

Abstract
Substantial reductions in hepatitis C virus (HCV) prevalence among people who inject drugs (PWID) cannot be achieved by harm reduction interventions such as needle exchange and opiate substitution therapy (OST) alone. Current HCV treatment is arduous and uptake is low, but new highly effective and tolerable interferon-free direct-acting antiviral (DAA) treatments could facilitate increased uptake. We projected the potential impact of DAA treatments on PWID HCV prevalence in three settings. A dynamic HCV transmission model was parameterized to three chronic HCV prevalence settings: Edinburgh, UK (25%); Melbourne, Australia (50%); and Vancouver, Canada (65%). Using realistic scenarios of future DAAs (90% sustained viral response, 12 weeks duration, available 2015), we projected the treatment rates required to reduce chronic HCV prevalence by half or three-quarters within 15 years. Current HCV treatment rates may have a minimal impact on prevalence in Melbourne and Vancouver (<2% relative reductions) but could reduce prevalence by 26% in 15 years in Edinburgh. Prevalence could halve within 15 years with treatment scale-up to 15, 40, or 76 per 1,000 PWID annually in Edinburgh, Melbourne, or Vancouver, respectively (2-, 13-, and 15-fold increases, respectively). Scale-up to 22, 54, or 98 per 1,000 PWID annually could reduce prevalence by three-quarters within 15 years. Less impact occurs with delayed scale-up, higher baseline prevalence, or shorter average injecting duration. Results are insensitive to risk heterogeneity or restricting treatment to PWID on OST. At existing HCV drug costs, halving chronic prevalence would require annual treatment budgets of US $3.2 million in Edinburgh and approximately $50 million in Melbourne and Vancouver. Conclusion: Interferon-free DAAs could enable increased HCV treatment uptake among PWID, which could have a major preventative impact. However, treatment costs may limit scale-up, and should be addressed. (Hepatology 2013;58:1598–1609)
The global burden of hepatitis C virus (HCV) infection continues to rise.1,2 The core of the HCV epidemic in the developed world occurs among people who inject drugs (PWID), who comprise the majority of new (80%) and existing (60%) cases.1 Globally, HCV seroprevalence (>60% in most countries)3 and incidence (5%-40% annually)4,5 remains high among PWID. Prevention strategies, such as needle and syringe programs (NSP) and opiate substitution therapy (OST), can reduce HCV transmission and have maintained low levels of human immunodeficiency virus (HIV) infection in many settings, but they are insufficient to achieve substantial reductions in HCV prevalence.6–9 This is partly because high HCV prevalence and long injecting duration among PWID in many settings combine such that the intervention coverage required for major prevalence reductions is unobtainable and unsustainable.9 Given that there is no HCV vaccine, alternative strategies for HCV prevention are urgently needed.
In HIV, the demonstration that antiretroviral therapy given to HIV-infected individuals can prevent secondary transmission has generated considerable excitement10 and suggests that we may have reached a tipping point for preventing HIV transmission.11 In contrast to HIV, HCV is curable and therapy is finite. Therefore, HCV treatment as prevention may provide even greater opportunity for preventing onward HCV transmission and directly reducing HCV chronic prevalence.
Mathematical modeling studies have suggested HCV treatment for PWID could be an effective12–16 and cost-effective17 intervention to prevent HCV transmission. However, these studies only considered treatment with pegylated interferon (PEG-IFN) and ribavirin (RBV). The feasibility of expanding this treatment regimen as a strategy for treatment as prevention is limited, given the poor tolerability and limited uptake of PEG-IFN+RBV therapy, particularly among PWID.18,19 However, therapeutic options for HCV are evolving rapidly. Preliminary data from IFN-free direct-acting antiviral (DAA) therapy phase 2 trials indicates that in the near future, regimens will be available with markedly reduced toxicity, high efficacy (>90% cure), improved dosing schedules (once or twice-daily), and shortened treatment duration (6-24 weeks).20–22 Such advances indicate that a HCV treatment as prevention strategy among PWID may be feasible in the very near future.
We project the potential impact of DAA therapy on HCV prevalence in three international settings with varied prevalence.
Subjects and Methods
Mathematical Model
A deterministic HCV transmission and treatment model among PWID12 was extended to incorporate additional biological and behavioral complexity (details in Fig. 1 and Supporting Information). The modeled population was stratified by infection state (uninfected, acute HCV, chronic HCV, on antiviral treatment, treatment failure), transmission risk (low/high), and current OST status (on/off). A fixed number (Φ(t)) of chronically infected PWID initiate treatment annually (or all chronic infections if fewer than Φ(t) are chronically infected), for a treatment duration of 1/ω(t). It was conservatively assumed treatment failures (those who do not attain sustained viral response [SVR]) could not be retreated due to potential resistance and reluctance to undergo further therapy. Furthermore, at baseline, few IFN+RBV treatment failures exist due to historically low treatment rates for PWID. In the base-case, PWID who are low-/high-risk and on/off OST are eligible for treatment; restricted treatment for only low-risk or those on OST was explored in the sensitivity analysis.
Figure 1.
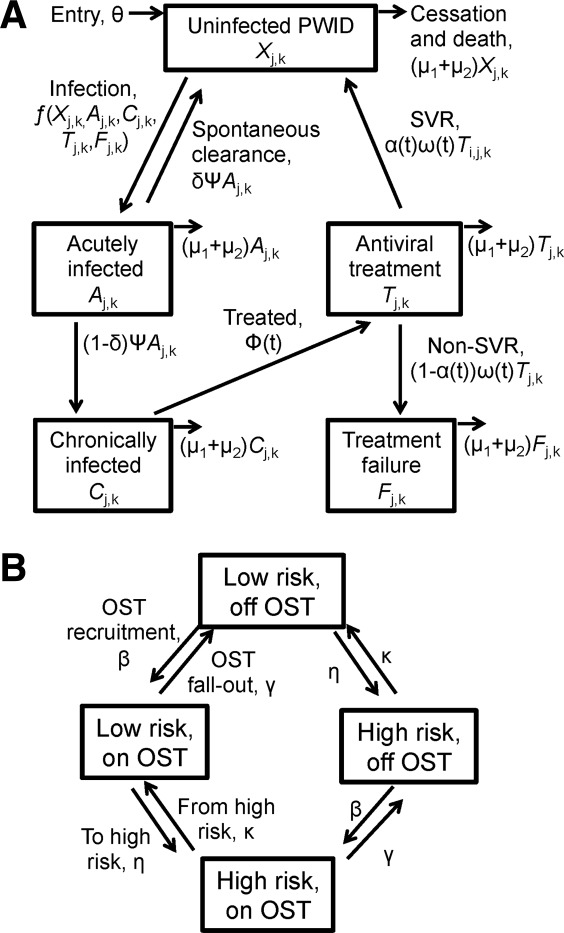
Model schematic showing HCV disease transmission and treatment states (A) and behavioral states (B). (A) Compartments for uninfected PWID (Xj,k), acutely infected PWID (Aj,k), chronically infected PWID (Cj,k), PWID on antiviral treatment (Tj,k), and PWID treatment failures (Fj,k). (B) The population was stratified by risk (low/high, j = 0 or 1, respectively), and OST (off/on, k = 0 or 1, respectively). New PWID enter the model at a constant rate (θ) as uninfected, off OST, and either low or high risk. Uninfected PWID can become acutely infected with HCV, where a proportion (δ) of individuals spontaneously clear their acute infection after a duration of time (1/ψ), and return to the uninfected compartment. Those who do not spontaneously clear the acute infection (1-δ) progress to chronic infection, where they are eligible for antiviral treatment. Because PWID are unlikely to be diagnosed during acute infection, it was assumed that they are not treated during the acute stage. If treated, a proportion [α(t)] achieve SVR and return to the uninfected compartment. Those who do not attain SVR [1-α(t)] move to the treatment failure compartment, where they cannot be retreated. PWID exit all compartments due to permanent cessation of drug use (μ1) or death due to drug or non–drug-related causes (μ2). The base-case analysis assumed PWID transition between high/low risk stages, as well as on/off OST. Additional details are provided in the Supporting Information.
