

Abstract
Epigallocatechin-3-gallate (EGCG), a powerful antioxidant and free ion scavenger found in green tea, exhibits inhibitory effects on different stages of tumorigenesis. Within gastric cancer cells, the transcription factor Kruppel-like factor 4 (KLF4) is downregulated, and it is possible that EGCG exerts its anti-tumorigenic function through modulation of KLF4 expression. In order to examine the effects of EGCG on KLF4 in a gastric tumor model, we treated the gastric cancer cell line NCI-N87 with EGCG. We found that EGCG treatment results in increased expression of KLF4 and alters expression of the KLF4 target genes p21, CDK4, and cyclin D1. EGCG inhibits the growth of NCI-N87 cells in a time- and dose-dependent manner through arresting the cell cycle in the G0/G1 phase. Furthermore, terminal deoxynucleotidyl transferase dUTP nick end labeling assay and 4′,6-diamidino-2-phenylindole staining revealed that EGCG is able to promote apoptosis of NCI-N87 cells. The suppressive effects of EGCG on cell growth and cell cycle protein expression are eliminated by decreasing KLF4 mRNA using siRNA and are magnified by overexpressing KLF4. Using KLF4 reporter constructs, we verified that the elevated expression induced by EGCG was mediated by increasing levels of activated MEF2A, which bound to the promoter region of KLF4. Taken together, this is the first time that EGCG is reported to increase the expression of KLF4, suggesting a novel mechanisms in gastric cancer treatment.
Keywords: Kruppel-like factor 4 (KLF4), Epigallocatechin-3-gallate (EGCG), Myocyte enhancer factor-2A (MEF2A), Gastric cancer, NCI-N87 cells
Introduction
Green tea made from the leaves of Camellia sinensis is one of the most popular beverages around the world and has been proven to have many important biological and pharmacological activities. Epigallocatechin-3-gallate (EGCG) is the major polyphenol present in green tea (Kurahashi et al. 2008); as a powerful antioxidant and free ion scavenger, it has demonstrated inhibitory effects on different stages of tumorigenesis in both in vitro and in vivo studies (Zhang et al. 2000a; Yang 1997). Additionally, researchers have also found that EGCG can modulate many biochemical and cellular processes that are involved in cancer progression. However, the mechanisms of how EGCG affects tumorigenesis of gastric cancer are still poorly understood.
Kruppel-like factor 4 (KLF4), a member of the mammalian KLF family, is a DNA-binding transcription factor that has been reported to regulate a variety of cellular processes including proliferation, differentiation, apoptosis, survival, motility, and morphogenesis (McConnell and Yang 2010). Previous studies have demonstrated that KLF4 shows oncogenic or tumor-suppressive functions depending on the tissue in which it is expressed (Rowland and Peeper 2006). A recent study revealed that the expression of KLF4 was significantly reduced in gastric cancer tissues compared with expression in normal tissues (Zhang et al. 2012). Furthermore, genetic and epigenetic analysis has indicated that a GGG→AGG (Gly→Arg) mutation at codon 107 in the third exon of the KLF4 gene, encoding a transcriptional activation domain of the protein, can occur in diffuse-type advanced gastric adenocarcinoma (Cho et al. 2007). Although these findings indicate an association between the expression of KLF4 and tumorigenesis of gastric cancer, the role of KLF4 in gastric cancer is still not fully understood.
Considering the previous findings of EGCG and KLF4, we therefore proposed the hypothesis that EGCG exerts its anti-tumorigenic function in gastric cancer through inducing the expression of KLF4. In order to test this hypothesis, we treated gastric cancer cells with EGCG and then examined the expression of KLF4. We found that treatment with EGCG could induce high KLF4 expression and slow the proliferation of gastric cancer cells. Additionally, this induction of KLF4 expression by EGCG treatment was mediated by the activation of myocyte enhancer factor-2A (MEF2A). Taken together, our results indicate a new mechanism of action for EGCG in suppressing the tumorigenesis of gastric cancer.
Materials and methods
Cell culture, treatment, and transfection
NCI-N87 cell line was purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were cultured in RPMI-1640 medium, supplemented with 10 % fetal bovine serum and antibiotics (100 μg/mL penicillin and 100 U/mL streptomycin) in a humid incubator with 5 % CO2 at 37 °C. Human KLF4 lentiviral vectors were acquired from Qiagen (Valencia, CA, USA). HEK 293T cells were used to package lentiviruses. Virus-containing supernatants were titrated on NCI-N87 cells as described previously (Dekker et al. 2005). Interference RNA (siRNA) for MEF2A and scrambled RNA (negative control) were obtained from Invitrogen (Carlsbad, CA, USA). KLF4-specific siRNA and the negative control were obtained from Dharmacon (Lafayette, CO, USA). Target siRNAs were transfected into NCI-N87 cells using Lipofectamine RNAiMAX (Invitrogen). EGCG was used to treat NCI-N87 cells for various periods of time with various doses as follows: to detect cell proliferation in response to EGCG in a dose-dependent manner, cells were treated with EGCG for 48 h at concentrations of 25, 50, and 100 μM; to detect cell proliferation in response to EGCG in a time-dependent manner, cells were treated with 50 μM EGCG for 24, 48, and 72 h; to detect alterations of KLF4 expression and cell cycle protein expression, cells were treated with EGCG for 48 h at concentrations of 25, 50, and 100 μM.
