low temperature. No N2H4 is observed. With 12[K(12-crown-4)2] as the catalyst, the NH3 yield is slightly lower at 3.5 ± 0.3 equiv. NH3 quantification was carried out by UV-Vis using the indophenol protocol38 as recently described in detail for the (TPiPrB)FeN2- catalyst system.21 The total NH3 product yield is lower for (CPiPr3)FeN2- than that which was obtained for (TPiPrB)FeN2- when acid was added prior to the reductant. The significance of these modest differences is unclear, especially given the extreme airsensitivity of the catalysts and the low turnover numbers. The order of addition of reagents has a minor effect; reversing the order and adding first acid, then reductant to 12[K(Et2O)0.5] decreases the yield to 3.8 ± 0.6 equiv. NH3 per Fe. In side-by-side comparisons using the same batches of reagents (KC8 and [H(Et2O)2][BArF4]) and the same order of addition (reductant added first), 12[K(Et2O)0.5] afforded 4.4 ± 0.2 equiv. NH3 per Fe, as compared to 5.0 ± 1.1 for (TPiPrB)FeN2- and 0.8 ± 0.4 for (SiPiPr3)FeN2-.
| (Eq. 1) |
Treatment of 12[K(Et2O)0.5] with 10 equivalents of [H(Et2O)2][BArF4] in the absence of added reductant generates negligible ammonia (<0.05 equivalents), verifying that both acid and reductant are necessary for the production of substantial amounts of NH3.
In order to examine possible reasons for the limited turnover for ammonia production with this system, we sought to determine the fate of the precatalyst over the course of the experiment. An analysis of the ironcontaining products of a reaction mixture using 10 equivalents of [H(Et2O)2][BArF4] and 12 equivalents of KC8 (Figure 7) identified the major iron-containing product as (CPiPr3)FeN2 (11), which is readily reduced by KC8 even at low temperature to reform the precatalyst 12. However, a significant amount of (CPiPr3)Fe(N2)(H) (9) is also present; 9 is not catalytically competent, generating no detectable ammonia when subjected to the catalytic conditions, and its formation is likely an important limiting factor in the catalyst performance. Another identifiable species by 1H NMR is (CPiPr3)FeCl (10). Despite our efforts to remove all Cl- in the preparation of [H(Et2O)2][BArF4], the large excess of acid employed in this experiment likely ensures a non-negligible Cl- impurity that may also attenuate catalyst activity. The identity of another diamagnetic hydride-bearing species apparent in the 1H NMR is not currently known.
Figure 7.
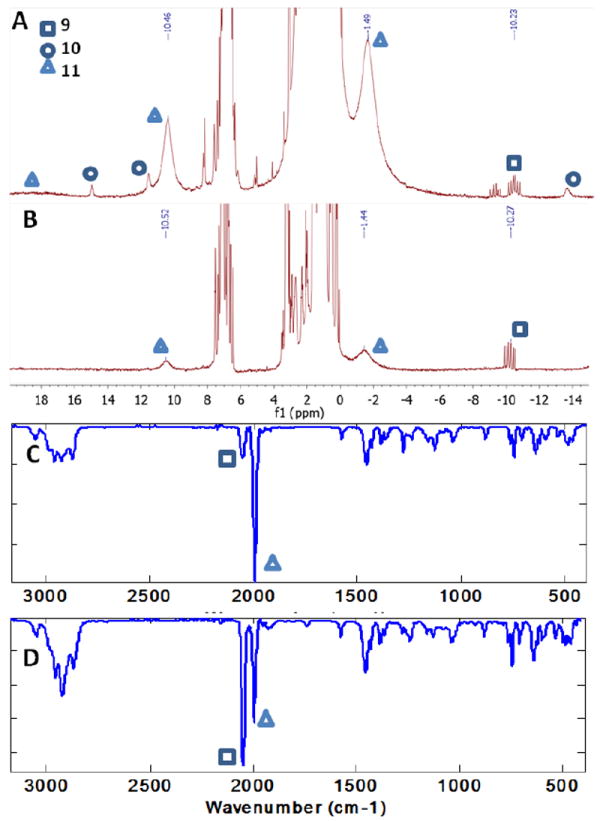
Spectroscopic analyses of reaction mixtures following the catalytic production of NH3 using 12[K(Et2O)0.5] as a catalyst. Symbols indicate characteristic resonances attributed to 9, 10, and 11. (A),(C) 1H NMR and IR spectra of a post-catalytic reaction mixture using 10 equiv. of [H(Et2O)2][BArF4] and 12 equiv. of KC8. (B),(D) 1H NMR and IR spectra of a post-catalytic reaction mixture using 38 equiv. of [H(Et2O)2][BArF4] and 40 equiv. of KC8.
Further product analysis using the full catalytic conditions (38 equivalents of [H(Et2O)2][BArF4] and 40 equivalents of KC8 with respect to the catalyst), showed that increasing amounts of (CPiPr3)Fe(N2)(H) (9) are formed as the system goes through more turnovers, corroborating the idea that this species serves as a catalytically inactive sink which builds up throughout the reaction. Integration of the NMR spectrum of such a reaction mixture against an internal standard suggests that approximately 70% of the catalyst has been converted to 9 (see SI); even at this point, however, some active catalyst remains in the form of 11 (Figure 7). The unknown hydride species present in the aforementioned reaction mixture derived from fewer equivalents of acid and reductant is no longer observed.
