

Abstract
This study aims to improve the drug oral bioavailability by co-administration with flavonoid inhibitors of the CYP2C isozyme and to establish qualitative and quantitative (QSAR) structure–activity relationships (SAR) between flavonoids and CYP2C. A total of 40 naturally occurring flavonoids were screened in vitro for CYP2C inhibition. Enzyme activity was determined by measuring conversion of tolbutamide to 4-hydroxytolbutamide by rat liver microsomes. The percent inhibition and IC50 of each flavonoid were calculated and used to develop SAR and QSAR. The most effective flavonoid was orally co-administered in vivo with a cholesterol-reducing drug, fluvastatin, which is normally metabolized by CYP2C. The most potent CYP2C inhibitor identified in vitro was tamarixetin (IC50 = 1.4 μM). This flavonoid enhanced the oral bioavailability of fluvastatin in vivo, producing a >2-fold increase in the area under the concentration–time curve and in the peak plasma concentration. SAR analysis indicated that the presence of a 2,3-double bond in the C ring, hydroxylation at positions 5, 6, and 7, and glycosylation had important effects on flavonoid–CYP2C interactions. These findings should prove useful for predicting the inhibition of CYP2C activity by other untested flavonoid-like compounds. In the present study, tamarixetin significantly inhibited CYP2C activity in vitro and in vivo. Thus, the use of tamarixetin could improve the therapeutic efficacy of drugs with low bioavailability.
KEY WORDS: bioavailability, CYP2C, flavonoid, structure–activity relationship, tamarixetin
INTRODUCTION
Compared with drugs with high and constant bioavailability, those with poor bioavailability show a larger therapeutic variation among patients and cause severe side effects more frequently. Drugs belonging to the Biopharmaceutics Drug Disposition Classification System (BDDCS) classes II and IV generally possess low oral bioavailability owing to their poor aqueous solubility. However, except for the influence of drug physicochemical properties on absorption, the extensive first-pass metabolism in the gut and liver represents another critical determinant of bioavailability, especially for drugs of BDDCS classes I and II (1).
According to previous reports (2–4), herbs or natural food products enriched with flavonoids have been shown to alter drug performance in humans. Flavonoids are constituents of frequently consumed foods, such as vegetables, fruits, cereal or plant-derived beverages, as well as components of herbal remedies. More than 6,500 naturally occurring plant flavonoids have been discovered (5). In addition to their well-known antioxidant effects, flavonoids also have great impacts on drug metabolism (6,7). The average dose of total flavonoids consumed in our daily life has been estimated to be several hundred milligrams (8,9). These dietary components therefore have the potential to inhibit the metabolism of co-administered drugs, especially those that are eliminated primarily via cytochrome P450 (P450) enzymes (10).
The P450 superfamily constitutes a major group of oxidative enzymes involved in the biotransformation of a wide range of endogenous substances, therapeutic drugs and xenobiotics (11,12). The human CYP2C subfamily accounts for approximately 18% of total P450s in the liver and catalyzes more than 20% of all marketed drugs (13–15). Several drugs with narrow therapeutic windows such as phenytoin, warfarin, and acenocoumarol are highly selectively metabolized by CYP2C. Additionally, inter-individual and inter-racial differences in the CYP2C genome may lead to either severe side effects, or inadequate drug response (16). Accordingly, studies are underway to identify compounds capable of enhancing or coordinating drug bioavailability through inhibition of CYP2C enzymes. However, in addition to enzyme regulation efficacy, an acceptable safety profile is of prime importance for a candidate CYP2C modulator. To avoid unwanted drug responses, CYP2C inhibition should be temporary and reversible (17). Ideally, the inhibition effects would be best confined to the gastrointestinal tract and liver, where CYP2C is abundantly and broadly distributed (18), rather than entering the body to produce systemic effects.
As herbal products have been used in health management for thousands of years, the active ingredients extracted from them are likely safe and harmless (10). A number of clinical trials have been conducted using large doses of flavonoids for disease prevention, and there were no serious side effects observed in these studies (19). However, it has been confirmed that several flavonoids are potent inhibitors of CYP2C-mediated drug metabolism (20,21) and thus possess the potential to alter the efficacy of some prescribed drugs (22). Although concomitant administration of high flavonoid-containing substances with therapeutic agents carries the risks of unwanted interactions, these plant-derived compounds may also markedly improve the bioavailability of some orally administered drugs. To our knowledge, however, purified flavonoids have not been used clinically to modulate oral drug bioavailability by inhibiting CYP2C.
The objective of this study was to screen 40 dietary flavonoids obtained from food or herbal medicines for their ability to inhibit CYP2C activity in vitro and to determine whether they could improve the drug oral bioavailability in vivo. Tolbutamide, a hypoglycemic agent with a single metabolic pathway, was used as a specific CYP2C substrate for in vitro screening. The enzyme activity of CYP2C was determined by measuring the hydroxylation of tolbutamide to 4-hydroxytolbutamide in rat liver microsomes (RLMs) (23). The effects of flavonoid on the bioavailability of fluvastatin, a hypolipidemic agent with approximately 20% oral bioavailability, were investigated in vivo in rats. This provided a suitable evaluation index because of fluvastatin’s highly effective intestinal absorption (90%) and its specificity for CYP2C metabolism (24,25). Furthermore, we examined the quantitative structure–activity relationships (QSAR) underlying these interactions to identify the structural features important for CYP2C inhibition. An optimal QSAR model was constructed using stepwise linear regression to predict the CYP2C inhibition activities of other untested flavonoids (26). Our findings suggested that this model could be used to direct the synthesis of flavonoid-like CYP2C inhibitors with higher potency for potential clinical application.
MATERIALS AND METHODS
Materials
All the organic solvents used for high-performance liquid chromatography (HPLC) were purchased from Merck (Darmstadt, Germany) or Nacalai Tesque (Kyoto Japan). Tolbutamide, chlorpropamide, 4-hydroxytolbutamide, ketoconazole, tetrabutylammonium fluoride (TBAF; 75 wt.%), glucose-6-phosphate (G6P), G6P dehydrogenase (G6PD), nicotinamide adenine dinucleotide phosphate (NADP+), and reagents for the determination of total protein were purchased from Sigma (St. Louis, MO, USA). The flavonoids screened in the study are shown in Fig. 1. All were at least 98% in purity and were purchased from Sigma, Indofine (Hillsborough, NJ, USA), Fujicco (Kobe, Japan), Wako Pure (Osaka, Japan), or Nacalai Tesque. Celecoxib served as the internal standard for animal studies and was generously supplied by Taiwan Biotech Co., Ltd. (Taipei, Taiwan). Fluvastatin and its reference standard were graciously provided by Tri-service General Hospital (Taipei, Taiwan) and Novartis Co., Ltd. (Taipei, Taiwan), respectively.
Fig. 1.
