

Abstract
Mifepristone (RU486) is marketed and used widely by women as an abortifacient, and experimentally for psychotic depression and anticancer treatments. After administration, metapristone is found to be the most predominant metabolite of mifepristone. We hypothesized that adhesion of circulating tumor cells (CTCs) to vascular endothelial bed is a crucial starting point in metastatic cascade, and that metapristone can serve as a cancer metastatic chemopreventive agent that can interrupt adhesion and invasion of CTCs to the intima of microvasculature. In the present study, we modified the synthesis procedure to produce grams of metapristone, fully characterized its spectral properties and in vitro cellular activities, including its cytostatic effects, cell cycle arrest, mitochondrial membrane potential, and apoptosis on human colorectal cancer HT-29 cells. Metapristone concentration dependently interrupted adhesion of HT-29 cells to endothelial cells. Metapristone may potentially be a useful agent to interrupt metastatic initiation.
KEY WORDS: cancer metastasis chemoprevention, in vitro cellular activities, metapristone, mifepristone, spectral properties
INTRODUCTION
Mifepristone (RU486) is a prototypical anti-progesterone agent used for termination of early pregnancy because of its capacity to work as an anti-progestin to block uterine progesterone receptors. It also has glucocorticoid receptor antagonist activity at higher concentrations, with more than three times the binding affinity for glucocorticoid receptors than dexamethasone (1). After being used by millions of women, and recently men, for more than 20 years, large amounts of preclinical and clinical information have been generated for this agent. The information clearly indicates that the compound has many more benefits for global public health than originally thought. In preclinical oncology studies, mifepristone was reported to have a potent anti-proliferative effect on cancer cell lines derived from tumors of the breast (2), endometrium (3), cervix (4), prostate (5), gastrointestinal tract (6), brain (7), bone (8), and ovary (9).
In clinical trials, mifepristone has been used for the treatment of breast cancer, unresectable meningioma, prostate cancer, uterine fibroids, endometriosis, and Cushing syndrome at doses ranging from 1 to 1,500 mg per patient, depending on the type of disease and for a period ranging from 1 to 168 months. The clinical trials of mifepristone, however, have not produced promising data to support its use as an anti-cancer drug (10), probably because the current post-metastatic chemotherapy setting is too late to stop circulating tumor cells (CTCs) from spreading to susceptible tissues, and thus unable to affect the metastatic process. More than 90% of cancer deaths come from cancer recurrence and metastasis after surgical removal of the primary cancer. Cancer metastasis is a serious economic, social, and scientific problem world-wide that needs an urgent solution. As the estimated number of cancer survivors will reach 18 million in the USA alone (11), cancer metastasis chemoprevention research becomes increasingly important and badly needed. Currently, there are few ongoing projects to identify safe and effective cancer metastasis chemopreventive agents. It is our belief that metastatic cancers may not be cured, but can be prevented.
In our ongoing effort to identify safe and effective cancer metastasis chemopreventives for asymptomatic cancer survivors, we thought metapristone, the major active metabolite of mifepristone, may be a good candidate for further investigation for the following reasons: (1) The patentable metapristone is the primary metabolite of mifepristone. It has a t1/2 and AUC about 2-fold more than the parent mifepristone and its other metabolites in humans (12–14). (2) The relative binding affinity of metapristone to human glucocorticoid receptors and progesterone receptors is 63% and 43% of mifepristone, respectively (15), suggesting that metapristone may produce lesser side effects than mifepristone because metapristone binds less avidly to the two receptors. (3) Progesterone receptor, with which mifepristone and metapristone can interact, is present on both colorectal cancer cells and vascular endothelial cells (16). Our preliminary data demonstrated that metapristone inhibited adhesion of colorectal cancer cells to endothelial cells in a dose-dependent manner, suggesting that metapristone may be able to prevent the invasion of CTCs through the endothelial cell layer on the microvasculature of distant metastatic organs. (4) Mifepristone undergoes enterohepatic cycling (17). The long-term recycling of mifepristone from liver to intestine suggests that mifepristone and metapristone are suitable for prevention of colorectal cancer metastasis because liver is the common dissemination route for colorectal cancer metastasis. (5) Both mifepristone and metapristone have an anti-inflammatory effect that may contribute to their cancer metastatic chemoprevention activity.
More interestingly, accumulating evidence suggests that both embryonic implantation and tumor metastasis share striking similarities in biological behaviors in terms of cell adhesion (18), immune escape (19), angiogenesis (20), invasion (21), and tumor metastasis-related gene expression (22). The cellular mechanisms used by the embryo during implantation are re-utilized by cancer cells to adhere, invade, and extravasate to form micrometastatic foci within the metastatic tissues (20).
In view of its metabolic stability, as well as safety and efficacy estimated from the existing information on parent mifepristone, we selected metapristone to develop as a novel cancer metastatic chemopreventive. The present studies report our synthetic route for metapristone, the characterization of its spectral properties, and molecular docking to receptors. In addition, we examined the molecular and cellular mechanisms by which metapristone exerts effects on adhesion and invasion of cancer cells to vascular endothelium.
MATERIALS AND METHODS
Materials and Reagents
Mifepristone [RU486, 17β-hydroxy-11β-(4-dimethylamino) phenyl-17α-(1-propynyl)-estra-4, 9-dien-3-one], MW 429.59, was purchased from Shanghai New Hualian pharmaceutical Co., with purity >98%. Silica gel (200–300 mesh) used in column chromatography was provided by Tsingtao Marine Chemistry Co. Other reagents were obtained from commercial suppliers in analytically pure or chemically pure forms.
Synthesis
The main chemical synthesis scheme for metapristone [N-monodemethyl RU486, (or 17β-hydroxy-11β-(4-monomethylamino) phenyl-17α-(1-propynyl)-estra-4,9-dien-3- one), MW 415.57] was similar to that reported previously for mifepristone synthesis (23,24). However, some key modifications to the synthesis scheme were made to improve the yield and purity. Briefly, mifepristone (800 mg) was dissolved in a 1:1 mixture (10 ml) of tetrahydrofuran and methanol. Freshly calcined calcium oxide (900 mg) was added to the mixture. Iodine (1,000 mg) dissolved in the same solvent (2 ml) was added dropwise at 0°C. The reaction mixture was stirred at 0°C for 70 min. After calcium oxide was removed, the mixture was diluted with 15% sodium thiosulfate and extracted with methylene chloride. The combined organic layers were washed with water and brine sequentially, and made dry by using sodium sulfate. The solvent was evaporated after filtration, and the crude product was purified on a silica gel column using petroleum ether/ethyl acetate (v/v 4:1) as the eluent to give metapristone.