Notably, in neither of these experiments was any free ligand 1 (nor any ligand decomposition product) detected; it appears that all of the iron present remains ligated by the CPiPr3 ligand. This lack of degradation is promising, and suggests that improvements to the N2 reduction catalysis, in terms of turnover number, may yet prove possible if the formation of terminal hydride 9 can be limited by modification of either the ligand scaffold and/or the catalytic conditions. Indeed, it may be that biological nitrogenases are designed to avoid catalytically inactive hydride sinks by being themselves modest hydrogenases.39 A cluster approach would be a particularly good design in this context.40
CONCLUSIONS
To conclude, we have synthetically introduced the tripodal (CPiPr3)H ligand and have prepared and structurally compared its {(CPiPr3)FeN2}n complexes (n = 0, -1, +1) with those of the isostructural series {(SiPiPr3)FeN2}n. The {(CPiPr3)FeN2}n complexes feature an axial N2 ligand bound trans to an axial C-atom in a trigonal bipyramidal geometry, a design meant to crudely model one plausible geometry for a single Fe-N2 binding site in the iron-molybdenum cofactor (FeMoco). The Calkyl-Fe interaction in the (CPiPr3)Fe system exhibits a substantially higher degree of ionic character, and is more flexible, than for the related Sisilyl-Fe interaction in the isostructural and isoelectronic (SiPiPr3)Fe system.17 We suggest that this type of Fe-C flexibility crudely models the flexibility one can intuit for an N2-Fe-Cinterstitial interaction within FeMoco. Whereas the N2 anion (SiPiPr3)FeN2- does not effectively facilitate the delivery of H-atoms to N2 to produce NH3 via proton/reductant equivalents, an Et2O solution of (CPiPr3)FeN2- under 1 atm of N2 releases ca. 4.6 equiv NH3 relative to Fe. The modest catalytic N2 reduction behavior of (CPiPr3)FeN2- at -78 °C is comparable to (TPiPrB)FeN2-.21
It is noteworthy that amongst the isostructural SiPiPr3, TPiPrB, and CPiPr3 series, the system with the most flexible axial linkage, (TPiPrB)Fe, gives the greatest catalytic yield under a common set of reaction conditions, while the least flexible, (SiPiPr3)Fe, gives only substoichiometric yields of ammonia; the (CPiPr3)Fe system falls in between the two both in terms of flexibility and catalytic competence. While we emphasize caution in interpreting these differences given the low overall turnover numbers, they are consistent with the previously advanced hypothesis that a flexible Fe-Cinterstitial interaction might facilitate N2 binding and reduction at a single Fe site within FeMoco. Our structural and DFT studies22 demonstrate that, in the right environment, a carbon atom can serve as a modestly flexible ligand trans to an Fe-N2 binding site, and that this flexibility is enhanced by the ability of the carbon to accommodate a significant ionic charge. It seems likely to us that the inorganic carbide ligand in FeMoco is similarly, and likely more, able to stabilize substantial ionic character in the Fe-Cinterstitial bond (Figure 1), resulting in a flexible interaction that initially exposes an N2 binding site that can be further modulated as a function of the NxHy reduction state.
At this early stage reliable conclusions concerning the influence of the carbon atom on the intimate step-wise mechanism of nitrogen reduction are premature. Even within our synthetic series, it may be that different catalysts follow different mechanistic pathways (distal vs. alternating, or some hybrid path);21,41 for instance the most flexible system, (TPiPrB)Fe, may be better suited to facilitate a distal pathway that samples strongly pibonded intermediates, while (CPiPr3)Fe, which we presume is less flexible, could instead be dominated by an alternating or hybrid pathway. Whether these structurally related iron systems mediate nitrogen reduction by a common or different mechanism will be challenging to determine but is a fascinating question. The work presented here adds to the context needed for further mechanistic studies on both synthetic and biological iron systems for catalytic nitrogen fixation.
EXPERIMENTAL METHODS
General
All manipulations were carried out using standard Schlenk or glovebox techniques under an N2 atmosphere. Unless otherwise noted, solvents were deoxygenated and dried by thoroughly sparging with N2 followed by passage through an activated alumina column in a solvent purification system by SG Water, USA LLC. Non-halogenated solvents were tested with a standard purple solution of sodium benzophenone ketyl in tetrahydrofuran in order to confirm effective moisture removal. O-iodotriphenylmethane,28 H(OEt2)2[B(3,5-(CF3)2-C6H3)4],42 KC8,43 [(TPB)FeN2][Na(12-crown-4)2],25 [(SiPiPr 3)FeN2][Na(12-crown-4)2]17 and [(CSiPPh3)FeN2][K(18-crown-6)2]22 were prepared according to literature procedures. [Decamethylferrocenium][B(3,5-(CF3)2-C6H3)4] was prepared by treating [ferrocenium][B(3,5-(CF3)2-C6H3)4]44 with decamethylferrocene and used without purification. FeI2(THF)2 was prepared by treating Fe powder with I2 in THF,45 and was dried to FeI2 by heating under vacuum at 80°C for 6 hours. All other reagents were purchased from commercial vendors and used without further purification unless otherwise stated.
Physical methods
Elemental analyses were performed by Robinson Microlit Laboratories (Ledgewood, NJ). Deuterated solvents were purchased from Cambridge Isotope Laboratories, Inc., degassed, and dried over active 3-Å molecular sieves prior to use. 1H and 13C chemical shifts are reported in ppm relative to tetramethylsilane, using residual proton and 13C resonances from solvent as internal standards. 31P and 19F chemical shifts are reported in ppm relative to 85% aqueous H3PO4 and CFCl3, respectively. Solution phase magnetic measurements were performed by the method of Evans.46 Optical spectroscopy measurements were taken on a Cary 50 UV-Vis spectrophotometer using a 1-cm two-window quartz cell. Electrochemical measurements were carried out in a glovebox under a dinitrogen atmosphere in a one compartment cell using a CH Instruments 600B electrochemical analyzer. A glassy carbon electrode was used as the working electrode and platinum wire was used as the auxiliary electrode. The reference electrode was Ag/AgNO3 in THF. The ferrocene couple Fc+/Fc was used as an internal reference. Solutions (THF) of electrolyte (0.2 M tetra-n-butylammonium hexafluorophosphate) and analyte were also prepared under an inert atmosphere.
X-ray Crystallography
XRD studies were carried out at the Beckman Institute Crystallography Facility on a Bruker Kappa Apex II diffractometer (Mo Kα radiation). Structures were solved using SHELXS and refined against F2 on all data by full-matrix least squares with SHELXL.47 The crystals were mounted on a wire loop. Methyl group hydrogen atoms not involved in disorder were placed at calculated positions starting from the point of maximum electron density. All other hydrogen atoms, except where otherwise noted, were placed at geometrically calculated positions and refined using a riding model. The isotropic displacement parameters of the hydrogen atoms were fixed at 1.2 (1.5 for methyl groups) times the Ueq of the atoms to which they are bonded. Further details for each structure can be found in the Supporting Information.