HepG2 cells were also obtained from the ATCC and grown in Dulbecco's Modified Eagle's medium.
Cell proliferation
Cell proliferation was assessed using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay. Following the completion of each experiment, cells were incubated with MTT (Sigma, St Louis, MO, USA) at a final concentration of 1.0 mg/mL for 4 h at 37 °C. The formed formazan crystal was solubilized in DMSO and then photometrically quantified at 570 nm using a microplate reader (He et al. 2006).
Fluorescence activated cell sorting analysis
NCI-N87 cells were collected after 48 h of treatment with or without various concentrations of EGCG. After being fixed in 70 % ethanol and washed three times with PBS, cells were stained with a 50 μg/mL propidium iodide (PI) solution containing 0.5 % Triton X-100, 0.1 mM EDTA, and 25 μg/ml RNase A. Fluorescence was measured and analyzed using a fluorescence activated cell sorting Calibur Flow Cytometer (Becton Dickinson Immunocytometry Systems, Palo Alto, CA, USA).
Quantitative real-time polymerase chain reaction
Following the completion of each respective experiment, total RNA was isolated from NCI-N87 cells using Trizol reagent (Invitrogen) according to the manufacturer's protocol. Two micrograms of total RNA was then used to synthesize cDNA. Quantitative real-time polymerase chain reaction (qRT-PCR) was performed using SYBR Green qPCR Master Mix (Roche AG, Basel, Switzerland). The following primer pairs of human origin were used: KLF4: forward, 5′-CAAGTCCCGCCGCTCCATTACCAA-3′ and reverse, 5′-CCACAGCCGTCCCAGTCACAGTGG-3′ (reverse); CDK2: forward, 5′-CATTCCTCTTCCCCTCATCA-3′ and reverse, 5′-CATCCTGGAAGAAAGGGTGA-3′; cyclin A: forward, 5′-GTTCCTTACCCAGTACTTCC-3′ and reverse, 5′-AGACAAGGCTTAAGACTCTC-3′; p21: forward, 5′-AAGACCATGTGGACCTGTCACTGT-3′ and reverse, 5′-GAAGATCAGCCGGCGTTTG -3′; cyclin E1: forward, 5′-TTTAC C CAAACTCAAC GTGCAAGC C-3′ and reverse, 5′TTCACACACCTCCATTAACCAATCC-3′; CDK6: forward, 5′-TCAGATGTTGATCAGCTAGGAAAAA-3′, reverse, 5′-TCATTAGGCCAGTCCTCTTCTTCT-3′; cyclin D1: forward, 5′-CCGTCCATGCGGAAGATC-3′; reverse, 5′-ATGGCC AGCGGGAAGAC-3′; CDK4: forward, 5′- TGGAGCGTTGGCTGTATCTTT-3′; reverse, 5′- TGGAGG CAATCCAATGAGATC-3′; and GAPDH: forward, 5′-CCACATCGCTCAGACACCAT-3′ and reverse, 5′-CCAGGCCCAATACG-3′.
Western blot analysis
Protein lysates were made by cell lysis buffer (Cell Signaling Technology, Beverly, MA, USA) and then separated using a 10 % SDS-PAGE gel followed by being transferred onto high-quality polyvinylidene difluoride membrane (Invitrogen). After blocking for 2 h at room temperature using 10 % nonfat dry milk in TBST buffer, membranes were sequentially incubated with primary antibodies for 3 h and horse radish peroxidase-conjugated secondary antibodies for 2 h at RT. Blots were developed by the enhanced chemiluminescence technique (Santa Cruz Biotechnology, Santa Cruz, CA USA) according to the manufacturer's instruction (Sheng et al. 2012). The following antibodies were used in this study: anti-cyclin D1, cyclin E, CDK2, CDK4, CDK6, and p21 (Cell Signaling Technology); anti-phosphorylated MEF2A (Thr312) and anti-KLF4, cyclin A (Abcam, Cambridge, MA, USA); and anti-MEF2A and anti-β-actin (Santa Cruz Biotechnology).
TUNEL assay and DAPI staining
Cells were plated on Lab-Tek® Chamber Slides (Nalgene Nunc, Naperville, IL, USA) and incubated in the indicated medium. After 1 day, the cells were exposed to 100 μM EGCG for a further 48 h. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay was assessed using a commercially available kit (Promega, Madison, WI, USA) to verify the process of apoptosis following the given protocol. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI).