Spectral Analyses
The melting point of metapristone was determined on an electrically heated X-4 digital visual melting point apparatus. Metapristone as a yellow powder was dissolved in distilled methyl alcohol at a final concentration of 1 mM. A spectrometer (Quewell™ Q5000; linear detection range: 1–5,000 ng/μl) was used to determine the peak wavelength and the corresponding extinction coefficients of metapristone. Infrared spectra were analyzed a using Nicolet 360 Fourier Transform IR spectrometer (Nicolet Instruments, Inc.). The polyethylene film (potassium bromide, KBr) was used to calibrate the full scale and features of the spectrometer. The purified metapristone in an NMR tube was dissolved in deuterated chloroform for 1H-NMR analysis with an AVANCE III 500 MHz NMR system (Bruker, Switzerland). Chemical shifts were recorded as parts per million relative to tetramethylsilane as the internal standard for 1H-NMR. Mass spectral analysis was carried out with an Agilent 1100 LC/MSD Trap XCT (Agilent, America).
Chromatographic Analyses
Thin-layer chromatography (TLC) was performed on silica gel F254 plates (0.25 mm thick) from Tsingtao Marine Chemistry Co. Ltd. Five microlitre of metapristone (1 mM in methanol) was spotted at the origin of the plates, which were developed to a distance of 2.2 cm in a solvent developing system consisting of petroleum ether-tetrahydrofuran (3:1, v/v). Metapristone and its parent compound mifepristone were analyzed using a reversed phase HPLC system (Shimadzu, Kyoto, Japan) consisting of a LC-20A liquid delivery module, a SPD-20AV detector, a CTO-20A column oven, and a Shim-pack VP-ODS chromatographic column (250 × 4.6 mm, 5-μm particle size). The system was controlled with a CBM-20A system controller under the following conditions: flow rate, 1.0 ml/min; column temperature, 35°C; UV detection wavelength, 302 nm. A 20 μl aliquot of metapristone was auto-injected into the injection loop, and chromatographed using the C18 column eluted isocratically with a mobile phase of 75% methanol and 25% water. The area under each peak was calibrated with a Shimadzu data processor. All samples were diluted with methanol immediately before injection.
Cell lines and Cell Culture
Human cancer cell lines including hepatoma HepG2, colon HT-29, melanoma A-375, and cervical HeLa, as well as human embryo lung fibroblast cell line HELF were purchased from Shanghai Cell Bank in China. HepG2, A-375, HeLa, and HELF cells were maintained in RPMI 1640 medium supplemented with 10% FBS, penicillin (100 U/ml), and streptomycin (100 μg/ml) in a humidified atmosphere of 5.0% CO2 at 37°C. HT-29 cells were cultivated in McCoy's 5a medium.
Assay for Cell Viability/Proliferation
The cytostatic effect of metapristone and mifepristone was determined by using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (25). Cells (1 × 104/well) were plated in 100 μl of the culture medium/well in the 96-well plates. After overnight incubation, the cells were treated with different concentrations of the drugs in culture medium with 10% FBS for 24 h. Finally, MTT was added, and the cells were incubated for another 4 h in the medium without phenol red and serum. The MTT-formazan formed by metabolically viable cells was dissolved in 150 μl of dimethyl sulfoxide (DMSO) and shaken for 10 min. The absorbance was then measured on an ELISA reader at the test wavelength of 570 nm. Each test was repeated at least three times. The concentration of metapristone which gives a 50% growth inhibition value was defined as the IC50.
Migration Assay
We used a marker pen to mark three parallel lines on the bottom of six-well plates when the seeded HT-29 cells reached a density of 5 × 105 cells per well in triplicate for 24 h. A perpendicular scratch wound was generated by scratching with a 10-μl pipette tip. After rinsing with PBS to remove the detached cells, the medium containing different concentrations of metapristone (0, 40, and 80 μM) was added to the plates. Photographic images were taken from each well at 0 and 24 h after drug treatment. The distance that cells migrated through the marked area was determined by measuring the wound width at 24 h after treatment, and comparing it with the wound width at 0 h. The experiment was repeated three times.
Adhesion Assay
We used two methods to explore if metapristone had anti-adhesion effect: the fluorescence microscope photographed method and the MTT method.
Human umbilical vein endothelial cells (HUVECs) were isolated as described elsewhere (26). HUVECs were utilized between passages two and five, and grown to confluence in 24-well plates. They were washed and rested for 24 h in the M199 medium in the presence of 20% FCS and vascular endothelial growth factor (VEGF). The colon cancer HT-29 cells (105 cells per well) were plated in a final volume of 0.5 ml M199 medium on HUVECs pre-treated with 10 μM TNF-α for 4 h. Metapristone (1–100 μM) was added. HT-29 cells and HUVECs were co-cultured for 1 h at 37°C in 5% CO2. After incubation, non-adhered HT-29 cells were removed by washing three times (drop-to-drop) with 1 ml phosphate buffer solution (PBS). We randomly selected ten visual fields for each well and took pictures under a fluorescence microscope (Zeiss, Germany). The mean inhibition of adhesion for ten visual fields was calculated by using the equation: percent of control adhesion = [the number of adhered cells in treated samples/the number of adhered cells in the control group] × 100%.
For the MTT method, each well in 96-well plates was coated with 2 μg of Matrigel and was dried in a laminar flow cabinet overnight at room temperature. After washing three times with PBS to remove excess and unbound Matrigel, the wells were blocked with 20 ml of a 20 mg/l bovine serum albumin (BSA, Sigma) solution in McCoy’s 5a medium for 1 h at 37°C. Aliquots of 8 × 104 cells in 100 μl of serum-free McCoy’s 5a medium containing various concentrations of metapristone (5, 20, 40 μM), were added to each well and the cells were allowed to adhere for 1 h at 37°C. When the incubation was completed, the wells were washed three times with PBS to remove unbound cells. Then, the remaining cells were continuously incubated with the MTT solution (40 μl/well) for 4 h at 37°C, followed by treatment with 150 μl of DMSO for 10 min. Finally, Absorbance at 570 nm of each well was measured using an ELISA plate reader. Results were expressed as percnet of the adhesion rate of the control by using the following equation: A570nm of the adhered cells/A570nm of the adhered cells of the control group) × 100%.