Computations
A single-point calculation and Natural Bond Orbital (NBO) analysis was carried out on [(CPiPr 3)FeN2][K(Et2O)3] (12) using the crystallographically determined atomic coordinates at the B3LYP/6-31++G(d,p) level of theory using the Gaussion03 suite of programs.48 NBO analysis located a polarized σ interaction between Fe and the C-atom anchor (C01). Further details of the computational results can be found in the Supporting Information.
10-phenyl-10H-dibenzo[b,e]iodininium bromide (2)
The procedure for the generation of 2 and 4 (below) was adapted from a reported method for the generation of diaryliodonium salts.29 3-chloroperoxybenzoic acid (9.0 g, ~70% by mass, ~0.037 mol) was dissolved in dichloromethane (150 mL) and cooled to 0 °C. 2-iodotriphenylmethane (11.7 g, 0.0316 mol) was added as a solid in portions over the course of 10 minutes, during which time there was no observable change to the reaction mixture. This mixture was stirred at 0 °C for 10 minutes and then neat trifluoromethanesulfonic acid (8.74 mL, 0.0990 mol) was added via syringe over the course of 5 minutes. The reaction mixture turned dark brown. After an additional 20 minutes, the reaction mixture was allowed to warm to room temperature and stirred for one hour, and then the solvent was removed in vacuo. The solid material was suspended in 200 mL of diethyl ether and 200 mL of water, and then solid sodium bromide (14 g, 0.136 mol) was added and the mixture was shaken vigorously for 5 minutes, during which time a fine off-white precipitate developed. The precipitate was collected atop a sintered glass frit and washed copiously with water and diethyl ether (14.2 g, 0.0316 mol, quant). 1H NMR ((CD3)2S=O, 300 MHz, 298 K, δ): 8.27 (dd, J = 8 Hz, 1 Hz, 2H), 7.68 (td, J = 8 Hz, 1 Hz, 2H), 7.46 (td, J = 8 Hz, 1 Hz), 7.27 (m, 3H), 6.78 (dm, J = 8 Hz, 2H), 6.09 (s, 1H) ppm. 13C NMR ((CD3)2S=O, 75.4 MHz, 298 K, δ): 140.3 (s), 138.3 (s), 135.0 (s), 132.7 (s), 131.7 (s), 129.6 (s), 128.9 (s), 127.9 (s), 127.4 (s), 117.4 (s), 57.7 (s) ppm. ESI-MS (positive ion, amu): Calc. 370.0; Found 370.0.
2-bromo-2’-iodotriphenylmethane (3)
10-phenyl-10H-dibenzo[b,e]iodininium bromide (16.11 g, 0.0358 mol) was suspended in dry, degassed acetonitrile (250 mL), and solid tetrabutylammonium bromide (25 g, 0.078 mol) and copper(I) bromide (8 g, .06 mol) were added. The mixture was heated to a vigorous reflux and stirred at reflux for five days. The dark brown reaction mixture was then concentrated to dryness in vacuo, extracted with toluene, and filtered through a silica plug. The pale yellow filtrate was concentrated to dryness and the resulting material was recrystallized from methanol to give the desired product as an off-white powder which was collected atop a sintered glass frit and washed with cold methanol (12.7 g, 0.0282 mol, 79%). 1H NMR (CDCl3, 300 MHz, 298 K, δ): 7.90 (dd, J = 8 Hz, 1 Hz, 1H), 7.60 (dd, J = 8 Hz, 1 Hz, 1H), 7.34-7.18 (m, 5H), 7.13 (td, J = 8 Hz, 1 Hz, 1H), 7.03 (dd, J = 8 Hz, 1 Hz, 2H), 6.95 (td, J = 8 Hz, 1 Hz, 1H), 6.79 (dd, J = 8 Hz, 1 Hz, 2H), 6.02 (s, 1H) ppm. 13C NMR (CDCl3, 75.4 MHz, 298 K, δ): 145.2 (s), 142.2 (s), 141.1 (s), 140.1 (s), 133.1 (s), 131.2 (s), 130.7 (s), 130.0 (s), 128.5 (s), 128.3 (s), 128.2 (s), 128.0 (s), 127.2 (s), 126.7 (s), 126.3 (s), 102.9 (s), 60.8 (s) ppm. MS (amu): Calc. 449.9, 447.9; Found 449.9, 447.9.
10-(2-bromophenyl)-10H-dibenzo[b,e]iodininium iodide (4)
3-chloroperoxybenzoic acid (5 g, ~70% by mass, ~0.0203 mol) was dissolved in dichloromethane (200 mL) and cooled to 0 °C. 2-bromo-2’-iodotriphenylmethane (8.2 g, 0.0182 mol) was added as a solid in portions over the course of 10 minutes, during which time there was no observable change in the reaction mixture. This mixture was stirred at 0 °C for 10 minutes and then neat trifluoromethanesulfonic acid (5.04 mL, 0.0571 mol) was added via syringe over the course of 5 minutes. The reaction mixture turned dark brown. After an additional 30 minutes, the reaction mixture was allowed to warm to room temperature and stirred for 30 minutes, and then the solvent was removed in vacuo. The solid material was suspended in 200 mL of diethyl ether and 200 mL of water, and then solid potassium iodide (15 g, 0.090 mol) was added and the mixture was shaken vigorously for 5 minutes, during which time a fine yellow precipitate developed. The precipitate was collected atop a sintered glass frit and washed copiously with water and diethyl ether (9.95 g, 0.0173 mol, 95%). 1H NMR ((CD3)2S=O, 300 MHz, 298 K, δ): 8.20 (dd, J = 8 Hz, 1 Hz, 2H), 7.83 (dd, J = 8 Hz, 1 Hz, 2H), 7.72 (dd, J = 8 Hz, 1 Hz, 1H), 7.60 (td, J = 8 Hz, 1 Hz, 2H), 7.47-7.39 (m, 3H), 7.33 (td, J = 8 Hz, 1 Hz, 1H), 7.23 (dd, J = 8 Hz, 1 Hz, 1H), 6.02 (s, 1H) ppm. 13C NMR ((CD3)2S=O, 75.4 MHz, 298 K, δ): 138.9 (s), 135.4 (s), 135.1 (s), 135.0 (s), 133.4 (s), 132.8 (s), 131.7 (s), 130.7 (s), 130.0 (s), 128.0 (s), 117.2 (s), 110.0 (s), 58.8 (s) ppm. ESI-MS (positive ion, amu): Calc. 446.9, 448.9; Found 446.9, 448.9.