Luciferase assay
The 800-bp region of the KLF4 promoter was generated by PCR and cloned into the pGL3.0 basic vector (Promega). MEF2A plasmid was obtained from E. N. Olson (University of Texas M.D. Anderson Cancer Center). NCI-N87 cells were plated at a density of 5 × 104/well in 12-well plates 1 day before transfection. Transient transfection was performed using Lipofectamine 2000 (Invitrogen) according to instructions by the manufacturer. Briefly, a total of 1 to 2 μg of plasmid DNA was used in transfections, and total DNA was always kept constant. Cotransfection of β-galactosidase (-Gal) was used to calculate transfection efficiencies. Twenty-four hours after transfection, cells were harvested and assayed for luciferase activity.
Statistics
All data are presented as mean ± standard deviation. Statistical significance was determined using one-way analysis of variance followed by Tukey's post hoc test. Differences between means were considered significant at p < 0.05.
Results
KLF4 has been implicated as a central regulator of proliferation and apoptosis in various cell types, and has been implicated as being dysregulated in certain types of cancer. It is unknown whether EGCG is capable of regulating the expression of KLF4 in gastric cancer cells. Therefore, we investigated the expression patterns of KLF4 in human NCI-N87 cells after treatment with EGCG. Our experimental data demonstrated that EGCG increases the expression level of KLF4 in a dose-dependent manner from 0 to 100 μM at both the mRNA and protein levels (Fig. 1a, b).
Fig. 1.

EGCG increases mRNA and protein expression of KLF4. NCI-N87 cells were treated with various concentrations of EGCG for 48 h and a qRT-PCR analysis revealed that mRNA levels of KLF4 increased in response to EGCG treatment in a dose-dependent manner; b western blot analysis revealed that protein levels of KLF4 increased in response to EGCG treatment in a dose-dependent manner (*p < 0.01 vs. non-treated control, n = 4)
It has been reported that KLF4 regulates the expression of cell cycle proteins such as cyclins and CDKs, and thus we investigated the effects of EGCG on the target genes of KLF4. As shown in Fig. 2, qRT-PCR analysis revealed that the level of CDK inhibitor p21 in NCI-N87 cells was upregulated by treatment with EGCG in a concentration-dependent manner, using doses between 0 and 100 μM EGCG. EGCG treatment reduced the expressions of cyclin D and CDK4 at both mRNA levels (Fig. 3a) and protein levels (Fig. 3b) under the same conditions. However, the mRNA and protein levels of cyclin A, cyclin E, CDK2, and CDK6 were not affected by EGCG treatment (Fig. 3a, b). These results suggest that EGCG might arrest cell cycle progression in G0/G1 phase through the accumulation of p21 and inhibiting cyclin D1 and CDK4 expression. In order to verify this hypothesis, we further examined the effects of EGCG on cell proliferation and cell cycle. MTT assay was used to test the effects of EGCG on the proliferation of human NCI-N87 cells. As shown in Fig. 4a, MTT results demonstrated that EGCG reduced cell proliferation in a dose-dependent manner. After that, we incubated NCI-N87 cells with 50 μM EGCG and measured the antiproliferative effect of EGCG on NCI-N87 cells in a time-dependent manner. As shown in MTT assay, a time-dependent reduction of cell viability was observed compared with the untreated cells (Fig. 4b). All these data suggest that EGCG exerts growth inhibitory effect in NCI-N87 cells.
Fig. 2.

EGCG regulates expression of p21, a cell cycle functional protein downstream of KLF4. Real-time PCR result revealed that EGCG increased the expression of p21 at mRNA level (*p < 0.01 vs. non-treated control, n = 4)
Fig. 3.

EGCG regulates expression of cyclins and CDKs, two genes that are downstream targets of KLF4. a qRT-PCR analysis revealed that EGCG reduced the expression of cyclin D and CDK 4 at mRNA level (*p < 0.01 vs. non-treated control, n = 4). b Western blot analysis revealed that EGCG reduced the expression of cyclin D and CDK 4 at protein level (*p < 0.01 vs. non-treated control, n = 3)
Fig. 4.

EGCG inhibits NCI-N87 cell proliferation. a NCI-N87 cells were treated with various concentrations of EGCG for 48 h and then tested by MTT assay; b NCI-N87 cells were treated with EGCG (50 μM) for 12 to 72 h and then tested by MTT assay (∆p < 0.05; #p < 0.01 vs. non-treated control, n = 4)
KLF4 has also been shown to regulate apoptosis in multiple cell lines. In order to determine whether EGCG can have a direct effect upon apoptosis, TUNEL staining was used to detect apoptosis following EGCG treatment.