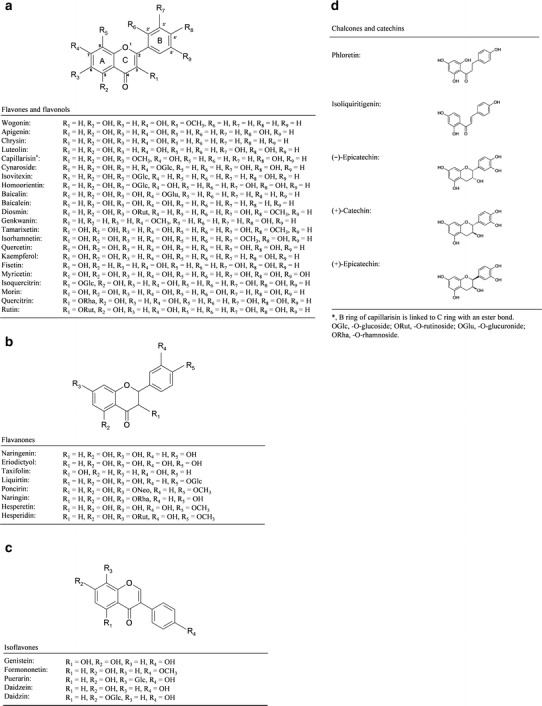
Chemical structures of the flavonoids investigated. All purified components were classified into six categories and illustrated as four groups, a flavones and flavonols, b flavanones, c isoflavones, and d chalcones and catechins. Each flavonoid molecule is composed of a core structure known as the three-ring flavan nucleus. The various functional groups of each compound on A–C rings are depicted as R substitutions
Methods
Screening for CYP2C Inhibitors
Animals
Male Sprague Dawley® (SD) rats weighing between 300 and 325 g were purchased from the National Laboratory Animal Center (Taipei, Taiwan). All rats were maintained under a 12-h light–dark cycle in a room with controlled temperature and humidity. They were given ad libitum access to food and water throughout the experiment. All experiments adhered to the National Institute of Health guidelines for the treatment of animals.
Rat Liver Microsome Preparation
RLMs were prepared as previously described, with slight modifications (27). Microsomes were obtained from 10 g liver samples of 8-week-old SD rats using the differential centrifugation technique. The final pellet was harvested and resuspended in an equal volume of 0.1 M phosphate buffer (pH 7.4). Liver microsomal protein levels were quantified by the Lowry method (28). The final RLM preparation had a protein concentration of approximately 25 mg/mL and was stored at −80°C prior to use.
CYP2C Enzyme Activity Assays
Enzyme activity assays were performed using RLMs to screen for CYP2C inhibitors. Prior to screening, in vitro assay conditions were established based on Michaelis–Menten kinetics. A 1-mL reaction mixture containing 0.25 or 0.5 mg microsomal protein was incubated with tolbutamide (0.025–2.5 mM) at 37°C for 2.5–90 min in the presence of cofactor cocktails (1 mM NADP+, 10 mM G6P, 2 IU G6PD, and 5 mM MgCl2 in phosphate buffer, 0.1 M; pH 7.4) (29). Incubations were terminated by addition of 0.1 mL 1 N hydrochloric acid. Chlorpropamide (25 μL of 10 μg/mL) was added to each reaction as an internal standard for subsequent HPLC analysis. The analytes were extracted using 2 mL methylene chloride, followed by centrifugation at 3,900×g for 10 min. The organic fraction was evaporated to dryness and reconstituted with 1 mL of acetonitrile before HPLC analysis.
Screening for CYP2C Modulators
For the screening tests, the reaction was initiated by the addition of tolbutamide (1 mM) with the test compound and was performed at 37°C for 7.5 min. Ketoconazole, a well-characterized inhibitor of CYP2C, was used as the positive control (30). On the basis of the inhibitory activity observed from the positive control, screening for CYP2C inhibition was carried out at three different concentrations (1, 10, and 100 μM) of each of the 40 flavonoids in triplicate. The data were expressed as a percentage of the control 4-hydroxytolbutamide formation in the absence of any test compound.
HPLC Analysis of Tolbutamide and 4-Hydroxytolbutamide
Tolbutamide, 4-hydroxytolbutamide, and the internal standard chlorpropamide were determined by HPLC (Shimadzu LC-10AD) with an ultraviolet detector at 230 nm. Chromatographic separation was carried out on a C18 column (150 × 4.6 mm, 5 μm) using a mobile phase composed of 10 mM sodium acetate buffer (pH 4.4) and acetonitrile (25:75, v/v) at a 1.3 mL/min flow rate. The retention times of 4-hydroxytolbutamide, chlorpropamide, and tolbutamide were 4.6, 14.2, and 26.5 min, respectively.
Pharmacokinetic Study in Rats
Fluvastatin and Flavonoid Oral Administration to Rats
The capacity of the CYP2C inhibitors to improve fluvastatin oral bioavailability in vivo was tested in SD rats. The rats were anesthetized by ether and surgically implanted with a jugular catheter for periodic blood sample collection. After recovery for 24 h, rats in the study group received an oral solution of the flavonol CYP2C inhibitor tamarixetin (10 mg/mL) in dimethylsulfoxide (30%, w/v) at a dose of 9.32 mg/kg while the control group received only vehicle. Immediately after administration of tamarixetin or vehicle, both groups were orally administered a 2-mg/mL solution of fluvastatin in water at a dose of 1.5 mg/kg. Twelve blood samples (0.5 mL) were collected over 24 h at 0 (pre-dose blank), 10, 20, 40, 60, 120, 240, 360, 480, 720, 1,080, and 1,440 min after administration. Each blood sample was collected in a microcentrifuge tube containing heparin (20 μL of 10 IU). Plasma samples were obtained by centrifugation at 13,000×g for 10 min, and the remaining blood sample was infused back into the rats with 0.25 mL 0.9% saline to maintain body fluid volume after sampling. The plasma samples were protected from light and stored at −80°C until assayed.
Assay Conditions for Fluvastatin Analysis
The plasma fluvastatin concentration was determined by HPLC with fluorescence detection (excitation, 309 nm; emission, 390 nm). The HPLC conditions were as follows: C18 reversed-phase column (150 × 4.6 mm, 5 μm); mobile phase, 0.1 M TBAF/0.1 M phosphate buffer (pH 6.0)/methanol (15:25:60, v/v/v); flow rate, 1.0 mL/min; and column temperature, 50°C. The analytical procedure was slightly modified from a previous report (31).
Quantitative Structure-Activity Relationship Analysis
QSAR Model Construction
The QSAR model was constructed using the method described by Sheu et al. (26) with a slight modification, and was based on a stepwise linear regression analysis with entry criteria set as F < 0.05 and removal criteria set as F > 0.1. SPSS statistical software (version 17.0; IBM Inc., Chicago, IL) was used to determine statistically significant relationships between the dependent variable (enzymatic inhibition) and the independent variables (flavonoid structural units or compound skeleton). The triplicate raw data, expressed as percentage enzyme inhibition, were set as dependent variables in a dose-dependent manner, namely Y1(1 μM), Y2(10 μM), and Y3(100 μM), or their respective logarithmically transformed variables, log Y1(1 μM), log Y2(10 μM) and log Y3(100 μM). The structural units (Χ1–X8 and X13–X16) and compound skeleton (X9–X12) were empirically chosen as independent variables where X1 = C3–OH; X2 = C5–OH; X3 = C6–OH; X4 = C7–OH; X5 = C3′–OH; X6 = C4′–OH; X7 = C4′–OCH3; X8 = glycosylation; X9 = flavone; X10 = flavonol; X11 = flavanone; X12 = isoflavone; X13 = hydroxylation at positions 3, 5, and 7; X14 = hydroxylation at positions 3 and 5; X15 = hydroxylation at positions 5 and 7; and X16 = hydroxylation at positions 3 and 7.