Cell Cycle Assay
The cell cycle was analyzed by flow cytometry (25). Briefly, HT-29 cells were treated with different concentrations of metapristone (0, 40, 60, and 80 μM) for 24 h. After incubation, a total of 1 × 106 cells was harvested from the treated and control samples. The cells were washed twice with PBS and fixed in 70% ice-cold ethanol overnight. The samples were concentrated by removing ethanol and the cellular DNA stained with fluorescent solution (1% (v/v) Triton X-100, 0.01% RNase, 0.05% PI) for 30 min at 37°C in darkness. The cell cycle distribution was then determined by flow cytometry. All experiments were performed three times.
Mitochondrial Membrane Potential Assay
The mitochondrial membrane potential (ΔΨm) assay was similar to that described previously (27,28). The assay is based on uptake of 3,3'-dihexyloxacarbocyanine iodide (DiOC6(3), a widely used lipophilic cationic cyanine dye) by mitochondria. ΔΨm was determined by the reduction in uptake of the potential sensitive dye DiOC6(3). Briefly, HT-29 cells were cultured on 6-well plates overnight and then incubated with metapristone (0, 20, 40, 60, and 80 μM) in 2.5 ml fresh culture medium. Cells were then incubated for 24 h, washed twice with PBS, and incubated with DiOC6(3) for 15 min. The percentage of DiOC6(3)-positive cells were determined by quantitative flow cytometric analysis.
Analysis of Distinction Between Apoptotic and Necrotic Cells
To distinguish the apoptotic cells from necrotic ones, the treated cells were stained with Annexin V-PE and 7-Amino-Actinomycin (7-AAD) double staining kit. HT-29 cells were treated with different concentrations of metapristone (0, 40, 60, 80 μM) for 24 h. After incubation, a total of 1 × 106 cells were collected from the treated and untreated control samples separately and washed twice with cold PBS. One hundred microlitre of the medium containing 1 × 105 cells was transferred to a 5 ml culture tube, then 5 μl of annexin V-PE and 7-AAD were added and incubated for 15 min in the dark. After adding 400 μl of 1X binding buffer to each tube, the cells were then analyzed by analytical flow cytometry (BD FACS Aria III). All experiments were performed three times.
Caspase-3 Activity Assay
HT-29 cells were preincubated with either 20 μM of a typical caspase-3 inhibitor Z-DEVD-FMK, or 20 μM of a negative sham caspase-3 inhibitor Z-FA-FMK for 30 min, and then different groups of HT-29 cells were treated with 80 μM of metapristone for 24 h. Following the incubation, the cells were collected and stained with PE Annexin V/7-AAD to identify cells that underwent apoptosis.
Computational Chemistry
A computer model was used to depict the structural docking between metapristone and its binding sites on glucocorticoid receptors and to examine whether metapristone could bind to the active sites of the receptors. The X-ray crystal structure of the receptors has been reported (29–31), and was utilized in the study as the docking protein (http://www.rcsb.org/pdb/). After molecular mechanical energy minimization, the ligand-receptor docking was optimized with the Hex 6.3 protocol (32) to refine protein structure and ligand protonated state.
Statistical Analysis
The data are presented as means ± standard deviations of three determinations. Statistical analysis was performed using the Student’s t test and one-way analysis of variance. Multiple comparisons between the means were done by the least significance difference (LSD) test. A probability value of <0.05 was considered significant. All computations were made by employing SPSS statistical software (version 19.0).
RESULTS
Synthesis and Spectral Characterization
Mifepristone was demethylated to give metapristone with yield of >30% (Fig. 1a). It was fully characterized by various spectroscopic methods, including infrared (IR), 1H-NMR, and mass spectra (MS).
Fig. 1.
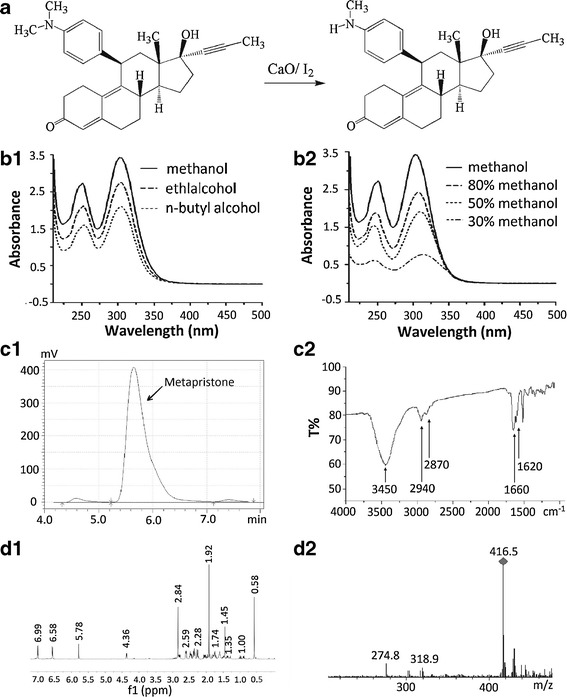
Synthesis scheme and spectral characterization of metapristone. a the synthetic route for metapristone; b UV–visible spectra of metapristone and the solvent effects: b1, metapristone in alcohol (b1) and methanol (b2) solvents; the UV–vis absorption maxima of metapristone decreased when the carbon chain of alcohol as well as the percentage of water in methanol increased. c representative HPLC analysis (c1) and infrared spectra (c2); d the 1H-NMR (d1) and mass (d2) spectra of metapristone
The metapristone powder was yellowish and not hygroscopic. The stability of the powder was significantly improved after purification. The yellowish powder was freely soluble in methylene dichloride, methyl alcohol, ethanol, chloroform, and acetone, but hardly soluble in water, sodium hydroxide(0.1 N), and hydrochloric acid (0.1 N). The melting point of metapristone, determined in an open capillary tube in a melting point apparatus, was found in the range of 85–87°C.