As shown in Fig. 5a, 100 μM EGCG significantly promoted apoptosis in NCI-N87 cells. Caspase 3 is a critical executioner of apoptosis. Our result indicated that administration of EGCG activated caspase 3 (Fig. 5b). To determine the effects of EGCG on cell cycle progression, NCI-N87 cells were treated with 0–100 μM EGCG for 48 h and subjected to flow cytometric analysis. Our data showed that EGCG reduced the number of cells in the S phase and increased the number of cells in the G0/G1 phase in a dose-dependent manner (p < 0.05) (Fig. 6). These results confirm that the cell cycle of NCI-N87 cells was indeed arrested in the G0/G1 phase by EGCG, which is in agreement with findings that EGCG could inhibit tumorigenesis (Chen et al. 2011). Taken together, these findings suggested a potential role of KLF4 in slowing cell proliferation of NCI-N87 induced by EGCG.
Fig. 5.
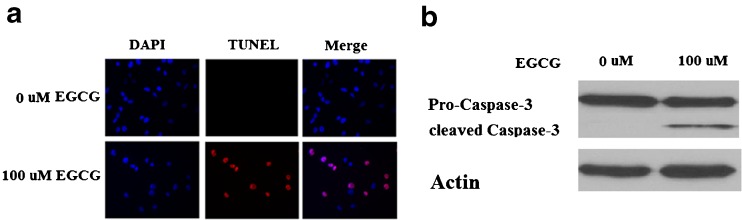
EGCG induces NCI-N87 cell apoptosis. a NCI-N87 cells were treated with EGCG for 48 h and stained using a TUNEL Assay Kit. Nuclear DNA was stained with DAPI; b western blot analysis revealed that EGCG activated caspase 3
Fig. 6.

EGCG arrests NCI-N87 cells in G0/G1 phase. After treatment with EGCG at the indicated concentration for 48 h, cell cycle was detected by flow cytometric analysis (∆p < 0.05 vs. non-treated control, n = 4)
As EGCG could induce the expression of KLF4 in NCI-N87 cells, induce apoptosis, and arrest cells in the G0/G1 phase, we evaluated whether EGCG was mediating its antiproliferative effects directly through KLF4. In order to do so, we used small interfering RNAs specific for KLF4 to knock down KLF4 expression and determine if EGCG could still mediate its effects in NCI-N87 cells. As shown in Fig. 7a, the successful knockdown of KLF4 was verified by western blot analysis. MTT assay revealed that inhibition of KLF4 by siRNA abolished the effects of treatment with EGCG, and cells grew at a rate comparable to cells that were not treated with EGCG (Fig. 7b). Accordingly, the increased expression of p21 and reduced expression of cyclin D1 and CDK4 induced by EGCG treatment were reversed by siRNA inhibition of KLF4 (Fig. 7c).
Fig. 7.

Knockdown of KLF4 eliminates the EGCG-induced growth inhibition of NCI-N87 cells. siKLF4 indicates the KLF4 siRNA group; NS indicates the nonspecific siRNA group. a Western blot analysis revealed the successful knockdown of KLF4; b cells were transfected with KLF4 siRNA, followed by incubation with 50 μM EGCG for 48 h; MTT assay revealed that knockdown of KLF4 could reduce the slowed cell growth caused by treatment with EGCG (*p < 0.01 vs. NS group; #p < 0.01 vs. NS + EGCG group); c western blot analysis revealed that knockdown of KLF4 reversed the alternations of p21, cycin D, and CDK4 expression induced by EGCG (*p < 0.01 vs. NS group; #p < 0.01 vs. NS + (EGCG) group)
Because knockdown of KLF4 reduced the effects of EGCG, we hypothesized that overexpression of KLF4 might amplify the effects of EGCG. To test this, we used a lentiviral plasmid to express KLF4 in NCI-N87 cells and then tested cell proliferation following incubation with EGCG. As shown in Fig. 8a, the successful overexpression of KLF4 was verified by western blot analysis. Our results indicated that overexpression of KLF4 magnified the inhibition of cell proliferation induced by EGCG (Fig. 8b). Importantly, overexpression of KLF4 magnified the effects of EGCG via the increased expression of p21 and reduced expression of cyclin D1 and CDK4 (Fig. 8c).
Fig. 8.