2-bromo-2’,2”-diiodotriphenylmethane (5)
Solid 10-(2-bromophenyl)-10H-dibenzo[b,e]iodininium iodide (4.54 g, 7.88 mmol) was sealed inside a Schlenk tube under N2 and heated to 200° C for 15 minutes, and then cooled to room temperature. The resulting dark violet residue was taken up in dichloromethane (50 mL) and washed with saturated aqueous sodium thiosulfate (50 mL) and then water (30 mL) and saturated aqueous sodium chloride (30 mL), then dried over magnesium sulfate, filtered, and concentrated to dryness in vacuo. The resulting off-white residue was recrystallized from methanol to give the desired product as a fine white powder, which was collected atop a sintered glass frit and washed with cold methanol (3.4 g, 5.90 mmol, 75%). 1H NMR (CDCl3, 300 MHz, 298 K, δ): 7.93 (d, J = 8 Hz, 2H), 7.64 (d, J = 8 Hz, 1H), 7.30-7.16 (m, 4H), 7.00 (t, J = 8 Hz, 2H), 6.72 (d, J = 8 Hz, 3H), 6.04 (s, 1H) ppm. 13C NMR (CDCl3, 75.4 MHz, 298 K, δ): 144.1 (s), 141.1 (s), 140.2 (s), 133.3 (s), 131.1 (s), 130.7 (s), 128.6 (s), 128.5 (s), 127.3 (s), 126.7 (s), 103.6 (s), 65.4 (s) ppm. MS (amu): Calc. 573.8, 575.8; Found 446.9, 448.9 ([M-I]+), 368.1 ([M-I-Br]+), 320.1, 322.1 ([M-2I]+).
Tris(2-(diisopropylphosphino)phenyl)methane (“(CiPrP3)H”) (1)
2-bromo-2’,2”-diiodotriphenylmethane (2.00 g, 3.48 mmol) was dissolved in diethyl ether (100 mL) and cooled to -78 °C while stirring. Solid t-butyllithium (1.36 g, 21.23 mmol) was added in portions over the course of 10 minutes and the reaction mixture was stirred at low temperature for 3 hours. Then chlorodiisopropylphosphine (1.96 g, 12.8 mmol) was dissolved in 10 mL of diethyl ether and added to the reaction mixture. The reaction mixture was allowed to warm slowly to room temperature overnight, resulting in the precipitation of a fine white solid. The reaction mixture was filtered through silica and the pale yellow-orange filtrate was concentrated to a sticky yellow solid which was triturated with acetonitrile to give an offwhite powder. The solid was washed copiously with acetonitrile and then dried under vacuum, giving 1.4 g (2.36 mmol, 68%) of the desired product. 1H NMR (C6D6, 300 MHz, 298 K, δ): 8.15 (q, J = 6 Hz, 1H), 7.44 (d, J = 7 Hz, 3H), 7.06 (td, J = 7 Hz, 2 Hz, 3H), 7.00-6.93 (m, 6H), 2.27 (septet of doublets, J = 4 Hz, 7 Hz, 3H), 1.73 (septet of doublets, J = 3 Hz, 7 Hz, 3H), 1.40 (dd, J = 7 Hz, 13 Hz, 9H), 1.32 (dd, J = 7 Hz, 12 Hz, 9H), 0.88 (dd, J = 7 Hz, 13 Hz, 9H), 0.44 (dd, J = 7 Hz, 12 Hz, 9H) ppm. 13C NMR (C6D6, 75.4 MHz, 298 K, δ): 159.0 (d, J = 29 Hz), 144.8 (d, J = 17 Hz), 140.0 (s), 139.3 (s), 132.4 (s), 59.1 (m), 32.7 (m), 30.0 (m), 29.4 (s), 27.3 (m), 21.0 (s) ppm. 31P NMR (C6D6, 121.4 MHz, 298 K, δ): -9.1 ppm. Anal. Calcd. for C37H55P3: C, 74.97; H, 9.35. Found: C, 74.73; H, 9.49.
{(CPiPr3)H}FeI2 (6)
(CPiPr3)H (500 mg, 0.843 mmol) was added to FeI2 (350 mg, 1.13 mmol) in 15 mL of toluene and stirred at 60 °C for 2 hours, at which point the reaction mixture was filtered through Celite and the yellow filtrate was concentrated to give a yellow powder (761 mg, 0.843 mmol, quant). Crystals suitable for X-ray diffraction were grown by layering of pentane over a saturated toluene solution. 1H NMR (C6D6, 300 MHz, 298 K, δ): 179.69, 26.00, 18.60, 14.92, 14.28, 13.62, 12.74, 9.96, 9.00, 8.29, 6.76, 6.16, 5.72, 5.48, 4.97, 4.28, 3.78, 0.30, 0.13, -0.48, -0.91, -2.02, -3.68, -5.09, -9.45 ppm. μeff (C6D6, Evans’ method, 298 K): 4.85 μB.
