

Abstract
Aromatase (CYP19) catalyzes the aromatization reaction of androgen substrates to estrogens, the last and rate-limiting step in estrogen biosynthesis. Inhibition of aromatase is a new and promising approach to treat hormone-dependent breast cancer. We present here the design and development of isoflavanone derivatives as potential aromatase inhibitors. Structural modifications were performed on the A and B rings of isoflavanones via microwave-assisted, gold-catalyzed annulation reactions of hydroxyaldehydes and alkynes. The in vitro aromatase inhibition of these compounds was determined by fluorescence-based assays utilizing recombinant human aromatase (baculovirus/insect cell-expressed). The compounds 3-(4-phenoxyphenyl)chroman-4-one (1h), 6-methoxy-3-phenylchroman-4-one (2a) and 3-(pyridin-3-yl)chroman-4-one (3b) exhibited potent inhibitory effects against aromatase with IC50 values of 2.4 μM, 0.26 μM and 5.8 μM, respectively. Docking simulations were employed to investigate crucial enzyme/inhibitor interactions such as hydrophobic interactions, hydrogen bonding and heme iron coordination. This report provides useful information on aromatase inhibition and serves as a starting point for the development of new flavonoid aromatase inhibitors.
Keywords: Breast cancer, Aromatase inhibitors, Isoflavanones
1. Introduction
Breast cancer is the leading cancer for women and is the second leading cause of the deaths after lung cancer. Women in the US have the highest incidence rate of developing breast cancer. Currently there are approximately 2.5 million women living with breast cancer in the US. The estimated number of new breast cancer cases for the year 2010 was over 200,000.1 One out of eight women will develop breast cancer over the course of a lifetime, a significant increase compared to 1 in 35 in the 1930s.2 The risk factors of breast cancer include age, personal health history, family health history, diet, exercise and obesity.3 Various forms of treatment are available for breast cancer, including surgery, radiation, chemotherapy and hormonal therapy.4 Hormonal therapy, unlike surgery, radiation and chemotherapy, is relatively facile and controllable, and is therefore embraced by a majority of patients with estrogen receptor positive breast cancer. The success of hormonal therapy is largely dependent on effective medicines that are potent, selective, and have favorable pharmacologic and pharmacokinetic profiles such as suitable absorption, distribution, metabolism and excretion (ADME) properties.
Breast cancer tumor cell proliferation is stimulated by the signal released when estrogen binds to estrogen receptors.5 In biosynthetic pathways, estrogens (estrone, estradiol and estriol) are synthesized from cholesterol through progestagens and androgens (e.g., androstenedione Fig. 1a). Aromatase (CYP19) catalyzes the aromatization reaction, the last and rate-limiting step in estrogen biosynthesis. Therefore, aromatase is a new and promising pharmaceutical target for hormonal-dependent breast cancer.6 Aromatase inhibitors (AIs) have many advantages over the traditional hormonal breast cancer therapy using selective estrogen receptor modulators (SERMs). These advantages include reversibility, total blockage and possibly reduced side effects.7 AI medicines are often categorized as steroidal or non-steroidal aromatase inhibitors (NSAIs) based on their structural similarity with steroids, or as 1st, 2nd and 3rd generations based on their evolution time.8 Intensive ongoing studies are focused on the third generation non-steroidal aromatase inhibitors (NSAIs), which can be further divided into the following classes based on their scaffold. The first class of NSAIs consists of analogs of third generation triazole AI drug molecules such as letrozole,9 anastrozole10 (Fig. 1b), which were approved by FDA and are used as first-line therapy in the treatment of breast cancer in postmenopausal women. These AIs are characterized by the presence of diarylmethyl and imidazole or triazole functionalities. Representative examples include arylimidazole and azolylbenzyl compounds.11 Since these aromatase inhibitors are structural modifications of synthetic NSAI drug molecules, they might have side effects similar to these of NSAI drugs when given on a long-term basis. The second class of NSAIs consists of natural products and their derivatives such as flavonoid, coumarin, resveratrol and lignan.12 Despite the scarcity of knowledge as to their mechanism of action, natural product drugs are believed to exhibit reduced side effects, which might be the results of the combined action of a mixture of constituents, or the multi-target function of one constituent.13 As the people are becoming aware of side effects of synthetic AI drugs, natural product NSAIs are attracting more and more attention recently for their future clinical uses.14 Some representative examples of naturally occurring flavonoid aromatase inhibitors are shown in Figure 1c (5,7-dihydroxyflavone) and 1d (5,7-dihydroxy -4′-methoxyisoflavone). In addition to the NSAIs mentioned above, new NSAI entities such as morpholino ethanone have recently been discovered using computational high-throughput docking studies.15
Figure 1.