Because the model is dynamic, the risk of infection or reinfection for a PWID is proportional to HCV prevalence, which changes over time. We do not assume any risk difference after treatment; reinfection risk is equal to primary infection risk. The forces of infection for each susceptible state were defined by the relative risk in that state, such that infectivity and susceptibility were altered by a factor Γ, Π, or Γ × Π if the PWID was on OST, high risk, or both, respectively. This was assumed to occur through a corresponding change in the relative frequency of transmission events with other PWID. The chance of a PWID having a transmission event with any PWID from another risk state and infectious status was proportional to the relative frequency of transmission events for PWID in that state. Due to rapid reductions of HCV RNA levels during treatment,23 we assumed the proportion on treatment who eventually achieve SVR (α(t)) are not infectious, whereas the remainder (1-α(t)) remain infectious. Some evidence indicates that acute infection may be associated with 2-log higher viral loads than during chronic infection24; however no studies have shown increased transmissibility during this stage. Therefore, we assumed equal infectivity for the base-case, but considered a five-fold higher transmissibility during acute HCV in the sensitivity analysis (assuming a similar relationship between viral load and transmissibility as for HIV25).
Modeling Treatment Scale-Up and Regimes
Treatment rates, durations, and SVR were varied over time to model scale-up and new treatments (see Supporting Information). No treatment prior to 2002 was modeled, because clinical guidance recommended against treatment of PWID. Due to the lack of reliable treatment data before 2007, a linear scale-up to current baseline treatment rates during 2002-2007 was modeled, with baseline rates constant during 2007-2012. Prior to 2012, we assumed all treatments used PEG-IFN+RBV. We assumed a continuation of baseline treatment rates from 2012-2015, during which time triple therapy with PEG-IFN+RBV and telaprevir/boceprevir will be available,22 although due to potential contraindications/drug interactions among PWID, we assumed only half of genotype 1 patients would be eligible for triple therapy. IFN-free DAAs were assumed to become available in 2015, followed by a 2-year linear scale-up in treatment (2015-2017) to scaled-up treatment rates (implemented from 2017-2027).
Multivariate Uncertainty Analyses
To consider the effect of uncertainty in the underlying parameters, we performed a multivariate probabilistic uncertainty analysis where 1,000 parameter sets were randomly sampled from setting specific parameter distributions in Table1. For each of the 1,000 parameter sets, the model was calibrated to the sampled HCV chronic prevalence in 2012 and proportion on OST/high risk by varying π, β, and η. The model was then used to project the prevalence reductions in each setting over 15 years (2012-2027) with no treatment scale-up, or scale-up to rates of 10, 20, 40, or 80 per 1,000 PWID annually. Additionally, the required scaled-up treatment rates to achieve prevalence reductions of ¼, ½, or ¾ within 15 years were projected. For all projections, 95% credibility intervals (CrI) were generated from the multivariate uncertainty sampling. A linear regression analysis of covariance was performed on the 15-year impact with scale-up to 10 per 1,000 PWID annually, and the proportion of the sum-of-squares contributed by each parameter was calculated to estimate the importance of individual parameters to the overall uncertainty.
Table 1.
Model Parameters and Sources
| Point Value (Sampling Bounds) |
||||||
|---|---|---|---|---|---|---|
| Parameter | Symbol | Edinburgh, UK | Melbourne, Australia | Vancouver, Canada | Units | References |
| HCV chronic prevalence among PWID in 2012* | Vary π to fit | 25% (22%-28%) | 50% (47%-53%) | 65% (63%-67%) | — | Edinburgh: Health Protection Scotland and the University of the West of Scotland,26 University of the West of Scotland, Health Protection Scotland, and West of Scotland Specialist Virology Centre36 |
| Melbourne: Iverson and Maher19 | ||||||
| Vancouver: Grebely et al.,18 Kim et al.28 | ||||||
| Sampled from a uniform distribution | ||||||
| PWID population size (used to calculate baseline treatment rate)† | 4,240 | 25,000 | 13,500 | — | Edinburgh: Hay et al.51 (∼0.9% of population) | |
| Melbourne: Iverson and Maher,19 Kirwan et al.52 (∼0.6% of the population) | ||||||
| Vancouver: McInnes et al.53 (∼2.1% of the population) | ||||||
| Baseline treatment rate (from 2007 onward) | Φ(t) for t ≥2007 | 8 (4-12) | 3 (1.5-4.5) | 5 (2.5-7.5) | Per 1,000 PWID per year | Edinburgh: Innes et al.,54 unpublished data |
| Melbourne: Iverson et al.,27 unpublished data | ||||||
| Vancouver: Grebely et al.,18 unpublished data | ||||||
| Sampled from a uniform distribution | ||||||
| Proportion G1 | 53% (49%-57%) | 56% (49%-63%) | 60% (56%-64%) | — | Edinburgh: Innes et al.,54 unpublished data | |
| Melbourne: Aitken et al.,55 McCaw et al.56 | ||||||
| Vancouver: Grebely et al.,18 unpublished data | ||||||
| Sampled from a uniform distribution | ||||||
| Death rate | μ2 | 1% | 0.83% | 3% | Per year | Edinburgh: Hickman et al.,30 Cornish et al.31 |
| Melbourne: Stoove et al.32 | ||||||
| Vancouver: Urban Health Research Initiative of the British Columbia Centre for Excellence in HIV/AIDS33 | ||||||
| Sampled from a Poisson distribution | ||||||
| Proportion PWID on OST | Vary β to fit | 57% (50%-64%) | 48% (44%-52%) | 45% (43%-47%) | Edinburgh: University of the West of Scotland, Health Protection Scotland, and West of Scotland Specialist Virology Centre,36 unpublished data‡ | |
| Melbourne: Iverson and Maher,19 Kirwan et al.52 | ||||||
| Vancouver: Urban Health Research Initiative of the British Columbia Centre for Excellence in HIV/AIDS33 | ||||||
| Sampled from a uniform distribution | ||||||
| Duration on OST | 12/γ | 8 (4-12) | 6.5 (3.25-8.75) | 7 (3.5-10.5) | Months | Edinburgh: Cornish et al.31 |
| Melbourne: Burns et al.57 | ||||||
| Vancouver: Nosyk et al.58 | ||||||
| Sampled from a uniform distribution | ||||||
| Proportion PWID high risk§ | φ, and vary η to fit | 33% (27%-39%) | 17% (14%-20%) | 64% (62%-66%) | — | Edinburgh: University of the West of Scotland, Health Protection Scotland, and West of Scotland Specialist Virology Centre,36 Allen et al.59 |
| Melbourne: O'Keefe et al.,60 unpublished data | ||||||
| Vancouver: Urban Health Research Initiative of the British Columbia Centre for Excellence in HIV/AIDS33 | ||||||
| Sampled from a uniform distribution | ||||||