Statistical Analysis
Student’s t test was performed on the enzyme inhibition data to assess the statistical significance of the observed effects, relative to control. Fluvastatin plasma concentrations versus time data were analyzed according to a non-compartmental pharmacokinetic model by WinNonlin 5.0.1 (Pharsight, Mountain View, CA). The statistical significance of the effects of tamarixetin on the fluvastatin plasma concentration was determined by one-way analysis of variance. Data were expressed as the mean ± standard error. Values of p < 0.05 were considered statistically significant.
RESULTS
4-Hydroxytolbutamide and Fluvastatin Assay Validation
The calibration curves for the 4-hydroxytolbutamide and fluvastatin assays were linear (r > 0.998) over the concentration ranges of 5–2,500 and 5–1,000 ng/mL, respectively. For the 4-hydroxytolbutamide analysis, the intra- and interday (n = 6) precision and accuracy showed acceptable levels of less than 4.7% and 5.5% as determined from the relative standard deviation and relative error. For fluvastatin, the intra- and interday (n = 6) precision and accuracy showed acceptable levels of less than 7.9% and 7.4% as determined from the relative standard deviation and relative error. The methods were therefore precise and sensitive enough for in vitro and in vivo sample analyses.
Optimization of the in Vitro Screening Assay
The CYP2C assay conditions were optimized such that product formation was linear with respect to incubation time and protein concentration. In addition, the initial substrate concentrations were selected based on kinetic parameters. As shown in Fig. 2a, the rate of 4-hydroxytolbutamide formation in the presence of 0.25 or 0.5 mg RLMs was linear at low tolbutamide concentrations and reached a plateau when the tolbutamide concentration was over 1 mM. This indicated that 1 mM tolbutamide saturated the enzyme present in 0.25 or 0.5 mg/mL of RLMs. The apparent Vmax and Km values for 4-hydroxytolbutamide formation obtained from the Eadie-Hofstee plot (Fig. 2b) were 0.23 nmol min−1 mg−1 and 0.27 mM in the presence of 0.25 mg/mL RLMs, as compared with 0.45 nmol min−1 mg−1 and 0.21 mM with 0.5 mg/mL RLMs. Assays conducted with the optimal conditions of 1 mM tolbutamide and 0.5 mg/mL RLMs yielded linear 4-hydroxytolbutamide formation over the first 10 min (Fig. 2c). Therefore, all screening tests were carried out using these conditions with 7.5 min incubation time to maintain the maximal reaction velocity.
Fig. 2.
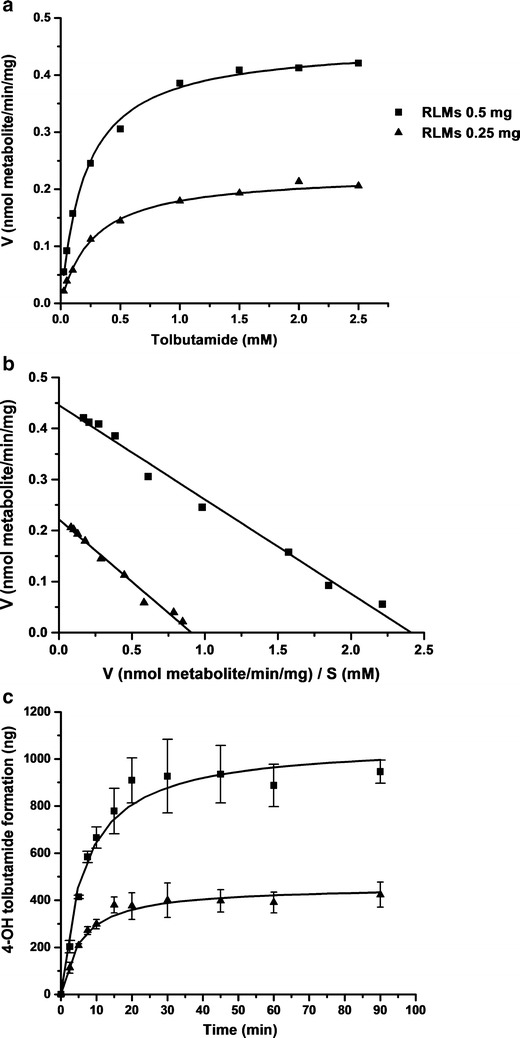
Representative velocity versus substrate concentration plot (a), Eadie–Hofstee plot (b), and the amount of 4-hydroxytolbutamide formation at different incubation times (c) in the presence of 0.25 and 0.5 mg/mL RLMs. Points are experimentally determined values (mean ± SD, n = 3), while solid lines represent the curves of best fit
Modulation Effects of Flavonoids on CYP2C Activity
The inhibition effects of the compounds tested in this study on CYP2C activity are summarized in Table I. Ketoconazole, a potent inhibitor of CYP2C, completely abolished enzyme activity at a concentration of 100 μM, and produced 81% and 46% inhibition at 10 and 1 μM, respectively. Among the 40 dietary flavonoids, the flavonol tamarixetin exhibited the most significant effects on enzyme activity at each concentration used. These effects were comparable to those of ketoconazole, and at 10 μM, tamarixetin actually produced slightly greater CYP2C inhibition than ketoconazole did. In addition to tamarixetin, flavonoids with an inhibitory potency higher than 80% at 10 μM included wogonin, luteolin, isorhamnetin, quercetin, naringenin, genistein, phloretin, and isoliquiritigenin. Although catechins are a subgroup of flavonoids, we did not observe any modulatory effects of (−)-epicatechin, (+)-catechin, or (+)-epicatechin on CYP2C. Interestingly, two chalcones (phloretin and isoliquiritigenin) were included in the study, and although they lacked the classic core flavonoid structure, they had marked effects on CYP2C activity at 100 μM. However, no significant effects were observed in the presence of 1 or 10 μM of these compounds.
Table I.