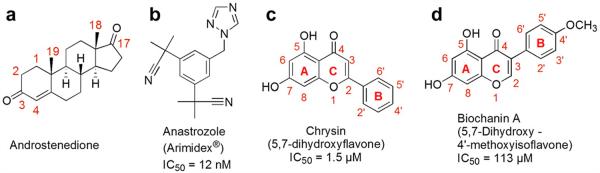
Molecular structures of the aromatase natural substrate and selected inhibitors. (a) Androstenedione, the natural substrate; (b) anastrozole (Arimidex®),10 a representative example of third-generation aromatase inhibitors in clinical use; (c), chrysin (from blue passion flower),16 a representative example of a naturally occurring flavone aromatase inhibitor; (d), biochanin A (from red clover),17 the most potent naturally occurring isoflavone aromatase inhibitor.
Flavonoid compounds, which include flavones, isoflavones, flavanones, and isoflavanones, display diverse biological activities such as antioxidant, anticancer, antiviral and anti-inflammatory activities.18 Natural flavones or their synthetic analogs have been reported to show inhibitory effects against aromatase (Fig. 1c). A common strategy to increase the potency of flavones is to introduce appropriate functional groups. Flavones with functionalities such as halides, nitro and amino have been synthesized and tested for their aromatase inhibitory effects by different groups working in this field.19 Specifically, methylated/hydroxylated flavanones,20 benzoflavanones,21 and imidazolyl flavones22 have been reported to be potent aromatase inhibitors. On the other hand, isoflavones, which differ from flavones in the phenyl substitution position on ring C, are not well studied probably due to their significantly poorer inhibitory effects against aromatase than flavones.23 Naturally occurring isoflavones such as daidzein, glycitein and genistein are either inactive or weak inhibitors against aromatase. Even though conflicting results exist, the most potent known natural isoflavone inhibitor is biochanin A (Fig. 1d) with reported IC50 values around 113 μM.17 Significant information on isoflavone AIs was provided by Brueggemeier, including first examples of isoflavone-based pyridyl-modified aromatase inhibitors24 in 2004 and subsequent improvements on isoflavone aromatase inhibitors with azoles, alkoxys, etc.25
Isoflavanones (Scheme 1) are a rare class of flavonoids that differ from isoflavones by having the C2–C3 bond saturated. We noticed that the isoflavone scaffold is inactive against aromatase, whereas the isoflavanone scaffold showed weak inhibition.20b A comparison of docking-predicted poses of isoflavones with isoflavanones revealed similar orientations in the enzyme pocket. However, due to the non-planarity of its scaffold, isoflavanone had an additional enzyme–ligand interaction with the highly hydrophobic enzyme pocket through its C2 methylene group. This CH2/π interaction26 might be responsible for the observed difference in the inhibitory activity between isoflavone and isoflavanone. We envisioned that with proper functional groups (see below) on this non-planar scaffold, isoflavanone could result in enhanced anti-aromatase activities. To the best of our knowledge, there is only one report that investigated aromatase inhibition by isoflavanone compounds such as 2-hydroxyisoflavanone (IC50 = 170 μM), 4′-hydroxyisoflavanone (IC50 = 160 μM) and 3′,4′-dihydroxyisoflavanone (IC50 ≥ 200 μM).20b Here we report the design and synthesis of a series of isoflavanone derivatives as aromatase inhibitors. A novel and convenient microwave-assisted gold-catalyzed annulation reaction was utilized to synthesize isoflavanone derivatives. Various substituents such as methyl, phenyl, methoxy and pyridyl were attached to the isoflavanone scaffold to explore hydrophobic interactions, hydrogen bonding and heme iron coordination. Computational docking of active isoflavanones into the aromatase active site was employed to detect crucial enzyme/inhibitor interactions. The calculated physicochemical properties of isoflavanone inhibitors are also discussed in order to obtain some preliminary insights on their long-term pharmacokinetic effects.