(CPiPr3)Fe(N2)H (9)
(CPiPr3)HFeI2 (370 mg, 0.410 mmol) was suspended in benzene (10 mL) and stirred vigorously over an excess of 0.7 % sodium/mercury amalgam (25 mg Na, 1.1 mmol) for two hours. The initially yellow suspension turned a deep brick red color during this time due to the formation of {(CPiPr3)H}FeI (7). The reaction mixture was filtered through Celite and concentrated to dryness in vacuo. The deep red residue was then suspended in diethyl ether (15 mL) at -78 °C and 3 mL of dimethoxyethane was added; this solution was vigorously stirred over excess sodium mirror for 4 hours at -78 °C, during which time the color lightened to orange. The reaction mixture was then filtered through Celite and concentrated to dryness. The residue was extracted into pentane and again filtered through Celite, giving a lighter yellow-orange filtrate which was concentrated to dryness again. This residue could be recrystallized from diethyl ether by slow evaporation to give yellow crystalline solids. These solids were washed with hexamethyldisiloxane and minimal cold diethyl ether, and then dried in vacuo to give 155 mg (0.229 mmol, 56%) of the desired product. Crystals suitable for X-ray diffraction were grown by evaporation of a concentrated pentane solution into hexamethyldisiloxane. 1H NMR (C6D6, 300 MHz, 298 K, δ): 7.57 (t, J = 6 Hz, 1H), 7.34 (m, 1H), 7.08 (m, 2H), 6.96 (m, 2H), 6.83-6.75 (m, 4H), 6.65 (m, 1H), 6.50 (m, 1H), 2.94 (septet, J = 8 Hz, 1H), 2.75 (m, 2H), 2.36 (septet, J = 6 Hz, 1H), 2.05 (septet, J = 7 Hz, 1H), 1.75-1.17 (m, 25H), 1.02 (dd, J = 7 Hz, 11 Hz, 3H), 0.65 (dd, J = 7 Hz, 15 Hz, 3H), 0.56 (dd, J = 7 Hz, 10 Hz, 3H), 0.27 (dd, J = 8 Hz, 13 Hz, 3H), -10.2 (ddd, J = 38 Hz, 53 Hz, 50 Hz) ppm. 31P NMR (C6D6, 121.4 MHz, 298 K, δ): 90.1 (dt, J = 100 Hz, 17 Hz, 1P), 67.0 (m, 1P), 63.4 (dt, J = 100 Hz, 17 Hz, 1P) ppm. IR (thin film; cm-1): 2046 (N-N), 1920 (Fe-H). Anal. Calcd. for C37H55FeP3N2: C, 65.68; H, 8.19; N, 4.14. Found: C, 65.91; H, 7.89; N, 3.94.
{(CPiPr3)H}FeBr (8)
{(CPiPr3)H}FeBr2 (5.0 mg, 0.0070 mmol, generated by treating CP3H with anhydrous FeBr2 in toluene) was dissolved in toluene, cooled to -78 °C, and treated with isopropyl magnesium chloride (3.5 μL, 2.0M in Et2O). The reaction mixture rapidly turned dark brick-red. It was stirred at low temperature for one hour and then allowed to warm to room temperature for thirty minutes before being filtered and concentrated. The dark red powder was not purified, but was analyzed by NMR in C6D6, and X-ray quality crystals were grown by layering pentane over a filtered benzene solution.
(CPiPr3)FeCl (10)
(CPiPr3)Fe(N2)H (61 mg, 0.0901 mmol) was dissolved in diethyl ether (8 mL) and cooled to -78 °C. HCl in diethyl ether (1.0 M, 108 μL, 0.108 mmol) was added to the solution in one portion. The reaction mixture was stirred at low temperature for one hour and then warmed to room temperature and stirred overnight. The color darkened to deep red-orange, and the reaction mixture was filtered through Celite and concentrated to dryness. The red residue was recrystallized by evaporation of a pentane solution into hexamethyldisiloxane and the resulting dark red crystals were washed sparingly with cold pentane and dried in vacuo, giving 46 mg (0.0673 mmol, 75%) of (CPiPr3)FeCl. Crystals suitable for X-ray diffraction were grown by evaporation of a concentrated pentane solution into hexamethyldisiloxane. 1H NMR (C6D6, 300 MHz, 298 K, δ): 179.93, 26.47, 23.05, 17.44, 17.22, 15.03, 11.66, 1.52, -10.27, -13.36, -16.82 ppm. μeff (C6D6, Evans’ method, 298 K): 4.92 μB. Anal. Calcd. for C37H54FeP3Cl: C, 65.06; H, 7.97. Found: C, 64.96; H, 8.01.
(CPiPr3)FeN2 (11)
(CPiPr3)FeCl (82 mg, 0.120 mmol) was dissolved in THF (2 mL) and stirred over sodium mirror for 20 minutes, or until NMR analysis showed complete consumption of the starting material, and then filtered and concentrated. The residue was extracted with pentane and filtered through Celite, and concentrated to a brownish-orange residue which was recrystallized by evaporation of a pentane solution into hexamethyldisiloxane. The dark brown-orange crystals were washed with hexamethyldisiloxane and cold pentane and dried in vacuo to give 39 mg (0.0581 mmol, 48 %) of (CPiPr3)FeN2. Crystals suitable for X-ray diffraction were grown by evaporation of a concentrated pentane solution into hexamethyldisiloxane. 1H NMR (C6D6, 300 MHz, 298 K, δ): 19.3 (very broad), 10.4, 6.8, 3.0, 2.0, 0.6, -1.4 ppm. μeff (C6D6, Evans’ method, 298 K): 1.75 μB. IR (thin film; cm-1): 1992 (N-N). Anal. Calcd. for C37H54FeP3N2: C, 65.78; H, 8.06; N, 4.15. Found: C, 66.03; H, 8.01; N, 3.86.
[(CPiPr3)FeN2][K(Et2O)0.5] (12[K(Et2O)0.5])
(CPiPr3)FeCl (40 mg, 0.0586 mmol) was dissolved in diethyl ether(5 mL) at room temperature and an excess of potassium graphite (KC8, 25 mg) was added. The reaction mixture was stirred for 10 minutes and then filtered through Celite. The dark brown solution was concentrated to about 2 mL and then pentane was layered over the ether solution and it was allowed to stand overnight during which time dark bluish-brown crystals formed. The supernatant was decanted and the crystals were washed thoroughly with pentane and thoroughly dried under vacuum, giving 26 mg of the desired product (0.0277 mmol, 47%). NMR analysis indicates the presence of 0.5 ether solvent molecules per anion. Crystals suitable for X-ray diffraction were grown by vapor diffusion of pentane into a diethyl ether solution; in these crystals the potassium cation is solvated by three diethyl ether molecules. 1H NMR (d8-THF, 300 MHz, 298 K, δ): 7.04 (s, 3H), 6.67 (s, 3H), 6.47 (s, 6H), 3.38 (q, J = 7 Hz, 2H, diethyl ether (CH3CH2)2O), 2.99 (br s, 3H), 2.14 (br s, 3H), 1.42 (d, J = 6 Hz, 9H), 1.36 (d, J = 5 Hz, 9H), 1.12 (t, J = 7 Hz, 3H, diethyl ether (CH3CH2)2O), 1.01 (d, J = 5 Hz, 9H), 0.12 (d, 9H) ppm. 31P NMR (5:1 C6D6/d8-THF, 121.4 MHz, 298 K, δ): 68.1 ppm. IR (thin film deposited from Et2O; cm-1): 1870 (N-N).