Inhibitory Effects of Various Dietary Flavonoids on Tolbutamide 4-Hydroxylation in Rat Liver Microsomes
| Flavonoids | Mean of selective inhibition of tolbutamide metabolism (%, n = 3) | Natural source | ||
|---|---|---|---|---|
| 1 μM | 10 μM | 100 μM | ||
| Ketoconazole (positive control) | 45.9* | 80.9** | 100.0** | – |
| Flavones | ||||
| Wogonin | 28.9* | 51.4** | 84.2** | Herba Scutellaria baicalensis |
| Apigenin | 7.4 | 21.7* | 76.9** | Parsley and celery |
| Chrysin | 6.9 | 60.8* | 75.3* | Passion flower |
| Luteolin | 13.5 | 38.0* | 93.5** | Parsley and broccoli |
| Capillarisin | 19.4 | 34.9* | 79.6** | Herba Artemisiae scopariae |
| Isovitexin | 35.6* | 37.5** | 45.5** | Barley grass juice, rice hull |
| Cynaroside | 9.4 | −0.3 | 25.1* | Parsley and celery |
| Homoorientin | 3.4 | 2.5 | 20.2 | Herba Swertia japonica |
| Baicalin | −2.0 | −5.4 | 7.8 | Herba Scutellaria baicalensis |
| Baicalein | 6.5 | 1.8 | 5.1 | Herba Scutellaria baicalensis |
| Diosmin | −7.1 | 4.7 | 2.5 | Citrus |
| Genkwanin | 30.9* | 37.4* | 48.1** | Aquilaria sinensis |
| Flavonols | ||||
| Tamarixetin | 42.0* | 88.3** | 90.4** | Herba Tamarix chinensis |
| Isorhamnetin | 18.7* | 61.2** | 87.7** | Sea buckthorn |
| Quercetin | 15.1* | 44.9** | 92.1** | Onion, cranberry |
| Kaempferol | 5.7 | 52.5* | 73.1** | Onion and Gingko biloba |
| Fisetin | 5.5 | 26.6 | 73.0* | Strawberries and onion |
| Myricetin | −3.2 | 10.6 | 89.0** | Onion and berries |
| Isoquercitrin | 21.0 | 27.1 | 62.0** | Onion and berries |
| Morin | 14.3 | 11.5 | 51.1* | Guava |
| Quercitrin | 24.2 | 21.1 | 13.5 | Onion and berries |
| Rutin | −2.6 | −4.0 | −6.5 | Buckwheat and berries |
| Flavanones | ||||
| Naringenin | 4.4 | 51.3* | 81.6** | Grapefruit juice |
| Eriodictyol | 32.8 | 28.7* | 69.8** | Yerba santa |
| Taxifolin | −1.3 | 31.2* | 50.6** | Milk thistle |
| Liquiritin | 18.1 | 18.4 | 24.8 | Licorice |
| Poncirin | −5.9 | 16.4 | 18.8 | Citrus |
| Naringin | −11.5 | −13.3 | −24.3 | Grapefruit juice |
| Hesperetin | −3.6 | 29.1 | 56.1* | Citrus |
| Hesperidin | 11.9 | 2.6 | 6.7 | Citrus |
| Isoflavones | ||||
| Genistein | 49.7* | 67.8** | 82.8** | Soya beans |
| Formononetin | 23.4 | 30.6* | 62.0* | Herba Radix astragali |
| Puerarin | 38.2 | 39.1* | 33.4 | Herba Radix puerariae |
| Daidzein | 1.4 | 13.5 | 24.6* | Soya beans |
| Daidzin | 1.0 | −2.5 | −1.2 | Soya beans |
| Chalcones | ||||
| Phloretin | 23.8 | 34.5 | 95.4** | Apple |
| Isoliquiritigenin | 0.9 | 29.7 | 95.7** | Licorice |
| Catechins | ||||
| (−)-Epicatechin | 1.1 | −8.6 | 5.5 | Green tea and wine |
| (+)-Catechin | −3.1 | −8.5 | −0.2 | Green tea and wine |
| (+)-Epicatechin | 17.0 | 6.3 | −12.0 | Green tea and wine |
*p < 0.05; **p < 0.01 vs. control
Effects of Flavonoids on Fluvastatin Oral Bioavailability in Rats
The in vitro screening described above identified tamarixetin as a potent inhibitor of rat liver CYP2C activity. Therefore, we orally administered tamarixetin to SD rats to determine whether this flavonoid significantly improved the oral bioavailability of fluvastatin. Pharmacokinetic parameters (Table II) were determined for animals given oral fluvastatin alone or with tamarixetin, and the fluvastatin plasma concentration versus time curves are depicted in Fig. 3. Tamarixetin significantly increased the bioavailability of fluvastatin as demonstrated by a 2.2-fold increase in its peak plasma concentration (Cmax) and a 2.4-fold increase in the area under the plasma concentration-time curve (AUCINF) whereas there was no apparent difference in the elimination half-life (T1/2) of fluvastatin between the groups. Taken together these pharmacokinetic data suggested that oral tamarixetin reduced fluvastatin metabolism by about 50% in vivo in the drug absorption phase but did not affect its distribution and disposition.
Table II.
Pharmacokinetic Parameters Following Oral Administration of 1.5 mg/kg Fluvastatin in the Presence or Absence of 9.32 mg/kg Tamarixetin in SD Rats
| PK parameter | Fluvastatin (B) control, n = 7 | Fluvastatin (A) with tamarixetin, n = 5 | A/B |
|---|---|---|---|
| C max (ng/mL) | 63.1 ± 10.4 | 141.4 ± 15.8* | 2.2 |
| AUC INF (h ng−1 mL−1) | 950 ± 133 | 2281 ± 387* | 2.4 |
| AUC t (h ng−1 mL−1) | 711 ± 82 | 1389 ± 166* | 2.0 |
| T max (h) | 4.7 ± 1.7 | 4.2 ± 1.1 | 0.9 |
| k (1/h) | 0.07 ± 0.01 | 0.07 ± 0.01 | 1.0 |
| T 1/2 (h) | 9.7 ± 0.7 | 11.3 ± 1.3 | 1.2 |
| CL/F (mL min−1 kg−1) | 29.1 ± 4.1 | 12.3 ± 2.1** | 0.4 |
| Vd/F (L/kg) | 24.8 ± 4.7 | 11.2 ± 0.9* | 0.5 |
Data = mean ± SE
*p < 0.05; **p < 0.01, significant difference compared with the control group
Fig. 3.
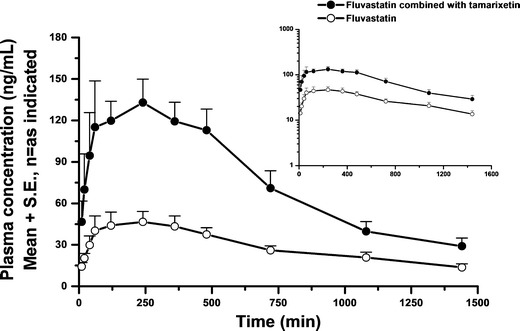
Mean plasma concentration-time curves of fluvastatin after oral administration of 1.5 mg/kg fluvastatin in the presence or absence of tamarixetin in SD rats. White circle, fluvastatin control (n = 7); black circle, co-administered with 9.32 mg/kg tamarixetin (n = 5). Values are means + SEM. Insert, semi-log scale
QSAR Analysis of Flavonoid Effects on CYP2C
The structure-activity relationships of the flavonoids that produced statistically significant CYP2C inhibition at all three reaction concentrations in the in vitro screening were analyzed. The regression models are summarized in Table III. The best correlation (adjusted R2 = 0.718) was observed for the QSAR model obtained by regressing the raw data of the inhibition at Y3(100 μM) against structural units X2 = C5–OH; X3 = C6–OH; X8 = glycosylation; and X15 = C5–OH and C7–OH, followed by Y2(10 μM) (adjusted R2 = 0.549) and finally Y1(1 μM) (adjusted R2 = 0.164). This analysis revealed that a hydroxyl group attached to either C5 or C5 and C7 increased the CYP2C-inhibitory effects of flavonoids, whereas hydroxylation at C6 or glycosylation at any position reduced their enzyme inhibition. Compared with the regression results obtained from the logarithmically transformed data shown in Table III, the raw data for the CYP2C modulatory effects were more appropriate for constructing the QSAR, with the Y3100 μM level being optimal. The regression analyses also indicated that the glycosylation of flavonoids resulted in the least favorable structure for obtaining an inhibitory effect. By contrast, hydroxylation of flavonoids at C5 or C7 appeared to be essential for inhibition of CYP2C.