| Duration high risk | 12/κ | 14 (7-21) | 13 (6.5-19.5) | 38 (19-57) | Months | Edinburgh: Vickerman et al.,9 Kemp et al.61 |
| Melbourne: O'Keefe et al.,60 unpublished data | ||||||
| Vancouver: Urban Health Research Initiative of the British Columbia Centre for Excellence in HIV/AIDS,33 unpublished data | ||||||
| Proportion spontaneously clear | δ | 26% | 26% | 26% | — | Micallef et al.29 |
| Duration of acute stage | 12/ψ | 6 (3-9) | 6 (3-9) | 6 (3-9) | Months | Mondelli et al.62 |
| Sampled from a uniform distribution | ||||||
| Duration of injecting until final cessation | 1/μ1 | 11 (6-20) | 11 (6-27) | 11 (6-23) | Years | Sweeting et al.,34 Kimber et al.35 (see Supporting Information for details) |
| Sampled from a uniform distribution | ||||||
| SVR | α(t)∥ | |||||
| PEG-IFN+RBV (G1) | 37% (26%-48%) | 37% (26%-48%) | 37% (26%-48%) | — | Aspinall et al.37 | |
| Sampled from a uniform distribution | ||||||
| PEG-IFN+RBV (G2/3) | 67% (56%-78%) | 67% (56%-78%) | 67% (56%-78%) | — | Aspinall et al.37 | |
| Sampled from a uniform distribution | ||||||
| Telaprevir/Boceprevir (G1) | 63% (44%-82%) | 63% (44%-82%) | 63% (44%-82%) | — | Jacobson et al.,38 Poordad et al.39 | |
| Sampled from a uniform distribution | ||||||
| IFN-free DAAs (all genotypes) | 90% | 90% | 90% | — | Gane et al.,20 Poordad et al.,21 Dore22 | |
| Estimated | ||||||
| Treatment duration | 52/ω(t)¶ | |||||
| PEG-IFN+RBV (G1 SVR) | 48 | 48 | 48 | Weeks | National Institute for Health and Clinical Excellence63 | |
| PEG-IFN+RBV (G1) | 12 | 12 | 12 | Weeks | National Institute for Health and Clinical Excellence63 | |
| Non-SVR | 24 | 24 | 24 | Weeks | National Institute for Health and Clinical Excellence63 | |
| PEG-IFN+RBV (G2/3) | 24# | 24# | 24# | Weeks | Jacobson et al.,38 Poordad et al.39 | |
| Weighted estimate based on stopping rules | ||||||
| Telaprevir/Boceprevir (G1) | 12 | 12 | 12 | Weeks | ||
| IFN-free DAAs (all genotypes) | Dore22 | |||||
| Estimated | ||||||
| Relative risk for acquiring HCV on OST | Γ | 0.41 (0.21-0.82) | 0.41 (0.21-0.82) | 0.41 (0.21-0.82) | — | Turner et al.8 |
| Sampled from a lognormal distribution | ||||||
| Relative risk for high risk | Π | 3.6 (1.5-8.7) | 3.6 (1.5-8.7) | 1.4 (1-2.1) | — | Edinburgh: Turner et al.,8 Allen et al.59 |
| Melbourne: Aitken et al.55 (assumed equal to Edinburgh) | ||||||
| Vancouver: Kim et al.28 | ||||||
| Sampled from a lognormal distribution | ||||||
Used to estimate the infection rate, π (vary π and fit to the HCV chronic prevalence). Note that π is not the incidence rate.
PWID population size was used to calculate baseline treatment rate per 1,000 PWID. Hence, for the model projections, the new injector entry rate (θ) was varied to fit to a total PWID population size of 1000.
From 2008/2009 NESI survey excluding those who attended a survey recruitment site for methadone.
When SVR rates vary by genotype, calculated using a weighted estimate based on population genotype distribution and SVR.
When treatment durations vary by genotype, calculated using a weighted estimated based on genotype distribution, SVR, and treatment duration.
Calculated based on early stopping rules and proportion achieving early viral response.
Abbreviations: G1, genotype 1; G2/3, genotype 2 or 3.
Sensitivity Analysis
To evaluate the impact of individual model assumptions, univariate sensitivity analyses were performed on projected prevalence reductions at 15 years with a treatment rate of 10 per 1,000 PWID annually using the point parameter values in Table1. The analysis determined the impact of: delayed scale-up initiation (starting 2019 versus 2015); longer scale-up duration (6 versus 2 years); lower/higher DAA SVR (80%/100% versus 90%); increased infectivity during acute infection (five-fold infectiousness compared with chronic infection, equal in base-case); restricting treatment to only those on OST or low risk, shorter/longer average duration of injecting career (6/20 years versus 11 years); shorter/longer duration on OST; no turnover from high to low risk; greater differences between high/low risk (six times the relative risk between high/low risk versus two times); and changes in mixing behavior between high/low risk (fully assortative versus proportional).
Model Parameterization
The model was parameterized to three international settings with a range of HCV chronic prevalence among PWID: Edinburgh, UK; Melbourne, Australia; and Vancouver, Canada. Model parameters and sources are given in Table1 and the Supporting Information.
HCV antibody prevalence estimates for Edinburgh, Melbourne, and Vancouver were 34%,26 66%,19,27 and 88%,18,28 respectively. Because 26% of individuals spontaneously clear acute infection,29 it was assumed 74% of HCV antibody-positive individuals were chronically infected, resulting in HCV chronic prevalence estimates of 25% in Edinburgh, 50% in Melbourne, and 65% in Vancouver.
Death and Cessation Rates
PWID death rates were similar in Edinburgh (1% per year30,31) and Melbourne (0.83% per year32) but higher in Vancouver (3% per year33). Site-specific unbiased estimates of the average duration of injecting until cessation are unavailable and difficult to obtain. We assumed an average injecting duration of 11 years,34 but varied this from 6 years up to 20 or 27 years in the uncertainty/sensitivity analyses based on seroprevalence survey data.19,28,35,36
Baseline Treatment Rates
Current annual numbers treated and treatment rates were estimated as: 32 PWID annually (8 per 1,000 PWID) in Edinburgh, 75 PWID annually (3 per 1,000 PWID) in Melbourne, and 68 PWID annually (5 per 1,000 PWID) in Vancouver.
SVR Rates
SVR rates for PEG-IFN+RBV were obtained from a meta-analysis of treatment among PWID (37% [95% confidence interval, 26%-48%] for genotype 1; 67% [95% confidence interval, 56%-78%] for genotypes 2/3).37 Telaprevir/boceprevir with PEG-IFN+RBV increases genotype 1 SVR rates by 70% over PEG-IFN+RBV,38,39 so a 63% SVR rate was modeled. It was assumed IFN-free DAAs will achieve 90% SVR for all genotypes with a duration of 12 weeks.20–22
Results
Base-Case
Without any treatment scale-up, low chronic HCV prevalence in Edinburgh (25%) combined with switching to new DAAs and moderate baseline levels of treatment (8 per 1,000 PWID annually) could lead to a 26% (95% CrI, 13%-45%) relative reduction in prevalence within 15 years. However, in Melbourne and Vancouver, higher chronic HCV prevalence (50% and 65%, respectively) combined with low current levels of treatment (<5 per 1,000 PWID annually) produce little impact (<2%) on prevalence over 15 years. Figure 2 shows HCV chronic prevalence reductions over time, and Fig. 3 shows relative prevalence reductions at year 15 (10 years after full scale-up).
Figure 2.