[(CPiPr3)FeN2][K(12-c-4)2] (12[K(12-c-4)2])
A sample of 12 (15 mg, 0.020 mmol) was dissolved in diethyl ether (1 mL) and 12-crown-4 (8.8 mg, 0.050 mmol) was added as a solution in diethyl ether (1 mL). The resulting solution was layered with pentane and allowed to stand overnight, resulting in the crystallization of 12[K(12-crown-4)2] as a very dark blue solid. The crystals were washed with pentane and dried under vacuum, giving 10 mg of material (53% yield). 1H NMR (d8-THF, 300 MHz, 298 K, δ) 6.86 (br s, 6H), 6.47 (s, 6H), 3.62 (s, 36H, 12-crown-4), 1.43 (s, 9H), 1.30 (s, 9H), 0.91 (s, 9H), 0.16 (s, 9H) ppm. 31P (C6D6, 121.4 MHz, 298 K, δ): 66 ppm. IR (thin film; cm-1) 1905 (N-N).
[(CPiPr3)FeN2][B(3,5-(CF3)2-C6H3)4] (13)
(CiPrP3)FeN2 (7.3 mg) was dissolved in diethyl ether (1 mL) and a solution of [Fe(C5Me5)2][B(3,5-(CF3)2-C6H3)4] in diethyl ether (1 mL) was added dropwise while stirring at room temperature. The reaction mixture was then concentrated to give an orange solid which was washed with benzene and then dried in vacuo. Crystals suitable for X-ray diffraction were grown by slow evaporation of a diethyl ether solution into hexamethyldisiloxane. 1H NMR (4:1 C6D6/THF-d8 under N2, 300 MHz, 298 K, δ): 16.65, 14.48, 8.15, 7.60, 2.71 ppm. (Note: the exact position of the paramagnetically shifted NMR peaks varies with the composition of the solvent due to the likely exchange of the N2 ligand with THF). μeff (d8-THF, Evans’ method, 298 K): 4.3 μB. IR (thin film; cm-1): 2128 (N-N). Satisfactory elemental analysis could not be obtained due to the lability of the coordinated N2 ligand.
(CPiPr3)FeN2SiMe3 (14)
[(CPiPr3)FeN2][K(Et2O)0.5] (35 mg, 0.0465 mmol) was dissolved in diethyl ether (2 mL) and cooled to -78 °C. Trimethylsilyl chloride (6 μL, 0.0473 mmol) was dissolved in diethyl ether (1 mL) and added dropwise to the stirring reaction mixture. The reaction was stirred at low temperature for one hour and then warmed to room temperature for one hour, concentrated to dryness, taken up in pentane, filtered through Celite, and concentrated. The red-orange residue was recrystallized by slow evaporation of a pentane solution into hexamethyldisiloxane, and the resulting red solids were washed with cold hexamethyldisiloxane and dried in vacuo to give 21 mg (0.0280 mmol, 60%) of solid material, which was contaminated with a small amount of CP3FeN2 (11) which we were unable to remove by repeated recrystallization. 14 decomposes slowly to 11 over time. 1H NMR (C6D6, 300 MHz, 298 K, δ) 7.33 (br m, 3H), 6.80 (t, J = 4 Hz, 6H), 6.63 (m, 3H), 2.67 (septet, J = 7 Hz, 3H), 1.97 (septet, J = 7 Hz, 3H), 1.45 (m, 18H), 0.96 (q, J = 7 Hz, 9H), 0.72 (q, J = 7 Hz, 9H), 0.12 (s, 3H) ppm. 31P (C6D6, 121.4 MHz, 298 K, δ): 80.1 ppm. IR (thin film; cm-1) 1736 (N-N).
Ammonia Quantification
A Schlenk tube was charged with HCl (4 mL of a 1.0 M solution in Et2O, 4 mmol). Reaction mixtures were vacuum transferred into this collection flask. Residual solid in the reaction vessel was treated with a solution of [Na][O-t-Bu] (40 mg, 0.4 mmol) in 1,2-dimethoxyethane (1 mL) and sealed. The resulting suspension was allowed to stir for 10 minutes before all volatiles were again vacuum transferred into the collection flask. After completion of the vacuum transfer, the flask was sealed and warmed to room temperature. Solvent was removed in vacuo and the remaining residue was dissolved in H2O (1 mL). An aliquot of this solution (20 μL) was then analyzed for the presence of NH3 (trapped as [NH4][Cl]) via the indophenol method.38 Quantification was performed with UV-Vis spectroscopy by analyzing absorbance at 635 nm.
Standard catalytic procedure with [(CPiPr3)FeN2][K(Et2O)0.5] (12)
[(CPiPr3)FeN2][K(Et2O)0.5] (1.9 mg, 0.0025 mmol) was dissolved in Et2O (0.5 mL) in a small Schlenk tube equipped with a stir bar. This solution was cooled to -78 °C in a cold well inside of the glove box. A suspension of KC8 (14 mg, 0.100 mmol) in Et2O (0.75 mL) was cooled to -78 °C and added to the reaction mixture with stirring. After five minutes, a similarly cooled solution of HBArF4 · 2 Et2O (93 mg, 0.092 mmol) in Et2O (1.0 mL) was added to the suspension in one portion with rapid stirring. Any remaining acid was dissolved in cold Et2O (0.25 mL) and added subsequently, and the Schlenk tube was sealed. The reaction was allowed to stir for 60 minutes at -78 °C before being warmed to room temperature and stirred for 15 minutes.
For further details of catalytic runs with other precatalysts and/or modified conditions, and complete tables of results, see Supporting Information.
Supplementary Material
Table 1.