Table III.
The Mathematical Equations and Statistical Parameters for the Quantitative Structure–Activity Relationship
| Structure parameter | Mathematical equations | Adjusted R 2 (overall F test) |
|---|---|---|
| Y1(1 μM) | =11.94 × (C7 − OH) + 9.56 × (isoflavone) + 3.60 | 0.164 (F 2, 104 = 11.20, p < 0.01) |
| Y2(10 μM) |
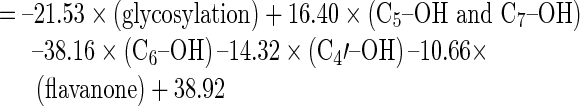
|
0.513 (F 5, 104 = 22.95, p < 0.01) |
| Y3(100 μM) |
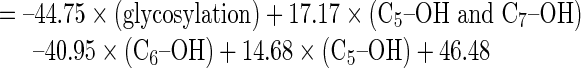
|
0.718 (F4, 104 = 67.06, p < 0.01) |
| Log Y1(1 μM) | =0.48 × (C7 − OH) + 0.54 | 0.126 (F 1, 104 = 15.93, p < 0.01) |
| Log Y2(10 μM) |
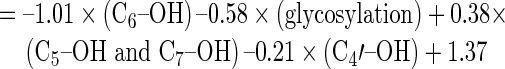
|
0.549 (F 4, 104 = 32.70, p < 0.01) |
| Log Y3(100 μM) | =−0.81 × (glycosylation) – 0.79 × (C6 −OH) + 0.32 × (C5 −OH) + 1.60 | 0.572 (F 3, 104 = 47.38, p < 0.01) |
DISCUSSION
We compared the predicted half-maximal inhibitory concentrations (IC50) for CYP2C activity in RLMs and conducted a preliminary structure–activity relationship (SAR) and QSAR analysis to elucidate the structural-determinants of flavonoid effects on this enzyme. The 40 dietary flavonoids tested in our in vitro screening can be classified into six categories on the basis of their structural features: flavones, flavonols, flavanones, isoflavones, chalcones, and catechins. The flavones and flavonols, such as wogonin, chrysin, tamarixetin, isorhamnetin, quercetin, and kaempferol potently inhibited CYP2C (IC50 1.2–16.8 μM) leading to the speculation that the planar structure with a 2,3-double bond in ring C was beneficial for CYP2C inhibition. This was supported by the finding that most of the flavanones or catechins lacked a double bond at that position and demonstrated lower CYP2C inhibition than flavones and flavonols. In addition, naringenin and eriodictyol had equal frame structures similar to those of apigenin and luteolin, respectively, but lacked the 2,3-double bond in their C rings and showed slightly inferior CYP2C inhibition. This suggested that the planar structure conferred on the flavone and flavonol molecules by the 2,3-double bond enabled them to intercalate with CYP2C more readily. The importance of the 2,3-double bond has also been highlighted in studies of flavonoid interactions with numerous membrane transporters and 15-lipoxygenases (32).
The CYP2C inhibitory effect was markedly reduced in flavonoids conjugated with a sugar moiety at any position on their core structure. In the QSAR model, stepwise regression analysis indicated that glycosylation of flavonoids was the least favorable structure for an inhibitory effect on CYP2C. None of the flavonoid glycosides tested in this study effectively inhibited CYP2C. With the exception of isoquercitrin, which exhibited some inhibition (IC50 = 43.4 μM), the IC50 values of all glycosylated flavonoids were over 100 μM. The stereo-structure of glycosides may create steric hindrance, weakening their binding affinity with CYP2C. In contrast to the glycosides, the aglycones are thought be responsible for enzyme modulation.
Simultaneous hydroxylation at positions 3, 5, and 7 or positions 5 and 7 could be considered essential for potent CYP2C inhibition. Genkwanin, which lacked hydroxyl substitutions at positions 3, 5, or 7, showed dramatically lower CYP2C inhibition than apigenin or kaempferol did (Fig. 4), although it had a 2,3-double bond in the C ring and was not glycosylated. Indeed, the statistical results demonstrated that when Y3(100 μM) was set as a dependent variable, hydroxylation at C5 alone or C5 and C7 were two critical independent factors for inhibition. By contrast, hydroxyl group attachment to position 6 significantly reduced inhibition of CYP2C activity. The only structural difference between baicalein and chrysin was that the former had an additional C6 hydroxylation, and it was an extremely weak inhibitor of CYP2C compared with chrysin. In the QSAR model, we also found that C6 hydroxylation reduced enzyme inhibition, whether the dependent variable was Y2(10 μM) or Y3(100 μM). Thus, the specific position of the hydroxyl substitution is important for enzyme inhibition. These findings were consistent with previous results demonstrating that C5 and C7 dihydroxylation of flavonoids was necessary for CYP2C inhibition (33).
Fig. 4.
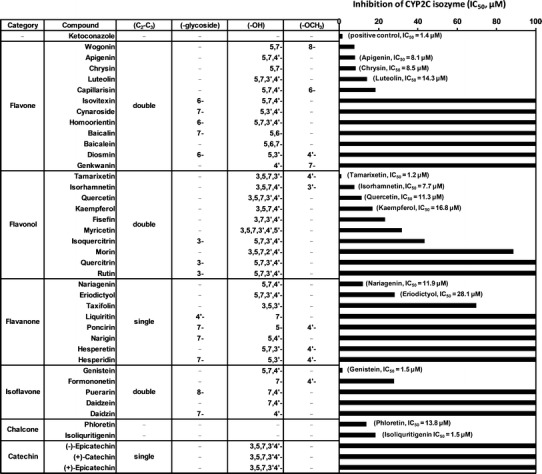
Inhibition of CYP2C-dependent tolbutamide 4-hydroxylation activity by flavonoids. Flavonoids were categorized into six groups according to their chemical structures. Compounds belong to flavones, flavonols, and isoflavones possessed the planar core structure with 2,3-double bond in the C ring. The IC50 values were obtained from experiments using varying concentrations
Otherwise, with the exception of glycosylated flavonols, compounds substituted with a methoxyl group at positions 3′ or 4′ were more effective CYP2C inhibitors, especially those with substitution at the latter position. For example, two primary metabolites of quercetin (tamarixetin and isorhamnetin) incorporated a methoxyl group at position 4′ and 3′, respectively, and were more potent inhibitors than quercetin. Lastly, although catechins have been reported to exert potent effects on multiple protein kinases (34), the three purified catechins screened in this study had virtually no inhibitory effects on CYP2C. Chow et al. have reported that repeated catechin administration is not likely to result in clinically significant effects on the disposition of drugs metabolized by CYP enzymes (35). Thus, we can reasonably speculate that the high level of catechins consumed daily in products such as green tea or its derivatives are unlikely to influence CYP2C activity.