Scheme 1.
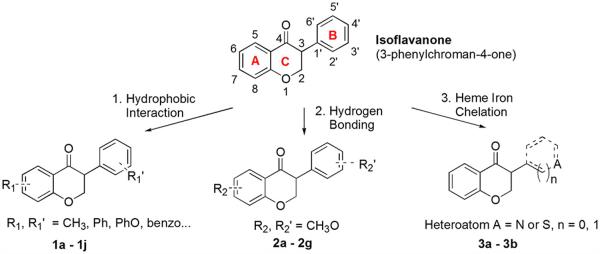
Structure and design strategy of isoflavanone aromatase inhibitors.
2. Results
2.1. Inhibitor design and synthesis
To explore the feasibility of isoflavanone compounds as aromatase inhibitors, a design strategy was adopted in which the isoflavanone core could be modified to yield diverse compounds with enhanced inhibitory potency. As revealed by the crystal structure of aromatase, there are three unique structural features of the aromatase active site.6b First, the enzyme binding pocket is highly hydrophobic. Therefore hydrophobic groups such as alkyl groups and aromatic rings may be favorable for enhanced binding. Second, hydrogen bonding plays an important role in enzyme–substrate interaction, and the 17-keto oxygen in the androstenedione substrate (Fig. 1a) forms a hydrogen bond with the Met 374 backbone amide. The hydroxyl of Ser 478 was also reported to participate in hydrogen bonding with carbonyl or cyano groups of the aromatase inhibitors.27 Third, the Fe2+ in the heme group of aromatase is capable of chelating to hetero atoms in inhibitors, especially N atoms. Many recent studies on aromatase inhibitors utilized pyridine,24,28 triazole29 or imidazole29,30 N-heterocyclic groups to enhance the coordination between the heme iron atom of the enzyme and the heterocyclic nitrogen lone pair. Based on these structural features, the design of isoflavanone aromatase inhibitors was centered on enhancement of hydrophobic interactions, hydrogen bonding and heme iron coordination (Scheme 1).
Isoflavanone compounds are normally synthesized in three steps from commercially available materials. A typical procedure involves a Lewis acid catalyzed Friedel–Crafts acylation of phenols, followed by an annulation of 2-hydroxyphenylketone and aldehyde to form isoflavone (3-phenylbenzopyrone), and finally hydrogenation of C2–C3 double bond to produce isoflavanone compounds.31 However, these synthetic routes are laborious, and harmful or sensitive reagents are utilized. Considering the commercial availability of various hydroxyaldehydes and phenylacetylenes, we employed a AuCN/nBu3P(1:25)-catalyzed annulation reaction of salicylaldehyde and phenylacetylene in toluene at 150 °C for 36 h to assemble the isoflavanone scaffold in one step.32 However, the long reaction time and the large amount of nBu3P ligand required motivated us to search for new reaction conditions. Therefore we investigated this reaction under microwave conditions and found that the reaction time was greatly reduced from 36 h to only 10 min when subjected to microwave irradiation at high temperatures. The reaction solvent, toluene, poorly absorbs microwave irradiation, hence the amount of AuCN catalyst was increased from 1% to 5% to facilitate temperature increase. The final optimized reaction conditions to synthesize isoflavanone derivatives under microwave irradiation were 5% AuCN, 25% nBu3P, 200 °C for 10 min (Scheme 2). Utilizing these new reaction conditions, twenty three isoflavanone derivatives were synthesized and evaluated for their inhibitory effects against aromatase (Table 1). These compounds were fully characterized by 1H NMR, 13C NMR and mass spectroscopy. Three proton signals in the range of 3.90–4.70 ppm were observed in the 1H NMR spectra, indicative of the aliphatic protons attached to C2 and C3 in the product isoflavanone scaffold.
Scheme 2.
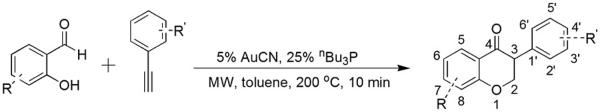
Microwave-assisted annulation reactions of hydroxyaldehydes and alkynes.
Table 1.