Select characterization data for the Fe-N2 adducts {(CPiPr)3FeN2}n and {(SiPiPr3)FeN2}n (n = -1, 0, 1).
| X = C, Sia | [X-Fe-N2]-b | X-Fe-N2 | [X-Fe-N2]+ |
|---|---|---|---|
|
| |||
| Fe-C (Å) | 2.1646(17) | 2.152(3) | 2.081(3) |
| Fe-Si (Å) | 2.2526(9) | 2.2713(6) | 2.298(7) |
|
| |||
| Fe-NX = C (Å) | 1.7397(16) | 1.797(2) | 1.864(7) |
| Fe-NX = Si (Å) | 1.763(3) | 1.8191(1) | 1.914(2) |
|
| |||
| ν(N2)X = C (cm-1) | 1870 | 1992 | 2128 |
| ν(N2)X = Si (cm-1) | 1891 | 2003 | 2143 |
|
| |||
| spin state | S = 0 | S = 1/2 | S = 1 |
Acknowledgments
This work was supported by the NIH (GM 070757) and the Gordon and Betty Moore Foundation, and through the NSF via a GRFP award to S.E.C. Larry Henling and Dr. Tzu-Pin Lin are thanked for their assistance with X-ray crystallography. Jon Rittle is thanked for testing the catalytic activity of [CSiPPh3]FeN2-.
Footnotes
The authors declare no competing financial interests.
Supporting Information
Spectroscopic data for new compounds, further experimental and computational details, and additional data on catalytic runs. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Smil V. Enriching the Earth. MIT Press; Cambridge: 2001. [Google Scholar]
- 2.(a) Burgess BK, Lowe DJ. Chem Rev. 1996;96:2983–3011. doi: 10.1021/cr950055x. [DOI] [PubMed] [Google Scholar]; (b) Eady RR. Chem Rev. 1996;96:3013–3030. doi: 10.1021/cr950057h. [DOI] [PubMed] [Google Scholar]
- 3.Peters JC, Mehn MP. In: Activation of Small Molecules: Organometallic and Bioinorganic Perspectives. Tolman WB, editor. Wiley-VCH; 2006. pp. 81–119. [Google Scholar]
- 4.Hazari N. Chem Soc Rev. 2010;39:4044. doi: 10.1039/b919680n. [DOI] [PubMed] [Google Scholar]
- 5.MacLeod KC, Holland PL. Nature Chem. 2013;5:559. doi: 10.1038/nchem.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crossland JL, Tyler DR. Coord Chem Rev. 2010;254:1883. [Google Scholar]
- 7.Yandulov DV, Schrock RR. Science. 2003;301:76. doi: 10.1126/science.1085326. [DOI] [PubMed] [Google Scholar]
- 8.Arashiba K, Miyake Y, Nishibayashi Y. Nature Chem. 2011;3:120. doi: 10.1038/nchem.906. [DOI] [PubMed] [Google Scholar]
- 9.Weare WW, Dai X, Byrnes M, Chin J-M, Schrock RR, Muller P. Proc Natl Acad Sci. 2006;103:17099. doi: 10.1073/pnas.0602778103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Saouma CT, Lu CC, Peters JC. Inorg Chem. 2012;51:10043. doi: 10.1021/ic301704f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Saouma CT, Kinney RA, Hoffman BM, Peters JC. Angew Chem Int Ed. 2011;50:1. doi: 10.1002/anie.201006299. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Saouma CT, Muller P, Peters JC. J Am Chem Soc. 2009;131:10358. doi: 10.1021/ja903967z. [DOI] [PubMed] [Google Scholar]
- 11.Smith JM, Lachiotte RJ, Pittard KA, Cundari TR, Lukat-Rodgers G, Holland PL. J Am Chem Soc. 2001;123:9222. doi: 10.1021/ja016094+. [DOI] [PubMed] [Google Scholar]
- 12.Field LD, Li H, Magill AM. Inorg Chem. 2001;48:5. doi: 10.1021/ic801856q. [DOI] [PubMed] [Google Scholar]
- 13.Li Y, Li Y, Wang B, Luo Y, Yang D, Tong P, Zhao J, Luo L, Zhou Y, Chen S, Cheng F, Qu J. Nature Chem. 2013;5:320. doi: 10.1038/nchem.1594. [DOI] [PubMed] [Google Scholar]
- 14.Rodriguez MM, Bill E, Brennessel WW, Holland PL. Science. 2011;334:780. doi: 10.1126/science.1211906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moret ME, Peters JC. J Am Chem Soc. 2011;133:18118. doi: 10.1021/ja208675p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuki M, Tanaka H, Sasaki K, Miyake Y, Yoshizawa K, Nishibayashi Y. Nature Comm. 2012;3:1254. doi: 10.1038/ncomms2264. [DOI] [PubMed] [Google Scholar]
- 17.Lee Y, Mankad NP, Peters JC. Nature Chem. 2012;2:558. doi: 10.1038/nchem.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Leigh GJ, Jimenez-Tenorio M. J Am Chem Soc. 1991;113:5862. [Google Scholar]; (b) Hall DA, Leigh GJ. J Chem Soc Dalton Trans. 1996:3539. [Google Scholar]; (c) Gilbertson JD, Szymczak NK, Tyler DR. J Am Chem Soc. 2005;127:10184. doi: 10.1021/ja053030g. [DOI] [PubMed] [Google Scholar]; (d) Yamamoto A, Miura Y, Ito T, Chen HL, Iri K, Ozawa F, Miki K, Sei T, Tanaka N, Kasai N. Organometallics. 1993;2:1429. [Google Scholar]; (e) George TA, Rose DJ, Chang Y, Chen Q, Zubieta J. Inorg Chem. 1995;34:1295. [Google Scholar]; (f) Borodko YG, Broitman MO, Kachapina LM, Shilov AE, Ukhin LY. J Chem Soc D. 1971:1185. [Google Scholar]
- 19.Einsle O, Tezcan A, Andrade SLA, Schmid B, Yoshida M, Howard JB, Reese DC. Science. 2002;297:1696. doi: 10.1126/science.1073877. [DOI] [PubMed] [Google Scholar]