Spectral analysis of metapristone by UV–visible scanning at every ±0.5 nm around the two maxima revealed the peak wavelengths of metapristone (prepared in methyl alcohol) at 251 and 302 nm, with molar extinction coefficients of 2,697 ± 20 and 3,432 ± 12 m2 mol−1, respectively. Solvent effects on the absorption maxima of metapristone were compared (Fig. 1b), demonstrating that the UV–vis absorption maxima of metapristone decreased when the carbon chain length of the alcohol solvent, or the percentage of water in methanol, increased. Increasing water content resulted in a bathochromic shift of the 302 nm peak, whereas it did not have the same effect on the 251 nm absorption peak. TLC spots were detected with UV light (254 nm) or visualized with iodine vapors. TLC showed metapristone and mifepristone with Rf values of 0.32 and 0.50, respectively (not shown).
The purity of metapristone was determined by using the established HPLC method. In each single run, only a major peak was observed, which dominated the chromatographic area, indicating that metapristone has a purity of >96% with minor mifepristone (1.2%) and di-demethylated mifepristone (0.8%) residues. The retention time of metapristone was at 5.65 min Fig. 1(c1). The IR absorption bands for active chemical groups were identified in KBr. The presence of the CH3 group in metapristone can be readily determined from the strong IR bands at 2,940 and 2,870 cm−1, which corresponds to the stretching vibration of the C-H bond. The absorption band at 1,660 cm−1 and 1,620 cm−1 are characteristic of the C=O bond and the C=C bond, respectively. A big absorption band at 3,450 cm−1 was assigned to N–H and O-H Fig. 1(c2).
The 1H-NMR spectrum and mass analysis of metapristone are shown in Fig. 1d. Corresponding characterization data were as follows: 1H-NMR (500 MHz, CDCl3): δ = 0.58 (s, 3H, C-18H), 1.35 (m, 1H, C-15H), 1.45 (m, 1H, C-7H), 1.74 (m, 2H, C-14H, C-15H), 1.92 (s, 3H, C-21H), 2.02 (td, J = 11.4, J = 3.2, 1H, C-16H), 2.07 (m, 1H, C-7H), 2.25 (m, 1H, C-16H), 2.28 (m, 1H, C-12H), 2.36 (m, 4H, C-1H, 2 × C-2H, C-12H), 2.47 (dd, J = 10.4, J = 0.8, 1H, C-8H), 2.59 (m, 2H, C-6H), 2.78 (m, 1H, C-1H), 2.84 (s, 3H, NCH3), 4.36 (d, J = 6.4, 1H, C-11H), 5.78 (s, 1H, C-4H), 6.58 (m, 2H, C-30/50H), 6.99 (m, 2H, C-20/60H); ESI-MS:m/z 416.5 [M + H]+, calculated for C28H33NO2: 415.25.
Cytostatic Activity
The cytostatic effect of metapristone was examined on A-375, HepG2, HT-29, and HELF cell lines by the MTT assay in parallel comparison with that of mifepristone. Metapristone and mifepristone produced anti-proliferative effects in a concentration dependent manner. The IC50 values for mifepristone were 38.5, 50.3, 59.5, and 35.8 μM. The corresponding values for metapristone were 55.7, 78.1, 83.5, and 53.8 μM, respectively (Fig. 2a), indicating that metapristone had somewhat less potent cytostatic effects on cancer cell lines and normal cells compared with mifepristone.
Fig. 2.
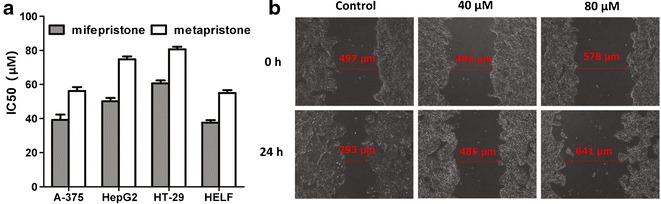
The anti-proliferative and anti-migration effects of metapristone. a In vitro activity of metapristone and RU486 (expressed as IC50 (μM)) against A-375, HepG2, HT-29, and HELF cell lines; b effect of metapristone on migration of HT-29 cells incubated in the cell wound scratch assay for 24 h
Migration Assay
The effect of metapristone on the migration of human colon HT-29 cells was evaluated by cell wound scratch assay. As shown in Fig. 2b, HT-29 cells migrated in the control group to partially close the wound, but there was a significant decrease in the number of migrated HT-29 cells in metapristone-treated groups. At 40 μM, metapristone prevented the cells from migrating within 24 h after treatment, while at 80 μM, metapristone treatment resulted in loss of some border cells and resultant enlargement of the wound.
Adhesion Assay
Mifepristone is known to dose dependently inhibit the heterotypic adhesion of cancer cells to basement membrane (33). To investigate whether metapristone has inhibitory effects on cell adhesion and invasion of HT-29 with HUVECs, the HT-29 cells were labeled with the fluorescent dye rhodamine 123. Fluorescence microscope observation revealed that metapristone interfered with adhesion of HT-29 cells to HUVECs in a concentration-dependent manner (Fig. 3a).
Fig. 3.
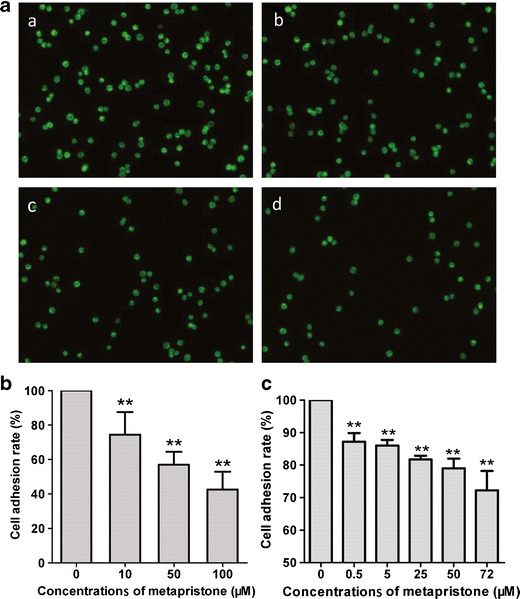
Inhibition by metapristone of HT-29 adhesion to HUVECs (a, b) and Matrigel (c). a representative microscopic observation of the inhibition by metapristone at 0, 10, 50, and 100 μM (a, b, c, and d) on adhesion of HT-29 to HUVECs; b quantitative analysis of the inhibition by metapristone on the adhesion of HT-29 to HUVECs; c quantitative MTT analysis of the inhibition by metapristone on the adhesion of HT-29 to Matrigel
To quantitatively examine metapristone inhibition of HT-29 cells adherence to HUVECs, 10 fields of each well were randomly selected, and the adhered spots were counted. Compared with the control, the adhesion rate of HT-29 cells was 73.6 ± 10.0%, 56.6 ± 5.9%, and 41.9 ± 8.0%, respectively, with 10, 50, and 100 μM of metapristone (Fig. 3b), indicating that it may reduce the risk factors for cancer metastasis.