Overexpression of KLF4 magnifies EGCG-induced growth inhibition of NCI-N87 cells. NEO indicates null control group; KLF4 indicates KLF4 overexpression group. a Western blot analysis revealed the successful overexpression of KLF4; b transiently transfected NCI-N87 cells with mock or full-length human KLF4 were incubated with 50 μM EGCG for 48 h; MTT assay revealed that overexpression of KLF4 could further the slowed cell growth induced by treatment with EGCG (*p < 0.01 vs. NEO group; #p < 0.01 vs. NEO + EGCG group); c western blot analysis revealed that overexpression of KLF4 promoted the effect of EGCG treatment upon the expression of p21, cycin D, and CDK4 (*p < 0.01 vs. NEO group; #p < 0.01 vs. NEO + EGCG group)
Recent studies demonstrated that MEF2A is critical for the resveratrol-mediated induction of KLF2 (Villarreal et al. 2010; Gracia-Sancho et al. 2010). We investigated the involvement of MEF2A in the induction of KLF4 by EGCG. The phosphorylation of MEF2A at Thr312 plays an important role in the transcriptional activity of MEF2A. As shown in Fig. 9, treatment with 50 μM EGCG for 48 h increased the levels of MEF2A phosphorylated at Thr312. NCI-N87 cells were transfected with siRNA specific for MEF2A or negative control siRNA and subsequently treated with EGCG. As shown in Fig. 10a, qRT-PCR results demonstrated that reduction of MEF2A using siRNA significantly blocked the upregulation of KLF4 by EGCG. This finding was confirmed at the protein level by western blot analysis (Fig. 10b).
Fig. 9.

Western blot analysis revealed that phosphorylated levels of MEF2A significantly increased after treatment with 50 μM EGCG for 48 h (*p < 0.01 vs. non-treated control, n = 4)
Fig. 10.

MEF2A participates in EGCG-induced KLF4 expression. a NCI-N87 cells were transfected with either MEF2A siRNA or nonspecific siRNA and subsequently treated with EGCG. qRT-PCR analysis revealed that knockdown of MEF2A significantly blocked the upregulation of KLF4 by EGCG (*p < 0.01 vs. NS + untreated group; #p < 0.01 vs. NS + EGCG group); b NCI-N87 cells were transfected with either MEF2A siRNA or nonspecific siRNA and subsequently treated with EGCG. Western blot analysis revealed that knockdown of MEF2A significantly blocked the upregulation of KLF4 by EGCG (*p < 0.01 vs. NS + non-treated group; #p < 0.01 vs. NS + EGCG group)
To further evaluate the relationship between MEF2A and KLF4, we assessed the effects of MEF2A on KLF4 promoter activity. As shown in Fig. 11, MEF2A induced KLF4 promoter (800 bp-Luc) activity in a dose-dependent manner.
Fig. 11.

EGCG and MEF2A increase KLF4 production via the KLF4 promoter. a NCI-N87 cells were transfected transiently with a reporter driven by KLF4 promoter, and then cells were treated by 50 μM EGCG for 24 h (*p < 0.01 vs. control); b NCI-N87 cells were cotransfected transiently with an expression plasmid of MEF2A (0.5 or 1.0 μg) and a reporter driven by KLF4 promoter (*p < 0.01 vs. control)
Finally, in order to verify the effects of EGCG on KLF4 expression in different cancer cell lines, another cell line, Hep G2, was used in this study. As shown in Fig. 12, experimental data demonstrated that EGCG increased the expression level of KLF4 at both the mRNA level and protein level in Hep G2 cells. These results indicate that the effects of EGCG on KLF4 expression are not isolated to the NCI-N87 on cell type, suggesting that this compound has the potential for broader anticancer application.
Fig. 12.

EGCG elevated the expression of KLF4. Hep G2 cells were treated by 50 μM EGCG for 48 h and a qRT-PCR analysis revealed that mRNA levels of KLF4 increased in response to EGCG treatment (*p < 0.01 vs. untreated control, n = 4); b western blot analysis revealed that protein levels of KLF4 increased in response to EGCG treatment (*p < 0.01 vs. untreated control, n = 4)
Discussion
In the present study, we have demonstrated that EGCG treatment increases the expression of KLF4 in NCI-N87 cells in a dose-dependent manner, thereby increasing expression of p21 and reducing expression of cyclin D and CDK4. Further study indicated that EGCG could inhibit the growth of NCI-N87 cells in a time- and dose-dependent manner by arresting the cells in the G0/G1 phase. To our knowledge, we are the first to report that KLF4 expression is regulated by EGCG. Importantly, reducing KLF4 mRNA transcript by the use of siRNA could abolish the EGCG-induced cell growth arrest, while overexpression of KLF4 by lentiviral constructs magnified the EGCG-induced cell growth arrest. These data strongly point to a role for KLF4 in mediating EGCG-induced arrest of NCI-N87 cell proliferation. Furthermore, these findings are consistent with studies indicating that KLF4 can block the transition from the G1 to S phase in cell cycle, slowing cell growth through the repression of several positive cell cycle regulatory gene promoters, including cyclin D1 and ornithine decarboxylase (Shie et al. 2000; Chen et al. 2000). We also demonstrated that activation of MEF2A at Thr312 is a critical upstream component of EGCG-induced KLF4 expression.