Chemical structures and aromatase inhibition of isoflavanone derivatives 1a–4d
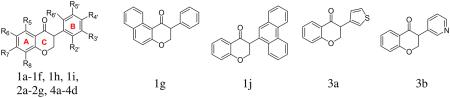
|
| Compd | R5 | R6 | R7 | R8 | R2′ | R3′ | R4′ | R5′ | R6′ | IC50 a (μM) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1a | H | H | H | H | H | H | H | H | H | 29 ± 4 |
| 1b | H | CH3 | H | H | H | H | H | H | H | 44 ± 11 |
| 1c | H | H | CH3 | H | H | H | H | H | H | 18 ± 7 |
| 1d | H | H | H | CH3 | H | H | H | H | H | N.A.b |
| 1e | H | H | H | H | H | CH3 | H | H | H | 20 ± 7 |
| 1f | H | H | H | H | H | H | CH3 | H | H | 41 ± 7 |
| 1g | Benzo | H | H | H | H | H | H | H | 20 ± 9 | |
| 1h | H | H | H | H | H | H | PhO | H | H | 2.4 ± 2.2 |
| 1i | H | H | H | H | H | H | Ph | H | H | Inactive |
| 1j | H | H | H | H | 9′-Phenanthryl | Inactive | ||||
| 2a | H | CH3O | H | H | H | H | H | H | H | 0.26 ± 0.06 |
| 2b | H | H | CH3O | H | H | H | H | H | H | 41 ± 16 |
| 2c | H | H | H | CH3O | H | H | H | H | H | N.A.b |
| 2d | H | H | H | H | CH3O | H | H | H | H | 97 ± 70 |
| 2e | H | H | H | H | H | CH3O | H | H | H | 99 ± 27 |
| 2f | H | H | H | H | H | H | CH3O | H | H | 24 ± 14 |
| 2g | H | H | H | H | H | CH3O | H | CH3O | H | 11 ± 7 |
| 3a | H | H | H | H | 3′-Thiophene | 30 ± 9 | ||||
| 3b | H | H | H | H | 3′-Pyridyl | 5.8 ± 1.8 | ||||
| 4a | H | F | H | H | H | H | H | H | H | 32 ± 7 |
| 4b | H | Cl | H | H | H | H | H | H | H | 5.1 ± 3.4 |
| 4c | H | Br | H | H | H | H | H | H | H | 29 ± 10 |
| 4d | H | tBu | H | H | H | H | H | H | H | 61 ± 15 |
IC50 values are average of at least three runs and standard deviations.
N.A. = not available. Compounds exhibit fluorescence which interferes with the bioassay.
2.2. Aromatase inhibition
The evaluation of the synthesized isoflavanones for aromatase inhibitory activity was performed using a fluorometric substrate (7-methoxy-trifluoromethylcoumarin) and human CYP19 aromatase with ketoconazole as a positive control.33 All experiments were performed at least in triplicate to determine the IC50 value (Fig. 2). Since racemic compounds were synthesized, the activities reported in this paper represent the lower limits of inhibitory potency. Interestingly, the parent molecule 1a showed moderate inhibition against aromatase with an IC50 value of 29 μM (Table 1). Under the same conditions, no significant inhibition was found with compounds 1i and 1j, in which the B ring was attached to a large aromatic group. All other compounds showed moderate to good inhibitory effects, with 2a being the most potent compound in this investigation (Table 1). Both enantiomers of synthesized isoflavanones were docked into the aromatase active site, and the enantiomer with the higher docking score was used to identify crucial enzyme/inhibitor interactions (Fig. 3).
Figure 2.

Aromatase inhibition by selected isoflavanone compounds and ketoconazole. Relative fluorescence is proportional to enzyme activity. Logistic fit reveals the IC50, a measure of potency (inhibitor concentration required to inhibit half of the enzyme’s activity).
Figure 3.

Representations of molecular docking performed in this study. The inhibitor is depicted as green sticks. The heme group is shown in orange. Selected relevant amino acid residues in aromatase are shown as sticks (white, H; black, C; blue, N; red, O). Lines are drawn between the atoms likely involved in hydrogen bonding or heme coordination. (a) 5,6-Benzoisoflavanone 1g; (b) 4′-phenoxyisoflavanone 1h; (c) 6-methoxyisoflavanone 2a; (d) 3′,5′-dimethoxyisoflavanone 2g; and (e) inhibitor 3-(pyridin-3-yl)chroman-4-one 3b.