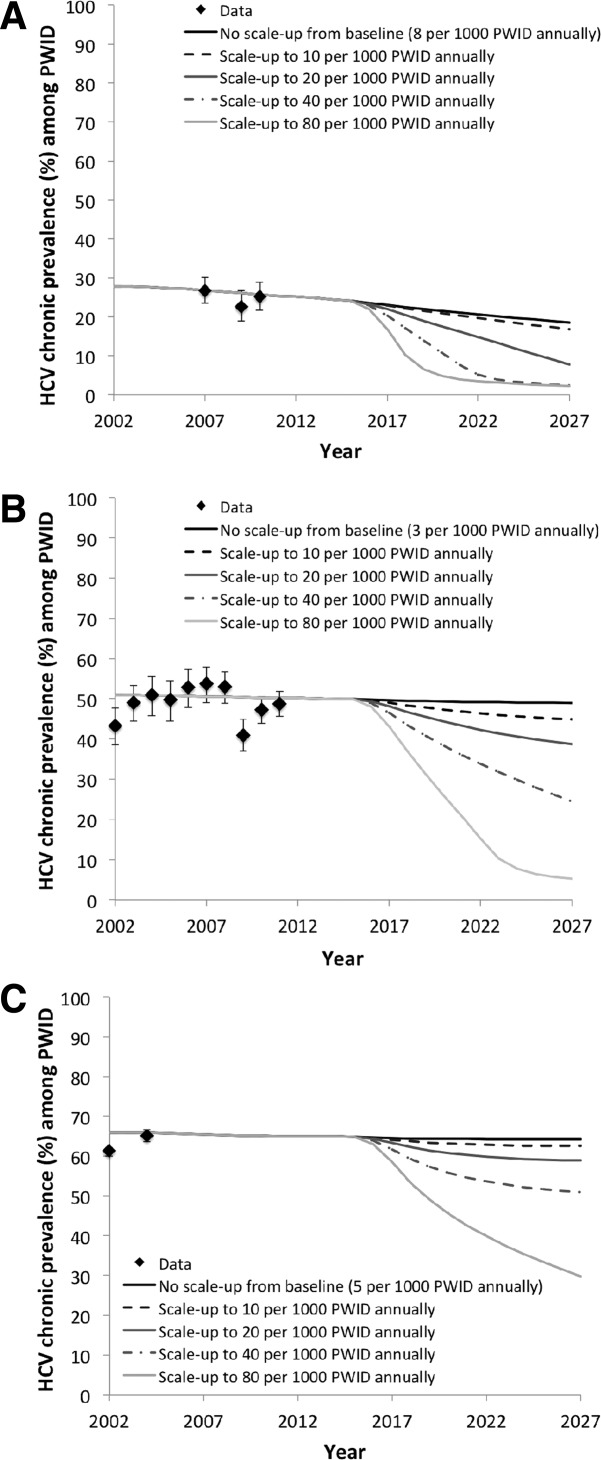
Chronic prevalence over time in (A) Edinburgh, (B) Melbourne, and (C) Vancouver. Simulations show no scale-up from baseline, or scale-up to 10, 20, 40, or 80 per 1,000 PWID treated annually. We assume no treatment prior to 2002, a linear scale-up to baseline treatment rates during 2002-2007, and baseline treatment rates during 2007-2012. A linear scale-up from baseline to scaled-up rate during 2015-2017 was modeled. HCV prevalence data points shown for comparison with 95% confidence intervals.
Figure 3.
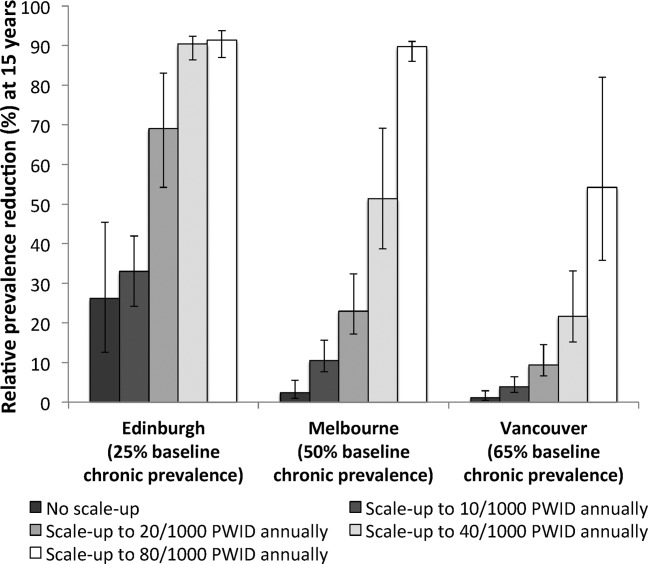
Relative prevalence reductions at 15 years (by 2027) with no treatment scale-up (8 per 1,000 PWID annually in Edinburgh, 3 per 1,000 PWID annually in Melbourne, and 5 per 1,000 PWID annually in Vancouver) and four treatment rate scenarios (10, 20, 40, and 80 per 1,000 PWID annually). Bars indicate the mean relative prevalence reductions, with whiskers representing the 95% CrI for the simulations.
Minimal and achievable levels of treatment scale-up result in substantial impact in Edinburgh and Melbourne. Scaling-up treatment to 20 per 1,000 PWID annually could result in relative prevalence reductions within 15 years of 69% (95% CrI, 54%-83%) and 23% (95% CrI, 17%-32%) in Edinburgh and Melbourne, respectively, but only 9% (95% CrI, 7%-15%) in Vancouver. Higher treatment rates (>40 per 1,000 PWID annually) are required to reduce prevalence by over >20% in Vancouver within 15 years. A scale-up to treating 80 per 1,000 PWID annually could reduce HCV chronic prevalence to below 5% in Edinburgh and Melbourne, and to 30% in Vancouver, within 15 years.
Figure 4 shows the levels of treatment necessary to reduce prevalence by ¼, ½, and ¾ within 15 years (10 years after full scale-up) in all settings. Halving current prevalence could be achieved through scaled-up treatment rates of 15 (95% CrI, 12-19), 40% (95% CrI, 30-50), and 76 (95% CrI, 56-102) per 1,000 PWID annually in Edinburgh, Melbourne, and Vancouver, respectively. This would require doubling treatment rates in Edinburgh (currently 32 PWID [8 per 1,000 PWID] annually). However, in Melbourne, it would require a 13-fold scale-up (currently 75 PWID [3 per 1,000 PWID] annually), and in Vancouver would require a 15-fold scale-up (currently 68 PWID [5 per 1,000 PWID] annually). Reducing prevalence by ¾ would require a scale-up of three-fold in Edinburgh (to 22 [95% CrI, 18-27] per 1,000 PWID annually), 18-fold in Melbourne (to 54 [95% CrI, 43-67] per 1,000 PWID annually), and 20-fold in Vancouver (to 98 [95% CrI, 74-127] per 1,000 PWID annually). This would result in HCV chronic prevalences of <10% in Edinburgh, <15% in Melbourne, and <20% in Vancouver, respectively.
Figure 4.
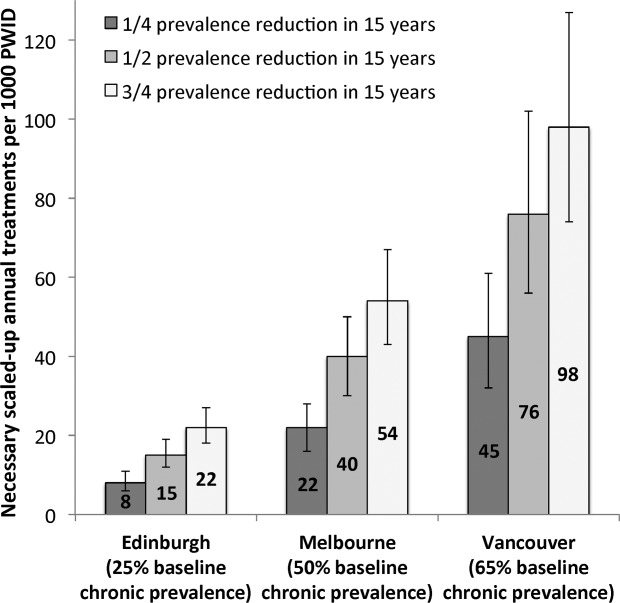
Annual scaled-up treatment rate required to reduce prevalence by ¼, ½, or ¾ in Edinburgh, Melbourne, and Vancouver within 15 years (by 2027). Bars (and numbers) indicate the mean value, with whiskers representing the 95% CrI.