EGCG, the most bioactive polyphenol in green tea, has been reported to be a broadly active chemopreventative anticancer agent in several animal models of cancer (Golden et al. 2009), but the underlying mechanisms have remained elusive. The studies presented here provide novel insights into the regulatory mechanisms of EGCG's anticancer characteristics through its induction of KLF4 expression. KLF4 is a transcription factor expressed in a wide variety of tissues in humans (Chiambaretta et al. 2004). Increasing evidence implicates KLF4 as a tumor suppressor in the gastric epithelium and this is supported by studies demonstrating that KLF4 is downregulated in gastric cancer, with evidence of loss-of-heterozygosity and hypermethylation (Wei et al. 2005). The loss of KLF4 might contribute to gastric cancer progression (Kanai et al. 2006). These studies form the basis of the hypothesis that induction of KLF4 expression might be a potential therapy in gastric cancer treatment. Consistent with its antiproliferative role, KLF4 exerts its roles in cell cycle arrest through regulating the expression of cell cycle proteins. It was reported that KLF4 could simultaneously induce the expression of cyclin-dependent kinase inhibitor proteins p21Cip1/WAF (Zhang et al. 2000b) and p57Kip2 (Chen et al. 2003), and repress the expression of cyclin D1 (Shie et al. 1999), cyclin D2 (Klaewsongkram et al. 2007), cyclin E (Yoon et al. 2005), and cyclin B1 (Yoon and Yang 2004), depending on the types of tissues. The ability of KLF4 to regulate these cell cycle proteins explains why we observe EGCG arresting NCI-N87 cells in the G0/G1 phase.
The mechanisms underlying transcriptional control and expression of KLF4 are complex. The precise mechanism of how various stimuli increase expression of KLF4 has been unclear (Evans and Liu 2008), but the possibilities include increased transcription of the KLF4 gene, increased mRNA stability, and/or increased protein stability. In the present study, we demonstrated that EGCG increases the expression of KLF4 at both the mRNA and protein levels, suggesting that the induction of KLF4 is at a transcriptional level. Moreover, this is the first time that EGCG is reported to activate MEF2A, which was confirmed to mediate the induction of KLF4. Until now, the role of MEF2A in gastric cancer has been unclear: MEF2A could regulate genes that control critical cellular processes including proliferation, differentiation, and survival (Heidenreich and Linseman 2004). Upstream signaling pathway for MEF2A activation is complex and further study will be needed to clarify the underlying mechanisms through which EGCG potentiates MEF2A activity. Mass spectrometric analysis experiment displayed that MEF2A has multiple p38MAPK phosphoacceptor sites (Cox et al. 2003). It was reported that p38MAPK critically interacts with and activates MEF2A (de Angelis et al. 2005). In addition, EGCG was demonstrated to significantly induce the ROS generation as well as p38 activation in Ishikawa cells, which appeared to be a critical mediator in EGCG-induced apoptosis (Manohar et al. 2013). Thus, we speculated that EGCG might promote MEF2A activity via activating p38MAPK. MEF2A is also a target of class II HDACs (Calalb et al. 2009). However, the effect of EGCG on class II HDACs is not known. Further study is needed to clarify the underlying mechanism.
In addition to development, proliferation, and apoptosis, KLF4 also play an important role in regulating cell differentiation. Previous studies have reported that KLF4 could regulate monocyte–macrophage differentiation and development (Feinberg et al. 2007). Importantly, KLF4 is highly expressed in epithelial tissues including the gastric and intestinal epithelium. Zebrafish Klf4a was demonstrated to be essential for the differentiation of different intestinal cell lineages (Li et al. 2011). KLF4 is required for proper goblet cell differentiation and maturation. Colonic goblet cells in KLF knockout mice do not show the normal goblet cell morphology, and the goblet cell marker MUC2 exhibits patchy expression throughout the colonic epithelium and is not confined to cells with “goblet-like” morphology (McConnell et al. 2007). Thus, we speculated that the increased expression of KLF4 by EGCG might affect the differentiation of normal gastric and intestinal epithelial cells, although we did not find that EGCG could induce differentiation of NCI-N87 cells.
In summary, our study has revealed a therapeutic effect of EGCG in gastric cancer and explored the underlying mechanism of how EGCG mediates this effect in cancer cell lines. To summarize based on our observations, we concluded that EGCG treatment in NCI-N87 cells activated MEF2A, leading to the increased expression of KLF4, which ultimately resulted in the alteration of cell cycle protein expression. The alteration of cell cycle protein expression arrested these cells in the G0/G1 phase and slowed cell proliferation. Importantly, these data provide further insight into how EGCG from green tea acts as an anticancer therapy and provide additional support for this compound to be considered in the treatment of cancer.
Footnotes
Yuwen Ma and Youkui Shi are the co-first authors.