3. Discussion
3.1. Hydrophobic interactions
The aromatase (CYP19) active site is highly hydrophobic and is dominated by aliphatic amino acid residues such as Met 374, Val 373, Val 370, Ile 305, Ala 306, Ile 133, Trp 224, Leu 372, Leu 477, Phe 134 and Thr 310. Hence inhibitors with alkyl or aromatic groups are expected to bind with high affinity.6b A methyl group was introduced to the A or B rings of isoflavanones with 1c and 1e being the most potent with IC50 values of 18 μM and 20 μM. There was no significant difference in activities between the methylated analogs and the model compound 1a. Overall, the presence and the position of the methyl substituent on the isoflavanone scaffold did not influence the inhibitory activity significantly. In addition to the methyl group, benzo and phenoxy groups were also installed for possible π–π stacking interactions. The extension of the isoflavanone A ring by a benzo group at positions 5 and 6 (compound 1g) increased the inhibitory potency slightly in comparison to 1a (IC50 = 20 μM). The docking pose of 5,6-benzoisoflavanone 1g in the aromatase active site (Fig. 3a) indicated that the primary interactions between 1g and the enzyme active site were the coordination between the carbonyl O atom in 1g and the iron in the heme group (distance = 2.5 Å). The benzo group attached to the isoflavanone A ring was predicted to have π–π stacking interactions with aromatase residue Trp 224.
Compounds 4′-phenoxyisoflavanone 1h and 4′-phenylisoflavanone 1i have some structural similarity by bearing an additional aromatic group on ring B, while their inhibitory potencies are surprisingly different. The inhibition potency of 1h increased 12-fold compared to the model compound 1a with an IC50 of 2.4 μM, while 1i was inactive. The differences in their activities may be attributed to the flexibilities of these two molecules. The presence of the oxygen bridge in compound 1h gives the molecule more flexibility so that it can bend and undergo π–π stacking interactions. On the other hand, compound 1i with a biphenyl group is rigid and cannot fit into the pocket. The same reason might also apply for compound 3-(phenanthren-9-yl)chroman-4-one 1j. Therefore, there were no anti-aromatase activities observed for 1h and 1j.
As mentioned above, the key interactions identified from computer modeling of 1h into the enzyme active site were π–π stacking interactions (Fig. 3b). The distance between the PhO benzene ring of 1h and aromatase residue Phe 221 was 2.7 Å, indicating strong π–π stacking interactions. The Asp 309 side chain was in close proximity with the oxygen atom of the 4′-phenoxide group. However, no H-bonds between 1h and Asp 309 were predicted by the docking results. The π–π stacking interactions between the A ring of inhibitor 1h and the heme group were also predicted (distance = 3.0 Å), as can be seen from the docking pose. Additionally, the carbonyl O atom in 1h was positioned to coordinate to the heme iron, similar to the heme coordination predicted for compound 1g (Fig. 3a).
3.2. Hydrogen bonds
In order to increase hydrogen bonding between inhibitor and enzyme, isoflavanone derivatives with methoxy groups capable of acting as H-bond acceptors at various positions were synthesized (compounds 2a–2f). The most potent inhibitor was 6-methoxyisoflavanone 2a which showed an about 100-fold increase in inhibition potency (IC50 = 0.26 μM). The Met 374 backbone NH and the 6-methoxy group in 2a is in close proximity (Fig. 3c), which might have favorable hydrogen bonding interactions. In addition, the π–π stacking interactions (distance = 3.2–4.1 Å) between the B ring of 2a and the heme porphyrin may also contribute to the observed high affinity. One should point out that both compounds 2a and 2b have increased electron density on ring A due to the presence of the electron-donating methoxy group. Since ring A was not involved in π–π stacking interactions with the enzyme active site for both compounds, there was no benefit from increasing the electron density on ring A.