In the present study, tamarixetin was identified as a potent inhibitor of CYP2C activity by in vitro screening in RLMs and was chosen for further evaluation in vivo. Tamarixetin can be extracted from Tamarix chinensis which is used as a traditional herbal antipyretic remedy. The structure of this flavonol was consistent with the structural requirements for CYP2C inhibition discussed above. It is also a specific CYP2C substrate and is metabolized into quercetin (36,37). Although tamarixetin has been reported to induce platelet dysfunction with an IC50 of approximately 40 μM in vitro, tamarixetin is unlikely to achieve such a high systemic concentration following oral administration (38). Williamson and Manach have indicated that the physiological concentrations of general flavonoids did not exceed 10 μM even after the intake of large doses (17). In addition, these dietary flavonoids undergo extensive first-pass metabolism in the liver prior to passage into the systemic circulation (6,39). Thus, we can speculate that under a reasonable dosing range, tamarixetin may not cause platelet toxicity in further studies.
Flavonoids have been shown to have a relatively low oral bioavailability (<10%) (6). In addition, previous pharmacokinetic studies of flavonoids in rats have demonstrated that these types of polyphenols underwent rapid metabolism and had large apparent volumes of distribution (6,40). The metabolism of oral flavonoids includes highly efficient glucuronidation, sulfation and CYP-mediated oxidation in enterocytes, and further metabolism via the same pathways in hepatocytes after absorption by the liver (39). Accordingly, we calculated the feeding dose of tamarixetin to rats on the basis of: (1) an estimation of 10% oral bioavailability, (2) the total volume of rat intestine and liver, and (3) ∼90% inhibition effect at 10 μM, as indicated by the in vitro screening test. We reasonably speculated that the given dose of tamarixetin had the potential to produce significant CYP2C inhibition in the liver and especially in the gastrointestinal tract, but the plasma concentration of tamarixetin would then decline rapidly once it entered the circulation, limiting its systemic effects.
The present study found that tamarixetin increased the Cmax and AUCINF of fluvastatin by 2.2- and 2.4-fold, respectively, without significantly changing the CL or T1/2 of fluvastatin (Table II). This indicated that tamarixetin may improve fluvastatin bioavailability by affecting the drug absorption phase, without altering pharmacokinetic properties relating to its distribution, metabolism, or elimination. These findings suggested that, as expected, tamarixetin may interacted with CYP2C in the gut and liver in a short-term and reversible manner before they passage into the circulatory system (17,41). Nevertheless, we still cannot rule out the possibility that the effects of tamarixetin on fluvastatin oral bioavailability could be mediated by inhibition of other P450 isozymes or even non-CYP metabolic enzymes (e.g., phase II enzymes or membrane transporters). However, considering the high intestinal absorption of fluvastatin and its marked specificity for CYP2C metabolism, these mechanisms appear less likely and could reasonably be considered minor.
A myriad of studies have been conducted with high-dose or long-term dietary flavonoid administration in humans to explore their anti-oxidant, anti-inflammatory, or anti-cancer activities and these compounds have generally been proven safe as well as efficacious (42–44). However, some studies have indicated that consumption of high-dose flavonoids may be associated with some potential adverse effects. Purified flavonoids given in high doses may affect trace element, folate, and vitamin C status. Furthermore, they may exhibit antithyroid and goitrogenic activities (45). These findings appear contradictory and further studies are required to understand how safe flavonoids are. Therefore, we cannot ignore the safety issues of high-dose flavonoids intake, even they were generally regarded as safe for health benefit. The careful estimation of flavonoid doses must be considered in future studies involving their concomitant administration to improve oral drug bioavailability.
The active ingredients in widely used nutraceuticals or herbal products may significantly influence the metabolic activities of P450 enzymes and lead to serious drug interactions. The present study sought to exploit this potential risk, using it as a strategy to improve the clinical usefulness of CYP2C-metabolized drugs. Apart from the expected benefit in improving drug oral bioavailability, preventing the production of toxic metabolites from CYP2C is another advantage of using inhibitors from these particular natural sources. The anticonvulsant drug valproic acid, is a specific substrate of CYP2C, and one of its toxic metabolites, 4-ene-valproate, has been reported to induce hepatotoxicity (46). Furthermore, 3′-azido-3′-deoxythymidine, an anti-HIV chemotherapeutic agent, is also catalyzed by CYP2C to a hematotoxic metabolite, 3′-amino-3′-deoxythymidine in human liver microsomes (47). Thus, we suggest that combined administration of such compounds with CYP2C enzyme inhibitors might not only improve their therapeutic efficacy, but also attenuate their toxic or undesired effects.
In addition, as suggested by the SAR and QSAR analyses, isorhamnetin, quercetin, and kaempferol were also found to exhibit potent CYP2C inhibition in our in vitro screening tests. With structural properties similar to tamarixetin, they may also prove useful for elevating the bioavailability of orally administered drugs.
CONCLUSIONS
In the present study, we identified tamarixetin as a potent inhibitor of CYP2C and demonstrated that this flavonoid significantly enhanced the bioavailability of the cholesterol-reducing drug fluvastatin. The SAR and QSAR analyses of flavonoids modulating CYP2C activity will inform future studies. Based on the results obtained, the strategy of employing flavonoids to elevate oral drug bioavailability was shown to be viable. Therefore, establishment of methods for the concomitant use of these dietary flavonoids to improve clinical outcomes or to diminish side effects is a promising area for further investigation. Indeed, preclinical human studies examining the ability of dietary flavonoids to improve the bioavailability of orally administered drugs are currently underway.
ACKNOWLEDGMENTS
This work was supported in part by Taiwan Biotech Co., Ltd. and Novartis Co., Ltd. in Taiwan. We are grateful to Dr. Ueng Yune-Fang for her technical assistance.
Statement of interest
No conflict of interest to be declared by authors.