Uncertainty/Sensitivity Analysis
Analysis of covariance indicated that uncertainty in average injecting duration contributed to the majority of variation (59%-78%) in impact at 15 years with a treatment scale-up to 10 per 1,000 PWID annually. The remaining variation was due to uncertainty in baseline treatment rate in Edinburgh, baseline prevalence in Melbourne, and baseline prevalence and death rate in Vancouver.
One-way sensitivity analyses showed baseline prevalence, injecting duration, and time to scale-up initiation had the most effect on model projections at 15 years with treatment scale-up to 10 per 1,000 PWID annually (Fig. 5, shown for Melbourne). Across the sites, if baseline chronic HCV prevalences were 5% lower, the impact of treatment scale-up increased by 24%-37%, whereas if baseline prevalences were 5% higher, impact decreased by 20%-27%. If the average injecting career was 20 rather than 11 years, then potential impact increased by 16%-53% (with greater impact at higher chronic prevalence), whereas if average injecting duration was shorter at 6 years, impact was reduced by 29%-43%. Delaying the initiation of scale-up by 4 years (2019 versus 2015) resulted in 7%-18% less impact. Decreasing/increasing IFN-free DAA SVR rates (to 80%/100% versus 90%) correspondingly decreased/increased impact by 12% to 15%. If acute infection was associated with a five-fold increase in transmissibility compared with chronic infection (equal for base-case), impact was reduced by 11%-16%.
Figure 5.
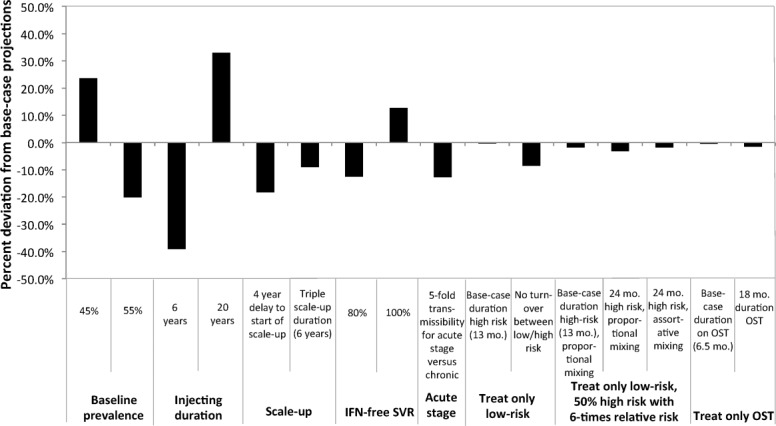
Results from the one-way sensitivity analyses; percent change from the base-case scenario of the predicted relative prevalence reduction at 15 years in Melbourne with scaled-up treatment rate of 10 per 1,000 PWID annually (from a baseline rate of 3 per 1,000 PWID annually). For the base-case, all chronically infected PWID (high/low risk or on/off OST) were eligible for treatment. Mo., months.
Changing other assumptions regarding treatment duration or population heterogeneity (e.g., average time in OST/high risk, proportion high-risk, relative transmission risk when in OST or high-risk, mixing assumptions between low and high-risk, restricting treatment to only those on OST or low-risk) had <10% impact on projections for a scaled-up treatment rate of 10/1,000 PWID annually. However, at higher treatment rates (e.g., 80 per 1,000 PWID for Melbourne), sustaining treatment at this level would require treating the non-OST population or expanding OST coverage.
Budgetary Impact
Previous cost-effectiveness analyses estimated the drug-only cost of triple therapy with protease inhibitors in the United States at approximately $50,000 USD per course.40 The cost of future IFN-free DAA regimens is unknown, but if they cost $50,000 ($25,000-$75,000), then the scaled-up treatment rates necessary to halve prevalence within 15 years (15, 40, and 76 per 1,000 PWID annually in Edinburgh, Melbourne, and Vancouver, respectively) would require an annual HCV treatment budget for PWID of $3.2 million ($1.6-$4.7 million) in Edinburgh, $50.0 million ($25-$75 million) in Melbourne, and $51.3 million ($25.7-$77.0 million) in Vancouver.
Discussion
This modeling study explored the feasibility of HCV treatment as prevention in the era of IFN-free DAA-based HCV therapy. Current levels of HCV treatment among PWID are projected to only achieve modest or negligible reductions in HCV chronic prevalence among PWID. However, scaling up treatment could lead to substantial reductions in HCV prevalence. In Edinburgh, a doubling of treatment rates (to 15 per 1,000 PWID annually) could halve prevalence; a three-fold increase could reduce chronic HCV prevalence to <7% within 15 years. Greater scale-up will be required in Melbourne and Vancouver, where current treatment rates are lower and chronic prevalence higher, but prevalence could be halved in 15 years with treatment rates of 40 per 1,000 PWID (a 13-fold increase from 3 per 1,000 PWID annually) in Melbourne and 76 per 1,000 PWID (a 15-fold increase from 5 per 1,000 PWID annually) in Vancouver. A 20-fold increase from baseline treatment rates could reduce chronic prevalence to <15% and <20% in Melbourne and Vancouver, respectively, in 15 years.
Such scale-up, though considerable in Melbourne and Vancouver, has been achieved and exceeded for HIV treatment in both resource rich and poor settings,41 and even amongst PWID in some settings.42,43 In addition, programs designed to address barriers to care among PWID have achieved yearly HCV treatment rates of 40-80 per 1,000 PWID with PEG-IFN+RBV in Australia, Canada, Europe, and the United States.44–47 Moreover, scale-up of IFN-free DAA in theory will be easier to implement and have greater impact than current treatment regimes. IFN-free DAA regimens will require shorter duration and less complex monitoring22 which in combination with higher SVR and reduced toxicity will markedly accelerate the current expansion of HCV treatment into the community, including integration with drug treatment, such as OST.
Limitations
These projections are based on a theoretical mathematical model, with several limitations. First, there is uncertainty in a number of parameters. These projections are predicated on assumptions of the effectiveness of IFN-free DAAs (based on phase 2 studies as evidence from large-scale evaluations are not yet available). Outcomes among PWID are unknown, but systematic reviews report similar response rates among PWID and non-PWID for IFN+RBV regimens.48,49 Additionally, active PWID are generally younger (a meta-analysis48 found a lower median age (38 years) for studies with HCV treatment among PWID compared with registration trials for PEG-IFN+RBV (43-45 years)) and have less advanced liver disease than the broader HCV population. We do not explicitly model HIV/HCV coinfection, as two of our settings have marginal (<1%) coinfection prevalences. However, in settings where a greater proportion of PWID are HIV/HCV-coinfected, lower SVR rates may be achieved. Sensitivity analyses revealed that a lower SVR of 80% would still achieve substantial impact, although slightly higher treatment rates would be required to achieve specific reductions in HCV prevalence.
Furthermore, better information on average injecting duration could substantially reduce uncertainty in the projections. The average age (and injecting duration) of people in drug treatment and serological surveys in the three sites suggest injecting durations between 11 and 27 years,19,28,35 but unbiased estimates are unavailable. An 11-year average injecting duration was assumed,34 but if it were longer, then greater impact would be achieved. Also, HCV risk and treatment uptake will vary between PWID subgroups, relating to injecting patterns or other factors such as homelessness. However, we considered scenarios where HCV treatment is delivered only in OST or when PWID are at low risk and show there is little impact on the outcome given movement between low-and high-risk states over an injecting career.