The B ring methoxylated isoflavanone did not show much promise at first (compounds 2d–2f). When a dimethoxylated compound 3′,5′-dimethoxyisoflavanone 2g was synthesized, however, the corresponding IC50 value was 11 μM, about a threefold increase compared to the parent molecule 1a. Docking indicated that the aromatase heme iron might coordinate to the carbonyl oxygen atom in 2g (Fig. 3d). A careful examination of the enzyme pocket around the heme group revealed that this was mostly a hydrophobic area which was composed of non-polar amino acids such as Ile 133, Leu 301, Ala 306, Ile 305, Phe 221, Trp 224 and Val 370. Therefore, the hydrophobic interactions between 3′,5′-dimethoxyisoflavanone 2g and the enzyme active site also contributed to the enhanced inhibitory potency. It should be pointed out that hydrogen bonding interactions were predicted between the other enantiomer of 2g and aromatase. Specifically, the NH in aromatase residue Arg 192 formed a hydrogen bond (2.0 Å) with the oxygen in 3′-methoxy group, and the -OH of the Ser 478 side chain formed a second hydrogen bond (2.1 Å) with the 5′-methoxy group. However, this enantiomer did not fit into the enzyme active site very well due to reduced hydrophobic interactions and received a lower docking score. Even though enhanced inhibitory potency was observed for the B-ring methoxylated compound 2g, one should keep in mind that the introduction of polar groups (e.g., –OH, –OCH3) on ring B of isoflavanone might also decrease activity, as it seems to be the case for 2′-methoxyisoflavanone 2d (IC50 = 97 μM), 3′-methoxyisoflavanone 2e (IC50 = 99 μM), 4′-hydroxyisoflavanone (IC50 = 160 μM)20b and 3′,4′-dihydroxyisoflavanone (IC50 = 200 μM).20b
3.3. Heme iron coordination
The coordination of the inhibitor with the iron atom of the heme moiety is an important feature of potent and selective inhibitors of aromatase. Functional groups such as imidazole, triazole and pyridine are able to form nitrogen heme iron coordination, and others such as phenol or thiophene may coordinate to the heme iron.34 3-(thiophen-3-yl)chroman-4-one 3a and 3-(pyridin-3-yl)chroman-4-one 3b were synthesized in order to probe the heme iron coordination. Compound 3a with a thiophene group which might coordinate to heme iron did not exhibit a significant increase in potency in the inhibition assay (Table 1). Compound 3b with a pyridine group showed fivefold increase in aromatase inhibitory potency (IC50 = 5.8 μM), likely due to the coordination to the heme iron. This observation was supported by our docking study, which suggested that the pyridine ring was about 2.4 Å away from heme group, and the pyridyl nitrogen coordinated to the iron in the heme group (Fig. 3e). Our attempts to synthesize 3-(pyridin-2-yl)chroman-4-one and 3-(1-methyl-1H-imidazol-5-yl)chroman-4-one were not successful, probably due to the reduced the activities of gold(I) catalysts upon their coordination to the N-heteroaromatic alkynes.35 Information on heme iron coordination with other N-heterocyclic rings such as pyridine and imidazole could not be provided for the current study. Since the enzyme pocket surrounding the heterocyclic ring of chroman-4-one inhibitors are hydrophobic and sterically restricted, further optimization on this series of molecules has to take these factors into consideration by avoiding large substituents (e.g., –OPh, biphenyl) or polar substituents (e.g., –OH, –OCH3).
3.4. Further exploration of 6-substituted isoflavanones
Prompted by the inhibitory effect of 6-methoxyisoflavanone, we examined the effects of different substituents at the C6 position of the parent molecule. Comparison of compounds 1b, 1g and 2a revealed that a relatively big polar group at this position might enhance the interactions between isoflavanone and the aromatase pocket. Specifically, introduction of an electron-donating substituent such as a methoxy group or electron-withdrawing substituent such as a benzo at the C5–C6 position on the A ring increased the aromatase inhibitory effect. Following these results, isoflavanones with F, Cl, Br and tbutyl at C6 position were prepared and tested for their aromatase inhibition (Table 1 4a–4d). Enhanced inhibitory effects were observed for compounds 4b which had a chlorine group. The presence of a tbutyl group at C6 (compound 4d) reduced the binding affinity. These observations could be explained from the electronic and steric properties of the amino acid residues around C6 position of isoflavanone. As mentioned previously, this region is spacious and polar due to the presence of Ser 478 and Arg 115 lining the wall of the pocket. A polar functional group with reasonable size might fit into this region and interact favorably by steric and electronic effects. Therefore, the modulation at C6 of isoflavanone with chlorine improved the binding affinity while that with the non-polar tbutyl decreased the binding interactions.