MTT assay revealed that metapristone inhibited cell adhesion to the artificial basement membrane, Matrigel in a dose-dependent manner. The adhesive rate of HT-29 cells was 87.2 ± 5.3%, 86.0 ± 3.4%, 81.7 ± 2.3%, 79.0 ± 6.0%, and 72.2 ± 11.9%, with metapristone at 0.5, 5, 25, 50, and 72 μM, respectively (Fig. 3c).
Investigation of Cell Cycle Distribution
To determine the mechanism by which metapristone modestly produced growth inhibition, cell cycle distribution was observed by flow cytometry after HT-29 cells were treated with different concentrations of the compound. To monitor cell cycle arrest, the DNA was stained with propidium iodide (PI). With increasing concentrations of metapristone, the cells accumulated in the G0/G1-phase and the S-phase was gradually reduced (Fig. 4a), indicating that metapristone arrested HT-29 cells mainly at the G0/G1 stage.
Fig. 4.
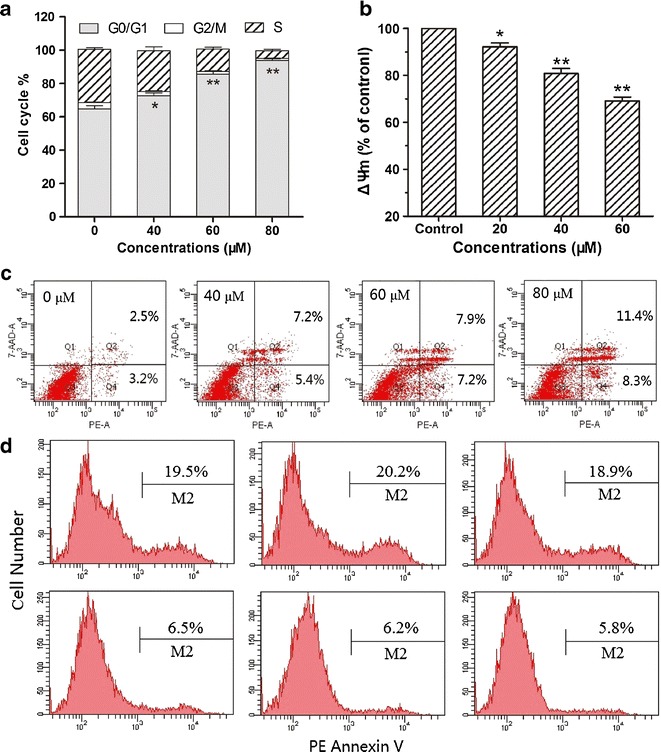
Investigation of anti-proliferative mechanisms for metapristone. a effects of metapristoe on cell cycle distribution in HT-29 cells; b effects of metapristone on ΔΨm in HT-29 cells; results are expressed as percentage change in metapristone treatment compared to the untreated control (*p < 0.05, **p < 0.01); c flow cytometric analysis of PE Annexin V/7-AAD staining for metapristone in HT-29 cells; d detection of caspase-3 activity by flow cytometric analysis of apoptosis in HT-29 cells, HT-29 cells were preincubated with the following: no inhibitor (upper left and bottom left panels), 20 μM Z-DEVD-FMK (upper center and bottom center panels) or 20 μM of a negative control inhibitor Z-FA-FMK (upper right and bottom right panels) for 30 min, and then either left untreated (bottom row) or treated with 80 μM of metapristone for 24 h (top row)
Metapristone-Induced Effects of ΔΨm
Mitochondrial membrane depolarization is a prelude of apoptosis. ΔΨm reflects activity of the electron transport chain and mitochondrial function. Treatment of HT-29 cells with metapristone at concentrations ranging from 0 to 60 μM produced loss of ΔΨm in a concentration-dependent manner (Fig. 4b). When the concentration of metapristone was increased to 80 μM, it induced modest hyperpolarization of mitochondrial membrane (82.4% of DiOC6(3)+ cells).
Annexin V-PE/7-AAD Double Staining Analysis
As shown in Fig. 4c, the untreated HT-29 were primarily PE Annexin V and 7-AAD negative, indicating that they were viable and not undergoing apoptosis. After a 24-h treatment with metapristone (40, 60, 80 μM), the cells were divided into four populations: cells that were viable and not undergoing apoptosis (PE Annexin V and 7-AAD negative, Q3); cells undergoing early apoptosis (PE Annexin V positive and 7-AAD negative, Q4); cells in end-stage apoptosis or already dead (PE Annexin V and 7-AAD positive, Q2); and some cell debris due to mechanical damage (PE Annexin V negative and 7-AAD positive, Q1). The protocol used for distinguishing apoptotic from necrotic cells was consistent with our previous publications (25,28), and the analysis showed that metapristone induced the cells into early apoptosis, advanced apoptosis, and necrosis in a concentration-dependent and sequential manner (Fig. 4c).
Caspase-3 Activity in the Treated Cells
To explore whether caspase-3 is involved in the cellular apoptosis induced by the high concentration of metapristone (80 μM) observed in the above-mentioned experiments, HT-29 cells were preincubated with the caspase-3 inhibitor Z-DEVD-FMK or the caspase-3 negative sham inhibitor Z-FA-FMK before metapristone was added to the tested cells. In comparison with both the positive apoptosis induced by metapristone alone and the apoptosis induced by metapristone in the presence of the sham inhibitor Z-FA-FMK, pretreatment of the cells with the caspase-3 inhibitor Z-DEVD-FMK did not significantly change the extent of cellular apoptosis (Fig. 4d), indicating caspase-3 is not involved in the apoptosis induced by high concentration of metapristone.
Analysis of Molecular Docking
Analysis of the computational simulation showed a striking similarity in structural docking and receptor binding of mifepristone and metapristone to glucocorticoid receptors. Both mifepristone and metapristone sit within a large hydrophobic binding cavity with key hydrogen-bonding contacts (Fig. 5). The amino acid residues around the N-dimethylamino group of mifepristone or the N-monomethylamino group of metapristone are Leu563, Asn564, Gly567, Gly568, Val571, and Ala605. Both drugs embed in the main active center (composed of Gln570, Arg611, and Gln642) of the receptors. The computational docking provided the evidence that the two drugs exert their pharmacological effects through basically the same mechanism of action.