Second, complexities involved in treatment scale-up are not modeled. Treatment scale-up will likely be delivered in the community alongside OST, but additional interventions may be required to increase the case-finding among PWID, including health care workforce training and interventions addressing stigma surrounding testing and treatment. Importantly, in our model, a fixed number of PWID are treated annually; therefore, as prevalence falls, an increasing proportion of infected PWID are treated. This will have implications for diagnosis and treatment retention, particularly among those PWID who are more difficult to reach. However, treatment recruitment may become easier as more PWID are treated.
Third, the model assumes a stable injecting population size that, although true in the settings examined, may not be applicable to all settings. For example, data from Amsterdam50 suggest a decline in the number of injectors. In these settings, as PWID prevalence falls, we would expect HCV prevalence to increase as the cohort ages, and detailed models of these settings would require age-specific information on prevalence of PWID and injecting duration to determine intervention impact.
Finally, the model incorporates current levels of OST, but it did not consider the impact of scale-up or targeting of interventions such as OST and NSP, which may contribute additionally toward reducing HCV transmission.9 As our aim was to explore the scale-up of antiviral treatment, we did not stratify the population by drug-type or explore OST eligibility criteria. Additionally, we do not explicitly model NSP, but account for existing levels of coverage in modeling the epidemic in each setting.
Implications and Comparison With Other Studies
This is the first analysis to explore the potential of new and future direct-acting HCV antiviral therapy as prevention in a range of global prevalence settings, and supports previous modeling studies indicating that HCV antiviral treatment could reduce transmission and HCV prevalence among PWID.12–16 In contrast, mathematical models have shown that scale-up of OST/NSP could have considerable impact in areas with historically low levels of OST/NSP; however, in many developed countries where coverage is already high (such as our sites), the scale-up required (e.g., 80% PWID on OST or high coverage NSP for 15 years) would be unachievable and unsustainable, and would achieve less impact than modest levels of HCV treatment.9
Overall, the projections suggest IFN-free DAA HCV treatment, as prevention is a feasible option for reducing the future burden of HCV-related disease, which is of critical public health importance given the lack of alternative effective HCV prevention strategies. HCV treatment is cost-effective, and in most settings treatment of PWID is highly cost-effective,17 primarily because of the potential prevention benefit and reduction in secondary transmission.
A question still remains, though, as to whether scale-up is affordable—especially if the drugs are marketed at similar cost to existing therapy. Expansion will be costly, and so any future scale-up of HCV treatment for prevention will require drug-price reform, especially for lower and middle income settings, but possibly also for developed countries that require high treatment rates.
Acknowledgments
We thank Avril Taylor and Norah Palmateer for Scottish Needle Exchange Surveillance Initiative data; Peter Hayes and Hamish Inness for Scottish HCV Clinical Database data; Kate Templeton and Allan McLeod for Edinburgh genotype data; Rebecca Jenkinson, Rachel Sacks-Davis, Campbell Aitken, Peter Higgs, Paul Dietze, and the participants of Networks and MIX for Melbourne data; Maryam Alavi for CHASE treatment uptake data; Evan Wood, Thomas Kerr, Julio Montaner, Kora DeBeck, and the study participants of VIDUS (supported by National Institutes of Health grant R01DA011591); and current and past researchers and staff for Vancouver data and comments.
Glossary
- CrI
credible interval
- DAA
direct-acting antiviral
- HCV
hepatitis c virus
- HIV
human immunodeficiency virus
- NSP
needle and syringe programs
- OST
opiate substitution therapy
- PEG-IFN
pegylated interferon
- PWID
people who inject drugs
- RBV
ribavirin
- SVR
sustained viral response.
Potential conflict of interest
N. K. M. has received an honorarium for speaking at a conference sponsored by Janssen. J. G. owns stock in Gilead and is a member of an advisory board for Merck. S. J. H. has received honoraria for speaking at conferences sponsored by MSD and Janssen and consults for Janssen. G. R. F. has received funding from Roche, Novartis, Janssen, Gilead, Bristol-Meyers Squibb, Boehringer Ingelheim, Idenix, Abbott, and Merck for consultancy and lectures. D. J. G. is a member of advisory boards and undertakes consultancy for Merck and Janssen. G. J. D. is a consultant/advisor and has received research grants from Roche, Merck, Janssen, Gilead, Bristol-Myers Squibb, and Abbott.
Additional Supporting Information may be found in the online version of this article.
References
- Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infec Dis. 2005;5:558–567. doi: 10.1016/S1473-3099(05)70216-4. [DOI] [PubMed] [Google Scholar]
- Ly KN, Xing J, Klevens RM, Jiles RB, Ward JW, Holmberg SD. The increasing burden of mortality from viral hepatitis in the United States between 1999 and 2007. Ann Int Med. 2012;156:271–278. doi: 10.7326/0003-4819-156-4-201202210-00004. [DOI] [PubMed] [Google Scholar]
- Nelson PK, Mathers BM, Cowle B, Hagan H, Des Jarlais DC, Horyniak D, et al. Global epidemiology of hepatitis B and hepatitis C in people who inject drugs: results of systematic reviews. Lancet. 2011;378:571–583. doi: 10.1016/S0140-6736(11)61097-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta SH, Astemborski J, Kirk GD, Strathdee SA, Nelson K, Vlahov D, et al. Changes in blood-borne infection risk among injection drug users. J Infec Dis. 2011;203:587–594. doi: 10.1093/infdis/jiq112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher L, Li J, Jalaludin B, Chant KG, Kaldor JM. High hepatitis C incidence in new injecting drug users: a policy failure? Aust New Zealand J Pub Health. 2007;31:30–35. doi: 10.1111/j.1753-6405.2007.00007.x. [DOI] [PubMed] [Google Scholar]
- Hagan H, Pouget ER, Des Jarlais DC. A systematic review and meta-analysis of interventions to prevent hepatitis C virus infection in people who inject drugs. J Infec Dis. 2011;204:74–83. doi: 10.1093/infdis/jir196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Den Berg C, Smit C, Brussel GV, Coutinho R, Prins M. Full participation in harm reduction programmes is associated with decreased risk for human immunodeficiency virus and hepatitis C virus: evidence from the Amsterdam Cohort Studies among drug users. Addiction. 2007;102:1454–1462. doi: 10.1111/j.1360-0443.2007.01912.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner K, Hutchinson S, Craine N, Hope V, Palmateer N, Vickerman P, et al. The impact of needle and syringe provision and opiate substitution therapy on the incidence of hepatitis C virus in injecting drug users: pooling of UK evidence. Addiction. 2011;106:1978–1988. doi: 10.1111/j.1360-0443.2011.03515.x. [DOI] [PubMed] [Google Scholar]