Financial Support
No
REFERENCES
- 1.Wu CY, Benet LZ. Predicting drug disposition via application of BCS: transport/absorption/elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm Res. 2005;22:11–23. doi: 10.1007/s11095-004-9004-4. [DOI] [PubMed] [Google Scholar]
- 2.Zhou S, Gao Y, Jiang W, Huang M, Xu A, Paxton JW. Interactions of herbs with cytochrome P450. Drug Metab Rev. 2003;35:35–98. doi: 10.1081/DMR-120018248. [DOI] [PubMed] [Google Scholar]
- 3.Foster BC, Arnason JT, Briggs CJ. Natural health products and drug disposition. Annu Rev Pharmacol Toxicol. 2005;45:203–226. doi: 10.1146/annurev.pharmtox.45.120403.095950. [DOI] [PubMed] [Google Scholar]
- 4.Zhou S, Koh HL, Gao Y, Gong ZY, Lee EJD. Herbal bioactivation: the good, the bad and the ugly. Life Sci. 2004;74:935–968. doi: 10.1016/j.lfs.2003.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boumendjel A, Pietro AD, Dumontet C, Barron D. Recent advances in the discovery of flavonoids and analogs with high-affinity binding to P-glycoprotein responsible for cancer cell multidrug resistance. Med Res Rev. 2002;22:512–529. doi: 10.1002/med.10015. [DOI] [PubMed] [Google Scholar]
- 6.Cermak R, Wolffram S. The potential of flavonoids to influence drug metabolism and pharmacokinetics by local gastrointestinal mechanisms. Curr Drug Metab. 2006;7:729–744. doi: 10.2174/138920006778520570. [DOI] [PubMed] [Google Scholar]
- 7.Cermak R. Effect of dietary flavonoids on pathways involved in drug metabolism. Expert Opin Drug Metab Toxicol. 2008;4:17–35. doi: 10.1517/17425255.4.1.17. [DOI] [PubMed] [Google Scholar]
- 8.Mullie P, Clarys P, Deriemaeker P, Hebbelinck M. Estimation of daily human intake of food flavonoids. Int J Food Sci Nutr. 2008;59:291–298. doi: 10.1080/09687630701539293. [DOI] [PubMed] [Google Scholar]
- 9.Scalbert A, Williamson G. Dietary intake and bioavailability of polyphenols. J Nutr. 2000;130(Suppl):2073–2085. doi: 10.1093/jn/130.8.2073S. [DOI] [PubMed] [Google Scholar]
- 10.Moon YJ, Wang X, Morris ME. Dietary flavonoids: effects on xenobiotic and carcinogen metabolism. Toxicol in Vitro. 2006;20:187–210. doi: 10.1016/j.tiv.2005.06.048. [DOI] [PubMed] [Google Scholar]
- 11.Krishna D, Klotz U. Extrahepatic metabolism of drugs in humans. Clin Pharmacokinet. 1994;26:144–160. doi: 10.2165/00003088-199426020-00007. [DOI] [PubMed] [Google Scholar]
- 12.Rendic S, di Carlo FJ. Human cytochrome P450s enzymes: a status report summarizing their reactions, substances, inducers, and inhibitors. Drug Metab Rev. 2001;29:413–580. doi: 10.3109/03602539709037591. [DOI] [PubMed] [Google Scholar]
- 13.Shimada T, Yamazaki H, Mimura M, Inui Y, Guengerich FP. Interindividual variations in human liver cytochrome P450s enzymes involved in the oxidation of drugs, carcinogens and toxic chemical: studies with liver microsomes of 30 Japanese and 30 Caucasians. J Pharmacol Exp Ther. 1994;270:414–423. [PubMed] [Google Scholar]
- 14.Goldstein JA. Clinical relevance of genetic polymorphisms in the human CYP2C subfamily. Brit J Clin Pharmacol. 2001;52:349–355. doi: 10.1046/j.0306-5251.2001.01499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee CR, Pieper JA, Frye RF, Hinderliter AL, Blaisdell JA, Goldstein JA. Tolbutamide, flurbiprofen, and losartan as probes of CYP2C9 activity in humans. J Clin Pharmacol. 2003;43:84–91. doi: 10.1177/0091270002239710. [DOI] [PubMed] [Google Scholar]
- 16.Xie HG, Prasad HC, Kim RB, Stein CM. CYP2C9 allelic variants: ethnic distribution and functional significance. Adv Drug Deliv Rev. 2002;54:1257–1270. doi: 10.1016/S0169-409X(02)00076-5. [DOI] [PubMed] [Google Scholar]
- 17.Kruijtzer CMF, Beijnen JH, Schellens JHM. Improvement of oral drug treatment by temporary inhibition of drug transporters and/or cytochrome P450 in the gastrointestinal tract and liver: an overview. Oncologist. 2002;7:516–530. doi: 10.1634/theoncologist.7-6-516. [DOI] [PubMed] [Google Scholar]
- 18.Ding X, Kaminsky LS. Human extrahepatic cytochromes P450: function in xenobiotic metabolism and tissue-selective chemical toxicity in the respiratory and gastrointestinal tracts. Annu Rev Pharmacol Toxicol. 2003;43:149–173. doi: 10.1146/annurev.pharmtox.43.100901.140251. [DOI] [PubMed] [Google Scholar]
- 19.Williamson G, Manach C. Bioavailability and bioefficacy of polyphenols in human. II. Review of 93 intervention studies. Am J Clin Nutr. 2005;81(Suppl 1):243–255. doi: 10.1093/ajcn/81.1.243S. [DOI] [PubMed] [Google Scholar]
- 20.Kim JY, Lee S, Kim DH, Kim BR, Park R, Lee BM. Effects of flavonoids isolated from Scutellariae radix on cytochrome P-450 activities in human liver microsomes. J Toxicol Environ Health A. 2002;65:373–381. doi: 10.1080/15287390252808046. [DOI] [PubMed] [Google Scholar]
- 21.Quintieri L, Bortolozzo S, Straliotto S, Moro S, Pavanetto M, Nassi A, et al. Flavonoids diosmetin and hesperetin are potent inhibitors of cytochrome P450 2C9-mediated drug metabolism in vitro. Drug Metab Pharmacokinet. 2010;25:466–476. doi: 10.2133/dmpk.DMPK-10-RG-044. [DOI] [PubMed] [Google Scholar]
- 22.Kimura Y, Ito H, Ohnishi R, Hatano T. Inhibitory effects of polyphenols on human cytochrome P450 3A4 and 2C9 activity. Food Chem Toxicol. 2010;48:429–435. doi: 10.1016/j.fct.2009.10.041. [DOI] [PubMed] [Google Scholar]
- 23.Yuan R, Madani S, Wei XX, Reynolds K, Huang SM. Evaluation of cytochrome P450s probe substrates commonly used by the pharmaceutical industry to study in vitro drug interaction. Drug Metab Dispos. 2002;30:1311–1319. doi: 10.1124/dmd.30.12.1311. [DOI] [PubMed] [Google Scholar]
- 24.Toda T, Eliasson E, Ask B, Inotsume N, Rane A. Roles of different CYP enzymes in the formation of specific fluvastatin metabolites by human liver microsomes. Basic Clin Pharmacol Toxicol. 2009;105:327–332. doi: 10.1111/j.1742-7843.2009.00453.x. [DOI] [PubMed] [Google Scholar]
- 25.Scripture CD, Pieper JA. Clinical pharmacokinetics of fluvastatin. Clin Pharmacokinet. 2001;40:263–281. doi: 10.2165/00003088-200140040-00003. [DOI] [PubMed] [Google Scholar]