Fig. 5.
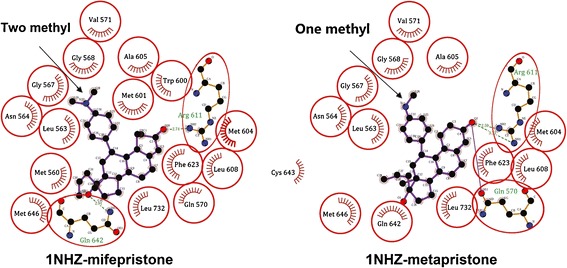
The molecular docking of mifepristone and metapristone with glucocorticoid receptors (PDB ID: 1NHZ). The purple structures representing either mifepristone (2-methyl) or metapristone (1-methyl) embedded in the active centers of the receptors linked by the key hydrogen bonds (dash lines)
DISCUSSION
The present studies provide a detailed report of the chemical synthesis and structural characterization of metapristone. The chemical structure was characterized and validated by UV/vis, IR, 1H-NMR and MS. The spectral data are consistent with those reported (24,34). The modified synthesis and purification procedures could produce metapristone in a yield of 30%. After further optimization of the synthesis procedure, we can now obtain a yield >50% in a pilot scale for both its animal and clinical studies. The computational docking model (Fig. 5) indicates that metapristone fits into the active centers of glucocorticoid receptors in a manner similar to mifepristone, suggesting strongly that metapristone can produce all pharmacological effects of mifepristone. Previous studies provided strong evidence that mifepristone, upon oral administration, was quickly metabolized to metapristone in animals and female human beings. Moreover, blood concentrations of metapristone quickly exceeded those of mifepristone and its other metabolites in humans (12–14). It is this information that led us to further examine metapristone as a potential cancer metastatic chemopreventive agent The present studies compared the cell growth inhibition activity between metapristone and mifepristone on four cancer cell lines and a normal cell line. The cytotoxicity of metapristone was somewhat lower than mifepristone, possibly making metapristone more suited as a cancer metastatic chemopreventive agent to be used for a long period to interfere adhesion of CTCs to endothelial vasculature.
Mifepristone has been shown to effectively inhibit the invasive and metastatic potential of the human gastric adenocarcinoma cell line MKN-45 via inhibition of heterotypic adhesion to basement membrane, cell migration, and angiogenesis (33). It also inhibits expression of NF-kappa B, and interferes with the activity of Beta-2 integrin (35). Based on flow cytometry analysis, we observed that metapristone arrested cancer cells mainly at the G0/G1 stage and induced the loss of mitochondrial membrane potential at low concentrations. Furthermore, at a high concentration (80 μM), metapristone might enhance mitochondrial membrane permeability for protons, resulting in partial uncoupling of electron transfer, and the leak of protons and membrane hyperpolarization (36). As the concentration increased, metapristone caused apoptosis/necrosis in the treated cells in a concentration-dependent manner. The difference between apoptotic and necrosis cell death is that during the initial stages of apoptosis, the cell membrane remains intact, while at the very moment that necrosis occurs the cell membrane loses its integrity and becomes leaky. Annexin V is a Ca2+-dependent phospholipid-binding protein with high affinity for phosphatidylserine. The translocation of the latter and its biomarker Annexin V to the cell surface is indicative of apoptosis (37). The metapristone-induced apoptosis did not seem to be related to caspase-3 activity.
Over the past two decades, many striking similarities have been revealed between implanted embryos and CTCs in terms of their migratory, invasive, and proliferative properties (22,38). Embryos are capable of implanting almost anywhere in the reproductive system, and possess an intrinsic ability to attach, migrate, and invade. This is evidenced in women by the occurrence of ectopic pregnancies with growth and invasion into the fallopian tube, ovary, or cervix. Although embryos have the capacity for invasion, the endometrium can either facilitate or limit this process. Similar to the embryonic implantation, the activated CTCs interact with vascular endothelium and then extravasate in the distant metastatic organs. We propose that the initiation of adhesion of CTCs to vascular endothelial cells is the first and important step for CTCs to start the metastatic cascade. Inhibition of the initial step may thus prevent consequential formation of the metastasis foci. In the present studies, we demonstrated that metapristone could interfere with adhesion of HT-29 cells to HUVECs in a concentration-dependent manner (Fig. 3a, b). The cell scratch test results showed that metapristone could slow down migration speed (Fig. 2b). Collectively, the results suggest that metapristone may inhibit the adhesion of CTCs to the intima of blood vessels in the distant metastatic organs (39). Thus, metapristone has the potential to reduce the risk of cancer metastasis.
The major drivers of early and late metastasis may be different. Early metastasis is more related to the interaction between CTCs and endothelium required for the adhesion and invasion cascade to occur than is the later process, in which angiogenesis and proliferation of the formed micrometastasis per se, as well as the spreading of CTCs to other tissues become prevailing and untreatable. It is our belief that a safe and effective chemoprevention targeted to the early metastatic process can interrupt the cascade. Importantly, the mifepristone- or metapristone-mediated interruption of CTC adhesion and invasion at the inner membrane of microcirculation in the distant metastasis tissues provides a molecular framework for future clinical evaluation of metapristone (or other antiprogesterones) as a potential cancer metastatic chemopreventive agent in an adjuvant setting in patients who have already undergone surgery to remove their primary breast and colorectal cancers.
CONCLUSIONS
The present studies describe a modified synthetic process for metapristone and the characterization of its spectral properties. Metapristone had a modest cytostatic effect on cancer cell lines compared with mifepristone. It caused HT-29 cells to be arrested at the G0/G1 stage, induced some apoptosis at high-dose, and interfered with adhesion of HT-29 cells to HUVECs. Metapristone is not a traditional anticancer drug. Rather, it may fit into a new class of cancer metastatic chemopreventives.
Acknowledgements
These studies were supported by grants from the National Science Foundation of China Nos. 81201709, J1103303 and 81273548. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Jichuang Wang and Jianzhong Chen These authors equally contributed to the work.
The study was presented in part at the 2013 annual meeting of American Association of Pharmaceutical Scientists, Antonio, TX, USA; November, 2013.