- Vickerman P, Martin N, Turner K, Hickman M. Can needle and syringe programmes and opiate substitution therapy achieve substantial reductions in HCV prevalence? Model projections for different epidemic settings. Addiction. 2012;107:1984–1995. doi: 10.1111/j.1360-0443.2012.03932.x. [DOI] [PubMed] [Google Scholar]
- Cohen M, Chen Y, McCauley M, Gamble T, Hosseinipour M, Kumarasamy N, et al. Prevention of HIV-1 infection with early antiretorival therapy. N Engl J Med. 2011;365:493–505. doi: 10.1056/NEJMoa1105243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J. Halting HIV/AIDS epidemics. Science. 2011;334:1338–1340. doi: 10.1126/science.334.6061.1338. [DOI] [PubMed] [Google Scholar]
- Martin NK, Vickerman P, Foster GR, Hutchinson SJ, Goldberg DJ, Hickman M. Can antiviral therapy for hepatitis C reduce the prevalence of HCV among injecting drug user populations? A modelling analysis of its prevention utility. J Hepatol. 2011;54:1137–1144. doi: 10.1016/j.jhep.2010.08.029. [DOI] [PubMed] [Google Scholar]
- Martin NK, Vickerman P, Hickman M. Mathematical modelling of hepatitis C treatment for injecting drug users. J Theor Biol. 2011;274:58–66. doi: 10.1016/j.jtbi.2010.12.041. [DOI] [PubMed] [Google Scholar]
- Zeiler I, Langlands T, Murray JM, Ritter A. Optimal targeting of hepatitis C virus treatment among injecting drug users to those not enrolled in methadone maintenance programs. Drug Alcohol Depend. 2010;110:228–233. doi: 10.1016/j.drugalcdep.2010.03.006. [DOI] [PubMed] [Google Scholar]
- Vickerman P, Martin N, Hickman M. Can hepatitis C virus treatment be used as a prevention strategy? Additional model projections for Australia and elsewhere. Drug Alcohol Depend. 2011;113:83–85. doi: 10.1016/j.drugalcdep.2010.08.001. [DOI] [PubMed] [Google Scholar]
- Hellard M, Jenkinson R, Higgs P, Stoové M, Sacks-Davis R, Gold J, et al. Modelling antiviral treatment to prevent hepatitis C infection among people who inject drugs in Victoria, Australia. Med J Aust. 2012;196:638–641. doi: 10.5694/mja11.10981. [DOI] [PubMed] [Google Scholar]
- Martin NK, Miners A, Vickerman P, Foster G, Hutchinson S, Goldberg D, et al. The cost-effectiveness of HCV antiviral treatment for injecting drug user populations. Hepatology. 2012;55:49–57. doi: 10.1002/hep.24656. [DOI] [PubMed] [Google Scholar]
- Grebely J, Raffa JD, Lai C, Krajden M, Kerr T, Fischer B, et al. Low uptake of treatment for hepatitis C virus infection in a large community-based study of inner city residents. J Viral Hepat. 2009;16:352–358. doi: 10.1111/j.1365-2893.2009.01080.x. [DOI] [PubMed] [Google Scholar]
- Iverson J, Maher L. Australian Needle and Syringe Program National Data Report 2007-2011. Kensington, Australia: The Kirby Institute; 2012. [Google Scholar]
- Gane EJ, Stedman CA, Hyland RH, Ding X, Svarovskaia E, Symonds WT, et al. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N Engl J Med. 2013;368:34–44. doi: 10.1056/NEJMoa1208953. [DOI] [PubMed] [Google Scholar]
- Poordad F, Lawitz E, Kowdley KV, Cohen DE, Podsadecki T, Siggelkow S, et al. Exploratory study of oral combination antiviral therapy for hepatitis C. N Engl J Med. 2013;368:45–53. doi: 10.1056/NEJMoa1208809. [DOI] [PubMed] [Google Scholar]
- Dore GJ. The changing therapeutic landscape for hepatitis C. Med J Aust. 2012;196:629–632. doi: 10.5694/mja11.11531. [DOI] [PubMed] [Google Scholar]
- Dahari H, Ribeiro RM, Perelson AS. Triphasic decline of hepatitis C virus RNA during antiviral therapy. Hepatology. 2007;46:16–21. doi: 10.1002/hep.21657. [DOI] [PubMed] [Google Scholar]
- Liu L, Fisher B, Thomas D, Cox A, Ray S. Spontaneous clearance of primary acute hepatitis C virus infection correlated with high initial viral RNA level and rapid HVR1 evolution. Hepatology. 2012;55:1684–1691. doi: 10.1002/hep.25575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn TC, Wawer M, Sewankambo N, Serwadda D, Li C, Wabwire-Mangen F, et al. Viral load and heterosexual transmission of human immunodeficiency virus type 1. N Engl J Med. 2000;342:921–929. doi: 10.1056/NEJM200003303421303. [DOI] [PubMed] [Google Scholar]
- Health Protection Scotland and the University of the West of Scotland. The Needle Exchange Surveillance Initiative (NESI): Prevalence of HCV, HIV and Injecting Risk Behaviours Among Injecting Drug Users Attending Needle Exchanges in Scotland, 2007. Glasgow, UK: Health Protection Scotland; 2008. [Google Scholar]
- Iverson J, Topp L, Maher L. Australian NSP Survey National Data Report 1995-2010. Kensington, Australia: The Kirby Institute; 2011. [Google Scholar]
- Kim C, Kerr T, Li K, Zhang R, Tyndall MW, Montaner J, et al. Unstable housing and hepatitis C incidence among injection drug users in a Canadian setting. BMC Public Health. 2009;9:270. doi: 10.1186/1471-2458-9-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micallef JM, Kaldor JM, Dore GJ. Spontaneous viral clearance following hepatitis C infection: a systematic review of longitudinal studies. J Viral Hepat. 2006;13:34–41. doi: 10.1111/j.1365-2893.2005.00651.x. [DOI] [PubMed] [Google Scholar]
- Hickman M, Hope V, Coleman B, Parry J, Telfer M, Twigger J, et al. Assessing IDU prevalence and health consequences (HCV, overdose and drug-related mortality) in a primary care trust: implications for public health action. J Public Health. 2009:1–9. doi: 10.1093/pubmed/fdp067. [DOI] [PubMed] [Google Scholar]
- Cornish R, Macleod J, Strang J, Vickerman P, Hickman M. Risk of death during and after opiate substitution treatment in primary care: prospective observational study in UK General Practice Research Database. BMJ. 2010;341 doi: 10.1136/bmj.c5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoove MA, Dietze PM, Aitken CK, Jolley D. Mortality among injecting drug users in Melbourne: a 16-year follow-up of the Victorian Injecting Cohort Study (VICS) Drug Alcohol Depend. 2008;96:281–285. doi: 10.1016/j.drugalcdep.2008.03.006. [DOI] [PubMed] [Google Scholar]
- Urban Health Research Initiative of the British Columbia Centre for Excellence in HIV/AIDS. Drug situation in Vancouver. Vancouver, Canada: 2009. http://uhri.cfenet.ubc.ca/images/Documents/dsiv2009.pdf. Accessed April 2013.
- Sweeting MJ, De Angelis D, Ades AE, Hickman M. Estimating the prevalence of ex-injecting drug use in the population. Stats Meth Med Res. 2009;18:381–395. doi: 10.1177/0962280208094704. [DOI] [PubMed] [Google Scholar]
- Kimber J, Copeland L, Hickman M, Macleod J, McKenzie J, De Angelis D, et al. Survival and cessation in injecting drug users: prospective observational study of outcomes and effect of opiate substitution treatment. BMJ. 2010;340:c3172. doi: 10.1136/bmj.c3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- University of the West of Scotland, Health Protection Scotland, and West of Scotland Specialist Virology Centre. 2010. The Needle Exchange Surveillance Initiative (NESI): Prevalence of HCV and Injecting Risk Behaviours Among Injecting Drug Users Attending Needle Exchanges in Scotland, 2008/2009; http://www.documents.hps.scot.nhs.uk/bbvsti/hepatitis-c/publications/nesi-needle-exchange.pdf. Accessed April 2013.