References
- Calalb MB, McKinsey TA, Newkirk S, Huynh K, Sucharov CC, Bristow MR. Increased phosphorylation-dependent nuclear export of class II histone deacetylases in failing human heart. Clin Transl Sci. 2009;2(5):325–332. doi: 10.1111/j.1752-8062.2009.00141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZY, Shie J, Tseng C. Up-regulation of gut-enriched 396 Krüppel-like factor by interferon-gamma in human colon carcinoma cells. FEBS Lett. 2000;477:67–72. doi: 10.1016/S0014-5793(00)01764-6. [DOI] [PubMed] [Google Scholar]
- Chen X, Whitney EM, Gao SY, Yang VW. Transcriptional profiling of Kruppel-like factor 4 reveals a function in cell cycle regulation and epithelial differentiation. J Mol Biol. 2003;326:665–677. doi: 10.1016/S0022-2836(02)01449-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Wan SB, Yang H, Yuan J, Chan TH, Dou QP. EGCG, green tea polyphenols and their synthetic analogs and prodrugs for human cancer prevention and treatment. Adv Clin Chem. 2011;53:155–177. doi: 10.1016/B978-0-12-385855-9.00007-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiambaretta F, De Graeve F, Turet G, Marceau G, Gain P, Dastugue B, Rigal D, Sapin V. Cell and tissue specific expression of human Kruppel-like transcription factors in human ocular surface. Mol Vis. 2004;10:901–909. [PubMed] [Google Scholar]
- Cho YG, Song JH, Kim CJ, Nam SW, Yoo NJ, Lee JY, Park WS. Genetic and epigenetic analysis of the KLF4 gene in gastric cancer. APMIS. 2007;115:802–808. doi: 10.1111/j.1600-0463.2007.apm_643.x. [DOI] [PubMed] [Google Scholar]
- Cox DM, Du M, Marback M, Yang EC, Chan J, Siu KW, et al. Phosphorylation motifs regulating the stability and function of myocyte enhancer factor 2A. J Biol Chem. 2003;278:15297–15303. doi: 10.1074/jbc.M211312200. [DOI] [PubMed] [Google Scholar]
- de Angelis L, Zhao J, Andreucci JJ, Olson EN, Cossu G, McDermott JC. Regulation of vertebrate myotome development by the p38 MAP kinase-MEF2 signaling pathway. Dev Biol. 2005;283:171–179. doi: 10.1016/j.ydbio.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Dekker RJ, van Thienen JV, Rohlena J, de Jager SC, Elderkamp YW, Seppen J, de Vries CJ, Biessen EA, van Berkel TJ, Pannekoek H, Horrevoets AJ. Endothelial KLF2 links local arterial shear stress levels to the expression of vascular tone-regulating genes. Am J Pathol. 2005;167:609–618. doi: 10.1016/S0002-9440(10)63002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans PM, Liu C. Roles of Krüpel-like factor 4 in normal homeostasis, cancer and stem cells. Acta Biochim Biophys Sin (Shanghai) 2008;40:554–564. doi: 10.1111/j.1745-7270.2008.00439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg MW, Wara AK, Cao Z, et al. The Kruppel-like factor KLF4 is a critical regulator of monocyte differentiation. EMBO J. 2007;26:4138–4148. doi: 10.1038/sj.emboj.7601824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golden EB, Lam PY, Kardosh A, Gaffney KJ, Cadenas E, Louie SG, Petasis NA, Chen T, Schönthal AH. Green tea polyphenols block the anticancer effects of bortezomib and other boronic acid-based proteasome inhibitors. Blood. 2009;113:5927–5937. doi: 10.1182/blood-2008-07-171389. [DOI] [PubMed] [Google Scholar]
- Gracia-Sancho J, Villarreal G, Jr, Zhang Y, Garcia-Cardena G. Activation of Sirt1 by resveratrol induces Klf2 expression conferring an endothelial vasoprotective phenotype. Cardiovasc Res. 2010;85:514–519. doi: 10.1093/cvr/cvp337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Zhou H, Tang H, Luo Y. Deficiency of disulfide bonds facilitating fibrillogenesis of endostatin. J Biol Chem. 2006;414:1048–1057. doi: 10.1074/jbc.M507745200. [DOI] [PubMed] [Google Scholar]
- Heidenreich KA, Linseman DA. Myocyte enhancer factor-2 transcription factors in neuronal differentiation and survival. Mol Neurobiol. 2004;29:155–166. doi: 10.1385/MN:29:2:155. [DOI] [PubMed] [Google Scholar]
- Kanai M, Wei D, Li Q, Jia Z, Ajani J, Le X, Yao J, Xie K. Loss of Kruppel-like factor 4 expression contributes to Sp1 overexpression and human gastric cancer development and progression. Clin Cancer Res. 2006;12:6395–6402. doi: 10.1158/1078-0432.CCR-06-1034. [DOI] [PubMed] [Google Scholar]