Contributor Information
Jingwei Shao, Email: shaojingwei@fzu.edu.cn.
Lee Jia, Phone: +86-151-59630201, FAX: +86-591-83792563, Email: pharmlink@gmail.com.
REFERENCES
- 1.Fleseriu M, Biller BM, Findling JW, Molitch ME, Schteingart DE, Gross C, et al. Mifepristone, a glucocorticoid receptor antagonist, produces clinical and metabolic benefits in patients with Cushing's syndrome. J Clin Endocr Metab. 2012;97(6):2039–2049. doi: 10.1210/jc.2011-3350. [DOI] [PubMed] [Google Scholar]
- 2.El Etreby MF, Liang YY, Wrenn RW, Schoenlein PV. Additive effect of mifepristone and tamoxifen on apoptotic pathways in MCF-7 human breast cancer cells. Breast Cancer Res Tr. 1998;51(2):149–168. doi: 10.1023/A:1006078032287. [DOI] [PubMed] [Google Scholar]
- 3.Fiscella J, Bonfiglio T, Winters P, Eisinger SH, Fiscella K. Distinguishing features of endometrial pathology after exposure to the progesterone receptor modulator mifepristone. Hum Pathol. 2011;42(7):947–953. doi: 10.1016/j.humpath.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fried G, Meister B, Râdestad A. Peptide-containing nerves in the human pregnant uterine cervix: an immunohistochemical study exploring the effect of RU 486 (mifepristone) Hum Reprod. 1990;5(7):870–876. doi: 10.1093/oxfordjournals.humrep.a137200. [DOI] [PubMed] [Google Scholar]
- 5.Ligr M, Li Y, Logan SK, Taneja S, Melamed J, Lepor H, et al. Mifepristone inhibits GRβ coupled prostate cancer cell proliferation. J Urol. 2012;188(3):981–988. doi: 10.1016/j.juro.2012.04.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goel N, Malik R, Rathi B, Bhaskaran S, Rajaram S, Mehta S, et al. Pregnancy with metastatic gastrointestinal stromal tumor (GIST) on imatinib chemotherapy: an oncologist’s nightmare and obstetrician’s dilemma. J Gastroint Cancer. 2013;44(1):115–117. doi: 10.1007/s12029-012-9464-4. [DOI] [PubMed] [Google Scholar]
- 7.Wang Y, Yang D, Song L, Li T, Yang J, Zhang X, et al. Mifepristone-inducible caspase-1 expression in mouse embryonic stem cells eliminates tumor formation but spares differentiated cells in vitro and in vivo. Stem Cells. 2012;30(2):169–179. doi: 10.1002/stem.1000. [DOI] [PubMed] [Google Scholar]
- 8.Cher ML, Towler DA, Rafii S, Rowley D, Donahue HJ, Keller E, et al. Cancer interaction with the bone microenvironment: a workshop of the National Institutes of Health Tumor Microenvironment Study Section. Am J Pathol. 2006;168(5):1405–1412. doi: 10.2353/ajpath.2006.050874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wempe SL, Gamarra-Luques CD, Telleria CM. Synergistic lethality of mifepristone and LY294002 in ovarian cancer cells. Cancer Growth Metast. 2013;6:1–13. doi: 10.4137/CGM.S11124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen JZ, Wang JC, Xu JG, Shao JW, Jia L. Pharmaceutical characteristics of mifepristone: from terminating pregnancy to preventing cancer metastasis. Med Res Rev 2013 (in press). [DOI] [PubMed]
- 11.de Moor JS, Mariotto AB, Parry C, Alfano CM, Padgett L, Kent EE, et al. Cancer survivors in the United States: prevalence across the survivorship trajectory and implications for care. Cancer Epidem Biomar. 2013;22(4):561–570. doi: 10.1158/1055-9965.EPI-12-1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heikinheimo O, Haukkamaa M, Lahteenmaki P. Distribution of RU 486 and its demethylated metabolites in humans. J Clin Endocrinol Metab. 1989;68(2):270–275. doi: 10.1210/jcem-68-2-270. [DOI] [PubMed] [Google Scholar]
- 13.Shi YE, Ye ZH, He CH, Zhang GQ, Xu JQ, Van Look PF, et al. Pharmacokinetic study of RU 486 and its metabolites after oral administration of single doses to pregnant and non-pregnant women. Contraception. 1993;48(2):133–149. doi: 10.1016/0010-7824(93)90004-Q. [DOI] [PubMed] [Google Scholar]
- 14.Teng YN, Dong RQ, Wang BJ, Liu HJ, Jiang ZM, Wei CM, et al. Determinations of mifepristone and its metabolites and their pharmacokinetics in healthy female Chinese subjects. Acta Pharm Sin. 2011;46(10):1241–1245. [PubMed] [Google Scholar]
- 15.Heikinheimo O, Kontula K, Croxatto H, Spitz I, Luukkainen T, Lahteenmaki P. Plasma concentrations and receptor binding of RU 486 and its metabolites in humans. J Steroid Biochem. 1987;26(2):279–284. doi: 10.1016/0022-4731(87)90083-5. [DOI] [PubMed] [Google Scholar]
- 16.Haier J, Nasralla M, Nicolson GL. Cell surface molecules and their prognostic values in assessing colorectal carcinomas. Ann Surg. 2000;231(1):11–24. doi: 10.1097/00000658-200001000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Permezel JM, Lenton EA, Roberts I, Cooke ID. Acute effects of progesterone and the antiprogestin RU 486 on gonadotropin secretion in the follicular phase of the menstrual cycle. J Clin Endocrinol Metab. 1989;68(5):960–965. doi: 10.1210/jcem-68-5-960. [DOI] [PubMed] [Google Scholar]
- 18.Lash GE, Fitzpatrick TE, Graham CH. Effect of hypoxia on cellular adhesion to vitronectin and fibronectin. Biochem Bioph Res Co. 2001;287(3):622–629. doi: 10.1006/bbrc.2001.5618. [DOI] [PubMed] [Google Scholar]
- 19.Arck P, Hagen E, Hildebrandt M, Klapp B, Hertwig K. Pregnancy as a model of controlled invasion might be attributed to the ratio of CD3/CD8 to CD56. Am J Reprod Immunol. 2000;44(1):1–8. doi: 10.1111/j.8755-8920.2000.440101.x. [DOI] [PubMed] [Google Scholar]