- 26.Sheu MT, Liou YB, Kao YH, Lin YK, Ho HO. A quantitative structure–activity relationship for the modulation effects of flavonoids on P-glycoprotein-mediated transport. Chem Pharm Bull. 2010;58:1187–1194. doi: 10.1248/cpb.58.1187. [DOI] [PubMed] [Google Scholar]
- 27.Pao LH, Hu OYP, Fan HY, Lin CC, Liu LC, Huang PW. Herb–drug interaction of 50 Chinese herbal medicines on CYP3A4 activity in vitro and in vivo. Am J Chin Med. 2012;40:57–73. doi: 10.1142/S0192415X1250005X. [DOI] [PubMed] [Google Scholar]
- 28.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 29.Miners JO, Smith KJ, Robson RA, McManus ME, Veronese ME, Birkett DJ. Tolbutamide hydroxylation by human liver microsomes: kinetic characterisation and relationship to other cytochrome P450 dependent xenobiotic oxidations. Biochem Pharmacol. 1988;37:1137–1144. doi: 10.1016/0006-2952(88)90522-9. [DOI] [PubMed] [Google Scholar]
- 30.Eagling VA, Tjia JF, Back DJ. Differential selectivity of cytochrome P450 inhibitors against probe substrates in human and rat liver microsomes. Br J Clin Pharmacol. 1998;45:107–114. doi: 10.1046/j.1365-2125.1998.00679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Toreson H, Eriksson BM. Determination of fluvastatin enantiomers and the racemate in human blood plasma by liquid chromatography and fluorometric detection. J Chromatogr A. 1996;729:13–18. doi: 10.1016/0021-9673(95)00897-7. [DOI] [PubMed] [Google Scholar]
- 32.Kitagawa S. Inhibitory effects of polyphenols on P-glycoprotein-mediated transporter. Biol Pharm Bull. 2006;29:1–6. doi: 10.1248/bpb.29.1. [DOI] [PubMed] [Google Scholar]
- 33.Shimada T, Tanaka K, Takenaka S, Murayama N, Martin MV, Foroozesh MK, et al. Structure–function relationships of inhibition of human cytochromes P450 1A1, 1A2, 1B1, 2C9, and 3A4 by 33 flavonoid derivatives. Chem Res Toxicol. 2010;23:1921–1935. doi: 10.1021/tx100286d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lorenz M, Wessler S, Follmann E, Michaelis W, Dusterhoft T, Baumann G, et al. A constituent of green tea, epigallocatechin-3-gallate, activates endothelial nitric oxide synthase by a phosphatidylinositol-3-OH-kinase-, cAMP-dependent protein kinase-, and Akt-dependent pathway and leads to endothelial-dependent vasorelaxation. J Biol Chem. 2004;279:6190–6195. doi: 10.1074/jbc.M309114200. [DOI] [PubMed] [Google Scholar]
- 35.Sherry Chow HH, Hakim I, Vining D, Crowell J, Cordova C, Chew W, et al. Effects of repeated green tea catechin administration on human cytochrome P450 activity. Cancer Epidemiol Biomarkers Prev. 2006;15:2473–2476. doi: 10.1158/1055-9965.EPI-06-0365. [DOI] [PubMed] [Google Scholar]
- 36.Nielsen SE, Breinholt V, Justesen U, Cornett C, Dragsted LO. In vitro biotransformation of flavonoids by rat liver microsomes. Xenobiotica. 1998;29:389–401. doi: 10.1080/004982598239498. [DOI] [PubMed] [Google Scholar]
- 37.Breinholt VM, Offord EA, Brouwer C, Nielsen SE, Brosen K, Friedberg T. In vitro investigation of cytochrome P450s-mediated metabolism of dietary flavonoids. Food Chem Toxicol. 2002;40:609–616. doi: 10.1016/S0278-6915(01)00125-9. [DOI] [PubMed] [Google Scholar]
- 38.Wright B, Moraes LA, Kemp CF, Mullen W, Crozier A, Lovegrove JA, et al. A structure basis for the inhibition of collagen-stimulated platelet function by quercetin and structurally related flavonoids. Br J Pharm. 2010;159:1312–1325. doi: 10.1111/j.1476-5381.2009.00632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Walle T. Absorption and metabolism of flavonoids. Free Radic Biol Med. 2004;36:829–837. doi: 10.1016/j.freeradbiomed.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 40.Lan K, Jiang XH, He JL. Quantitative determination of isorhamnetin, quercetin and kaempferol in rat plasma by liquid chromatography with electrospray ionization tandem mass spectrometry and its application to the pharmacokinetic study of isorhamnetin. Rapid Commun Mass Spectrom. 2007;21:112–120. doi: 10.1002/rcm.2814. [DOI] [PubMed] [Google Scholar]
- 41.Si D, Wang Y, Zhou YH, Guo Y, Wang J, Zhou H, et al. Mechanism of CYP2C9 inhibition by flavones and flavonols. Drug Metab Dispos. 2008;37:629–634. doi: 10.1124/dmd.108.023416. [DOI] [PubMed] [Google Scholar]
- 42.Dohadwala MM, Holbrook M, Hamburg NM, Shenouda SM, Chung WB, Titas M, et al. Effects of cranberry juice consumption on vascular function in patients with coronary artery disease. Am J Clin Nutr. 2011;93:934–940. doi: 10.3945/ajcn.110.004242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rizza S, Muniyappa R, Iantorno M, Kim JA, Chen H, Pullikotil P, et al. Citrus polyphenol hesperidin stimulates production of nitric oxide in endothelial cells while improving endothelial function and reducing inflammatory markers in patients with metabolic syndrome. J Clin Endocrinol Metab. 2011;96:E782–E792. doi: 10.1210/jc.2010-2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gates MA, Tworoger SS, Hecht JL, De Vivo I, Rosner B, Hankinson SE. A prospective study of dietary flavonoids intake and incidence of epithelial ovarian cancer. Int J Cancer. 2007;121:2225–2232. doi: 10.1002/ijc.22790. [DOI] [PubMed] [Google Scholar]
- 45.Egert S, Rimbach G. Which sources of flavonoids: complex diets or dietary supplements? Adv Nutr. 2011;2:8–14. doi: 10.3945/an.110.000026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Amini-Shirazi N, Ghahremani MH, Ahmadkhaniha R, Mandegary A, Dadgar A, Abdollahi M, et al. Influence of CYP2C9 polymorphism on metabolism of valproate and its hepatotoxin metabolite in Iranian patients. Toxicol Mech Method. 2010;20:452–457. doi: 10.3109/15376516.2010.497977. [DOI] [PubMed] [Google Scholar]
- 47.Pan-Zhou XR, Cretton-Scott E, Zhou XJ, Yang MX, Lasker JM, Sommadossi JP. Role of human liver P450s and cytochrome b5 in the reductive metabolism of 3′-azido-3′-deoxythymidine (AZT) to 3′-amino-3′-deoxythymidine. Biochem Pharmacol. 1998;55:757–766. doi: 10.1016/S0006-2952(97)00538-8. [DOI] [PubMed] [Google Scholar]