- Klaewsongkram J, Yang Y, Golech S, Katz J, Kaestner KH, Weng NP. Kruppel-like factor 4 regulates B cell number and activation-induced B cell proliferation. J Immunol. 2007;179:4679–4684. doi: 10.4049/jimmunol.179.7.4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurahashi N, Sasazuki S, Iwasaki M, Inoue M, Tsugane S, JPHC Study Group Green tea consumption and prostate cancer risk in Japanese men: a prospective study. Am J Epidemiol. 2008;167:71–77. doi: 10.1093/aje/kwm249. [DOI] [PubMed] [Google Scholar]
- Li IC, Chan CT, Lu YF, et al. Zebrafish Kruppel-like factor 4a represses intestinal cell proliferation and promotes differentiation of intestinal cell lineages. PLoS One. 2011;6:e20974. doi: 10.1371/journal.pone.0020974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manohar M, Fatima I, Saxena R, Chandra V, Sankhwar PL, Dwivedi A. (−)-Epigallocatechin-3-gallate induces apoptosis in human endometrial adenocarcinoma cells via ROS generation and p38 MAP kinase activation. J Nutr Biochem. 2013;24:940–947. doi: 10.1016/j.jnutbio.2012.06.013. [DOI] [PubMed] [Google Scholar]
- McConnell BB, Yang VW. Mammalian Kruppel-like factors in health and diseases. Physiol Rev. 2010;90:1337–1381. doi: 10.1152/physrev.00058.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell BB, Ghaleb AM, Nandan MO, Yang VW. The diverse functions of Krüppel-like factors 4 and 5 in epithelial biology and pathobiology. Bioessays. 2007;29:549–557. doi: 10.1002/bies.20581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland BD, Peeper DS. KLF4, p21 and context-dependent opposing forces in cancer. Nat Rev Cancer. 2006;6:11–23. doi: 10.1038/nrc1780. [DOI] [PubMed] [Google Scholar]
- Sheng B, Wang X, Su B, Lee HG, Casadesus G, Perry G, Zhu X. Impaired mitochondrial biogenesis contributes to mitchondrial dysfunction in Alzheimer's disease. J Neurochem. 2012;120:419–429. doi: 10.1111/j.1471-4159.2011.07581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shie JL, Pestell RG, TC C. Repression of the cyclin D1 promoter by gut-enriched Kruppel-like factor. Gastroenterology. 1999;11:A520. [Google Scholar]
- Shie JL, Chen ZY, Fu M, Pestell RG, Tseng CC. Gut-enriched Krüppel-like factor represses cyclin D1 promoter activity through Sp1 motif. Nucleic Acids Res. 2000;28:2969–2976. doi: 10.1093/nar/28.15.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarreal G, Jr, Zhang Y, Larman HB, Gracia-Sancho J, Koo A, García-Cardeña G. Defining the regulation of KLF4 expression and its downstream transcriptional targets in vascular endothelial cells. Chem Biophys Res Commun. 2010;391:984–989. doi: 10.1016/j.bbrc.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei D, Gong W, Kanai M, Schlunk C, Wang L, Yao JC, Wu TT, Huang S, Xie K. Drastic down-regulation of Kruppel-like factor 4 expression is critical in human gastric cancer development and progression. Cancer Res. 2005;65:2746–2754. doi: 10.1158/0008-5472.CAN-04-3619. [DOI] [PubMed] [Google Scholar]
- Yang CS. Inhibition of carcinogenesis by tea. Nature. 1997;389:134–135. doi: 10.1038/38154. [DOI] [PubMed] [Google Scholar]
- Yoon HS, Yang VW. Requirement of Kruppel-like factor 4 in preventing entry into mitosis following DNA damage. J Biol Chem. 2004;279:5035–5041. doi: 10.1074/jbc.M307631200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon HS, Ghaleb AM, Nandan MO, Hisamuddin IM, Dalton WB, Yang VW. Kruppel-like factor 4 prevents centrosome amplification following gamma-irradiation-induced DNA damage. Oncogene. 2005;24:4017–4025. doi: 10.1038/sj.onc.1208576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Miura Y, Yagasaki K. Suppression of adhesion and invasion of hepatoma cells in culture by tea compounds through antioxidative activity. Cancer Lett. 2000;159:169–173. doi: 10.1016/S0304-3835(00)00545-0. [DOI] [PubMed] [Google Scholar]
- Zhang W, Geiman DE, Shields JM, Dang DT, Mahatan CS, Kaestner KH, Biggs JR, Kraft AS, Yang VW. The gut-enriched Kruppel-like factor (Kruppel-like factor 4) mediates the transactivating effect of p53 on the p21WAF1/Cip1 promoter. J Biol Chem. 2000;275:18391–18398. doi: 10.1074/jbc.C000062200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Zhang J, Wang ZW, Zha L, Huang Z. Altered expression of Krüppel-like factor 4 and β-catenin in human gastric cancer. Oncol Lett. 2012;3:1017–1022. doi: 10.3892/ol.2012.619. [DOI] [PMC free article] [PubMed] [Google Scholar]