- 20.Murray MJ, Lessey BA. Embryo implantation and tumor metastasis: common pathways of invasion and angiogenesis. Semin Reprod Endocrinol. 1999;17(3):275–290. doi: 10.1055/s-2007-1016235. [DOI] [PubMed] [Google Scholar]
- 21.Fitzpatrick TE, Lash GE, Yanaihara A, Charnock-Jones DS, Macdonald-Goodfellow SK, Graham CH. Inhibition of breast carcinoma and trophoblast cell invasiveness by vascular endothelial growth factor. Exp Cell Res. 2003;283(2):247–255. doi: 10.1016/S0014-4827(02)00044-7. [DOI] [PubMed] [Google Scholar]
- 22.Janneau JL, Maldonado-Estrada J, Tachdjian G, Miran I, Motté N, Saulnier P, et al. Transcriptional expression of genes involved in cell invasion and migration by normal and tumoral trophoblast cells. J Clin Endocrinol Metab. 2002;87(11):5336–5339. doi: 10.1210/jc.2002-021093. [DOI] [PubMed] [Google Scholar]
- 23.Hödl C, Raunegger K, Strommer R, Ecker GF, Kunert O, Sturm S, et al. Syntheses and antigestagenic activity of Mifepristone derivatives. J Med Chem. 2009;52(5):1268–1274. doi: 10.1021/jm800985z. [DOI] [PubMed] [Google Scholar]
- 24.Hödl C, Strauss WS, Sailer R, Seger C, Steiner R, Haslinger E, et al. A novel, high-affinity, fluorescent progesterone receptor antagonist. Synthesis and in vitro studies. Bioconjug Chem. 2004;15(2):359–365. doi: 10.1021/bc034169o. [DOI] [PubMed] [Google Scholar]
- 25.Shao J, Dai Y, Zhao W, Xie J, Xue J, Ye J, et al. Intracellular distribution and mechanisms of actions of photosensitizer zinc(II)-phthalocyanine solubilized in Cremophor EL against human hepatocellular carcinoma HepG2 cells. Cancer Lett. 2013;330(1):49–56. doi: 10.1016/j.canlet.2012.11.017. [DOI] [PubMed] [Google Scholar]
- 26.Jaffe EA, Nachman RL, Becker CG, Minick CR. Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J Clin Invest. 1973;52(11):2745–2756. doi: 10.1172/JCI107470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zamzami N, Marchetti P, Castedo M, Decaudin D, Macho A, Hirsch T, et al. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J Exp Med. 1995;182(2):367–377. doi: 10.1084/jem.182.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shao J, Xue J, Dai Y, Liu H, Chen N, Jia L, et al. Inhibition of human hepatocellular carcinoma HepG2 by phthalocyanine photosensitiser PHOTOCYANINE: ROS production, apoptosis, cell cycle arrest. Eur J Cancer. 2012;48(13):2086–2096. doi: 10.1016/j.ejca.2011.10.013. [DOI] [PubMed] [Google Scholar]
- 29.Schoch GA, D'Arcy B, Stihle M, Burger D, Bär D, Benz J, et al. Molecular switch in the glucocorticoid receptor: active and passive antagonist conformations. J Mol Biol. 2010;395(3):568–577. doi: 10.1016/j.jmb.2009.11.011. [DOI] [PubMed] [Google Scholar]
- 30.Honer C, Nam K, Fink C, Marshall P, Ksander G, Chatelain RE, et al. Glucocorticoid receptor antagonism by cyproterone acetate and RU486. Mol Pharmacol. 2003;63(5):1012–1020. doi: 10.1124/mol.63.5.1012. [DOI] [PubMed] [Google Scholar]
- 31.von Geldern TW, Tu N, Kym PR, Link JT, Jae HS, Lai C, et al. Liver-selective glucocorticoid antagonists: a novel treatment for type 2 diabetes. J Med Chem. 2004;47(17):4213–4230. doi: 10.1021/jm0400045. [DOI] [PubMed] [Google Scholar]
- 32.Macindoe G, Mavridis L, Venkatraman V, Devignes MD, Ritchie DW. HexServer: an FFT-based protein docking server powered by graphics processors. Nucleic Acids Res. 2010;38(Web Server issue):W445–W449. doi: 10.1093/nar/gkq311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li DQ, Wang ZB, Bai J, Zhao J, Wang Y, Hu K, et al. Effects of mifepristone on invasive and metastatic potential of human gastric adenocarcinoma cell line MKN-45 in vitro and in vivo. World J Gastroenterol. 2004;10(12):1726–1729. doi: 10.3748/wjg.v10.i12.1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saha P, Hödl C, Strauss WS, Steiner R, Goessler W, Kunert O, et al. Synthesis, in vitro progesterone receptors affinity of gadolinium containing mifepristone conjugates and estimation of binding sites in human breast cancer cells. Bioorgan Med Chem. 2010;18(5):1891–1898. doi: 10.1016/j.bmc.2010.01.048. [DOI] [PubMed] [Google Scholar]
- 35.Ma Y, Katiyar P, Jones LP, Fan S, Zhang Y, Furth PA, et al. The breast cancer susceptibility gene BRCA1 regulates progesterone receptor signaling in mammary epithelial cells. Mol Endocrinol. 2006;20(1):14–34. doi: 10.1210/me.2004-0488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang L, Wang XM, Jiao XM, Liu SS. The effect of superoxide ainon on transmembrane potential and proton transfer of myocardial mitochondria. Chinese J Pathophysiol. 1996;12(2):181–184. [Google Scholar]
- 37.Vermes I, Haanen C, Steffens-Nakken H, Reutellingsperger C. A novel assay for apoptosis flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled annexin V. J Immunol Methods. 1995;184(1):39–51. doi: 10.1016/0022-1759(95)00072-I. [DOI] [PubMed] [Google Scholar]
- 38.Ferretti C, Bruni L, Dangles-Marie V, Pecking A, Bellet D. Molecular circuits shared by placental and cancer cells, and their implications in the proliferative, invasive and migratory capacities of trophoblasts. Hum Reprod Update. 2007;13(2):121–141. doi: 10.1093/humupd/dml048. [DOI] [PubMed] [Google Scholar]
- 39.Ellis LM, Radinsky R, Fidler IJ. Recent advances in the biology of cancer invasion and metastasis. Surgical Oncology: Contemporary Principles and Practice. 2001. p. 101–22.