- 20.(a) Spatzal T, Aksoyoglu M, Zhang L, Andrade SLA, Schleicher E, Weber S, Rees DC, Einsle O. Science. 2011;334:940. doi: 10.1126/science.1214025. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lancaster KM, Roemelt M, Ettenhuber P, Hu Y, Ribbe MW, Neese F, Bergmann U, DeBeer S. Science. 2011;334:974–7. doi: 10.1126/science.1206445. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lancaster KM, Hu Y, Bergmann U, Ribbe MW, DeBeer S. J Am Chem Soc. 2013;135:610–2. doi: 10.1021/ja309254g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wiig JA, Hu Y, Lee CC, Ribbe MW. Science. 2012;337:1672–1675. doi: 10.1126/science.1224603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anderson JS, Rittle J, Peters JC. Nature. 2013;501:84. doi: 10.1038/nature12435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rittle J, Peters JC. Proc Natl Acad Sci U S A. doi: 10.1073/pnas.1310153110. [Online early access]. Published Online: Sept 16, 2013. [DOI] [Google Scholar]
- 23.Macbeth CE, Harkins SB, Peters JC. Can J Chem. 2005;83:332. [Google Scholar]
- 24.Mankad NP, Whited MT, Peters JC. Angew Chem Int Ed. 2007;46:5768. doi: 10.1002/anie.200701188. [DOI] [PubMed] [Google Scholar]
- 25.Moret M-E, Peters JC. Angew Chem Int Ed. 2011;50:2063. doi: 10.1002/anie.201006918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bontemps S, Bouhadir G, Dyer PW, Miqueu K, Bourissou D. Inorg Chem. 2007;46:5149. doi: 10.1021/ic7006556. [DOI] [PubMed] [Google Scholar]
- 27.Lesueur W, Solari E, Floriani C, Chiesi-Villa A, Rizzoli C. Inorg Chem. 1997;36:3354. doi: 10.1021/ic9700819. [DOI] [PubMed] [Google Scholar]
- 28.Bickelhaupt F, Jongsma C, de Koe P, Lourens R, Mast NR, van Mourik GL, Vermeer H, Weustink RJM. Tetrahedron. 1976;32:1921. [Google Scholar]
- 29.Bielawski M, Olofsson B. Chem Commun. 2007:2521. doi: 10.1039/b701864a. [DOI] [PubMed] [Google Scholar]
- 30.Ciclosi M, Lloret J, Estevan F, Lahuerta P, Sanau M, Perez-Prieto J. Angew Chem Int Ed. 2006;45:6741. doi: 10.1002/anie.200601084. [DOI] [PubMed] [Google Scholar]
- 31.Schlosser M. Pure Appl Chem. 1988;60:1627. [Google Scholar]
- 32.Hoffmann R, Bissel R, Farnum DG. J Phys Chem. 1969;73:1789. [Google Scholar]
- 33.Lee Y, Kinney RA, Hoffman BM, Peters JC. J Am Chem Soc. 2011;133:16366. doi: 10.1021/ja207003m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Field LD, Guest RW, Vuong KQ, Dalgarno SJ, Jensen P. Inorg Chem. 2009;48:2246. doi: 10.1021/ic8019827. [DOI] [PubMed] [Google Scholar]
- 35.Anderson JS, Moret M-E, Peters JC. J Am Chem Soc. 2013;135:534. doi: 10.1021/ja307714m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weinhold F, Landis C. Valency and Bonding: A Natural Bond Orbital Donor-Acceptor Perspective. Cambridge, Univ. Press; Cambridge: 2005. [Google Scholar]
- 37.Hills A, Hughes DL, Jimenez-Tenorio M, Leigh GF, Rowley AT. J Chem Soc Dalton Trans. 1993;25:3041. [Google Scholar]
- 38.Weatherburn MW. Anal Chem. 1967;39:971. [Google Scholar]
- 39.(a) Yang Z-Y, Khadka N, Lukoyanov D, Hoffman BM, Dean DR, Seefeldt LC. Proc Natl Acad Sci U S A. 2013;110:16327. doi: 10.1073/pnas.1315852110. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Simpson FB, Burris RH. Science. 1984;224:1095. doi: 10.1126/science.6585956. [DOI] [PubMed] [Google Scholar]
- 40.(a) Tard C, Liu X, Ibrahim SK, Maurizio B, DeGioia L, Davis SC, Yang X, Wang L-S, Sawers G, Pickett CJ. Nature. 2005;433:610. doi: 10.1038/nature03298. [DOI] [PubMed] [Google Scholar]; (b) Gloaguen F, Rauchfuss TB. Chem Soc Rev. 2009;38:100. doi: 10.1039/b801796b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Darensbourg MY, Lyon EJ, Smee JJ. Coord Chem Rev. 2000;206-207:533. [Google Scholar]; (d) Peters JW, Lanzilotta WN, Lemon BJ, Seefeldt LC. Science. 1998;282:1853. doi: 10.1126/science.282.5395.1853. [DOI] [PubMed] [Google Scholar]; (e) Volbeda A, Charon M-H, Piras C, Hatchikian EC, Frey M, Fontecilla-Camps JC. Nature. 1995;373:580. doi: 10.1038/373580a0. [DOI] [PubMed] [Google Scholar]; (f) Vincent KA, Parkin A, Armonstrong FA. Chem Rev. 2007;107:4366. doi: 10.1021/cr050191u. [DOI] [PubMed] [Google Scholar]
- 41.(a) Chatt J, Dilworth JR, Richards RL. Chem Rev. 1978;78:589. [Google Scholar]; (b) Seefeldt L, Hoffman BM, Dean DR. Annu Rev Biochem. 2009;78:701. doi: 10.1146/annurev.biochem.78.070907.103812. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lukoyanov D, Dikanov SA, Yang Z-Y, Barney BM, Samoilova RI, Narasimhulu KV, Dean DR, Seefeldt LC, Hoffman BM. J Am Chem Soc. 2011;133:11655. doi: 10.1021/ja2036018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brookhart M, Grant B, Volpe AF., Jr Organometallics. 1992;11:3920. [Google Scholar]
- 43.Wietz IS, Rabinovitz MJ. J Chem Soc Perkin Trans. 1993;1:117. [Google Scholar]
- 44.Chavez I, Alvarez-Carena A, Molins E, Roig A, Maniukiewicz W, Arancibia A, Arancibia V, Brand H, Manriquez JM. J Organomet Chem. 2000;601:126. [Google Scholar]
- 45.Job R, Earl R. Inorg Nuc Chem Lett. 1979;15:21. [Google Scholar]
- 46.Evans DF. J Chem Soc. 1959:2003. [Google Scholar]
- 47.Sheldrick GM. Acta Cryst A. 2008;82:169. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 48.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03, Revision C.02. Gaussian, Inc.; Wallingford CT: 2004. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.