- Aspinall A, Corson S, Doyle J, Grebely J, Hutchinson SJ, Dore GJ, et al. Treatment of HCV infection among people who are actively injecting drugs: a systematic review and meta-analysis. Clin Infect Dis. In press. [DOI] [PubMed]
- Jacobson IM, McHutchison JG, Dusheiko G, Di Bisceglie AM, Reddy KR, Bzowej NH, et al. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med. 2011;364:2405–2416. doi: 10.1056/NEJMoa1012912. [DOI] [PubMed] [Google Scholar]
- Poordad F, McCone J, Bacon BR, Bruno S, Manns MP, Sulkowski MS, et al. Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med. 2011;364:1195–1206. doi: 10.1056/NEJMoa1010494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Cipriano LE, Holodniy M, Owens DK, Goldhaber-Fiebert JD. New protease inhibitors for the treatment of chronic hepatitis CA cost-effectiveness analysis. Ann Intern Med. 2012;156:279–290. doi: 10.1059/0003-4819-156-4-201202210-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO/UNAIDS/UNICEF. 2011. Global HIV/AIDS Response: Epidemic Update And Health Sector Progess Towards Universal Access.
- Mathers BM, Degenhardt L, Ali H, Wiessing L, Hickman M, Mattick RP, et al. HIV prevention, treatment, and care services for people who inject drugs: a systematic review of global, regional, and national coverage. Lancet. 2010;375:1014–1028. doi: 10.1016/S0140-6736(10)60232-2. [DOI] [PubMed] [Google Scholar]
- Wood E, Kerr T, Marshall BDL, Li K, Zhang R, Hogg RS, et al. Longitudinal community plasma HIV-1 RNA concentrations and incidence of HIV-1 among injecting drug users: prospective cohort study. BMJ. 2009:338. doi: 10.1136/bmj.b1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grebely J, Knight E, Genoway KA, Viljoen M, Khara M, Elliott D, et al. Optimizing assessment and treatment for hepatitis C virus infection in illicit drug users: a novel model incorporating multidisciplinary care and peer support. Eur J Gastroenterol Hepatol. 2010;22:270–277. doi: 10.1097/meg.0b013e32832a8c4c. [DOI] [PubMed] [Google Scholar]
- Hallinan R, Byrne A, Agho K, Dore GJ. Referral for chronic hepatitis C treatment from a drug dependency treatment setting. Drug Alcohol Depend. 2007;88:49–53. doi: 10.1016/j.drugalcdep.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Lindenburg CE, Lambers FA, Urbanus AT, Schinkel J, Jansen PL, Krol A, et al. Hepatitis C testing and treatment among active drug users in Amsterdam: results from the DUTCH-C project. Eur J Gastroenterol Hepatol. 2011;23:23–31. doi: 10.1097/MEG.0b013e328340c451. [DOI] [PubMed] [Google Scholar]
- Harris KA, Jr, Arnsten JH, Litwin AH. Successful integration of hepatitis C evaluation and treatment services with methadone maintenance. J Addict Med. 2010;4:20–26. doi: 10.1097/ADM.0b013e3181add3de. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimova RB, Zeremski M, Jacobson IM, Hagan H, Des Jarlais DC, Talal AH. Determinants of hepatitis C virus treatment completion and efficacy in drug users assessed by meta-analysis. Clin Infect Dis. 2013;56:806–816. doi: 10.1093/cid/cis1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellard M, Sacks-Davis R, Gold J. Hepatitis C treatment for injection drug users: a review of the available evidence. Clin Infect Dis. 2009;49:561–573. doi: 10.1086/600304. [DOI] [PubMed] [Google Scholar]
- Lindenburg C, Krol A, Smit C, Buster M, Coutinho R, Prins M. Decline in HIV incidence and injecting, but not in sexual risk behaviour, seen in drug users in Amsterdam: a 19-year prospective cohort study. AIDS. 2006;20:1771–1775. doi: 10.1097/01.aids.0000242824.59377.53. [DOI] [PubMed] [Google Scholar]
- Hay G, Gannon M, McKeganey N, Hutchinson S, Goldberg D. Estimating the National and Local Prevalence of Problem Drug Misuse in Scotland. University of Glasgow Centre for Drug Misuse Research and Health Protection Scotland. http://www.drugmisuse.isdscotland.org/publications/local/prevreport2004.pdf. Accessed on April 2013.
- Kirwan A, Dietze D, Lloyd B Victorian Drug Trends. Findings from the Illicit Drug Reporting System (IDRS) Sydney, Australia: National Drug and Alcohol Research Centre, University of New South Wales; 2011. 2012. [Google Scholar]
- McInnes C, Druyts E, Harvard S, Gilbert M, Tyndall M, Lima V, et al. HIV/AIDS in Vancouver, British Columbia: a growing epidemic. Harm Reduct J. 2009;6:5. doi: 10.1186/1477-7517-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innes HA, Hutchinson SJ, Allen S, Bhattacharyya D, Bramley P, Carman B, et al. Ranking predictors of a sustained viral response for patients with chronic hepatitis C treated with pegylated interferon and ribavirin in Scotland. Eur J Gastroenterol Hepatol. 2012;24:646–655. doi: 10.1097/MEG.0b013e32835201a4. [DOI] [PubMed] [Google Scholar]
- Aitken CK, Lewis J, Tracy SL, Spelman T, Bowden DS, Bharadwaj M, et al. High incidence of hepatitis C virus reinfection in a cohort of injecting drug users. Hepatology. 2008;48:1746–1752. doi: 10.1002/hep.22534. [DOI] [PubMed] [Google Scholar]
- McCaw R, Moaven L, Locarnini S, Boden D. Hepatitis C virus genotypes in Australia. J Viral Hepat. 1997;4:351–357. doi: 10.1046/j.1365-2893.1997.00060.x. [DOI] [PubMed] [Google Scholar]
- Burns L, Randall D, Hall W, Law M, Butler T, Bell J, et al. Opioid agonist pharmacotherapy in New SouthWales from 1985 to 2006: patient characteristics and patterns and predictors of treatment retention. Addiction. 2009;104:1363–1372. doi: 10.1111/j.1360-0443.2009.02633.x. [DOI] [PubMed] [Google Scholar]
- Nosyk B, Marsh D, Sun H, Schechter M, Anis A. Trends in methadone maintenance treatment participation, retention, and compliance to dosing guidelines in British Columbia, Canada: 1996-2006. J Subst Abuse Treat. 2010;39:22–31. doi: 10.1016/j.jsat.2010.03.008. [DOI] [PubMed] [Google Scholar]
- Allen EJ, Palmateer NE, Hutchinson SJ, Cameron S, Goldberg DJ, Taylor A. Association between harm reduction intervention uptake and recent hepatitis C infection among people who inject drugs attending sites that provide sterile injecting equipment in Scotland. Int J Drug Policy. 2012;23:346–352. doi: 10.1016/j.drugpo.2012.07.006. [DOI] [PubMed] [Google Scholar]
- O'Keefe D, Aitken CK, Higgs P, Dietze P. Concordance between self-reported and actual hepatitis C infection status in a cohort of people who inject drugs. Drug Alcohol Rev. 2013;32:208–210. doi: 10.1111/j.1465-3362.2012.00502.x. [DOI] [PubMed] [Google Scholar]
- Kemp PA, Neale J, Robertson M. Homelessness among problem drug users: prevalence, risk factors and trigger events. Health Soc Care Community. 2006;14:319–328. doi: 10.1111/j.1365-2524.2006.00624.x. [DOI] [PubMed] [Google Scholar]
- Mondelli M, Cerino A, Cividini A. Acute hepatitis C: diagnosis and management. J Hepatol. 2005;42:5108–5114. doi: 10.1016/j.jhep.2004.10.017. [DOI] [PubMed] [Google Scholar]
- National Institute for Health and Clinical Excellence. 2006. Peginterferon alfa and Ribavirin for the Treatment of Mild Chronic Hepatitis C. NICE Technology Appraisal Guidance 106;
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.