3.5. Calculated physicochemical properties
In order to explore the bioavailability of isoflavanone derivatives, theoretical calculations were carried out to predict some physicochemical properties of synthesized compounds. Lipinski’s rule of five36 and the later addition of other parameters such as polar surface area (PSA),37 well known in pharmaceutical industry, outline the basic principles for orally active small molecules: molecular weight (MW) should be under 500, lipophilicity which is expressed as logarithm of partition coefficient between n-octanol and water (cLogP) should be less than 5, the number of hydrogen bond donors (Hd) should be no more than 5, the number of hydrogen bond acceptors (Ha) should be no more than 10, and the topological polar surface area (TPSA) should be smaller than 90 Å.2 Among these parameters, LogP, the measure of the compound’s solubility and permeability, is believed to be very important. Very high lipophilicity and the resulted large LogP values cause poor absorption or permeation and should be avoided.
The calculated physicochemical properties38 of some of the most active isoflavanone inhibitors together with those for anastrozole are listed in Table 2. Based on these calculations, compounds 5,6-benzoisoflavanone 1g and 4′-phenoxyisoflavanone 1h had very large cLogP values, 4.55 and 5.15, respectively, which might be disadvantageous with regard to the pharmacokinetic properties of these molecules in biological systems. Compounds 2a, 2g and 3b, exhibited favorable cLogP values. The polar surface areas of these molecules were relatively small ranging from 26.3 to 44.8 Å2 in comparison with the average value (90 Å2) for approved drug molecules. This provides an excellent opportunity to modify these isoflavanone derivatives with additional functional groups.
Table 2.
Calculated physicochemical properties of isoflavanone derivatives and anastrozole®
| Compd Id. | cLogPa | MWb | Hac | Hdd | TPSAe (Å2) | IC50 (μM) |
|---|---|---|---|---|---|---|
| 1g | 4.55 | 274.3 | 2 | 0 | 26.3 | 20 |
| 1h | 5.15 | 316.4 | 3 | 0 | 35.5 | 2.4 |
| 2a | 3.43 | 254.3 | 3 | 0 | 35.5 | 0.26 |
| 2g | 3.44 | 284.3 | 4 | 0 | 44.8 | 11 |
| 3b | 1.69 | 225.3 | 3 | 0 | 39.2 | 5.8 |
| Anastrozole® | 2.85 | 293.4 | 5 | 0 | 78.3 | 0.01010 |
Log P, calculated logarithm of partition coefficient between n-octanol and water.
MW, molecular weight.
Ha, hydrogen bond acceptor.
Hd, hydrogen bond donor.
TPSA, topological polar surface area.
Figure 4 summarizes our current understanding as to how isoflavanones bind to the active site of aromatase. These compounds bind to the active site in an orientation such that their A, C and B rings mimic D, C and A rings of the steroids, respectively. The amino acids responsible for important interactions with isoflavanone derivatives are Phe 221, Met 374, Arg 192, Ser 478 and Asp 309. The modification on the A and B rings of isoflavanone compounds has resulted in aromatase inhibitors exhibiting stronger binding affinity and enhanced activities.
Figure 4.
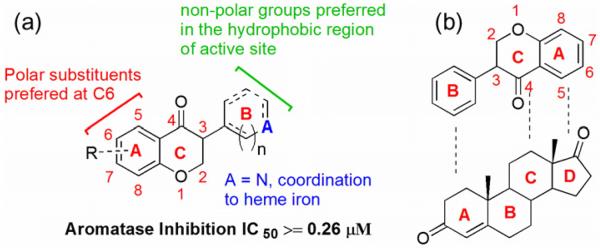
(a) Structural features of isoflavanones required for effective inhibition of aromatase; (b) structural similarity between isoflavanone scaffold and aromatase natural substrate—androstenedione.
4. Conclusion
In conclusion, we have demonstrated in this work that isoflavanones can be used as aromatase inhibitors through appropriate structural modulation. For the first time, a small set of isoflavanones with various functionalities was synthesized via a one-step annulation reaction and tested for their aromatase inhibitory effects. Compared to other flavanoid aromatase inhibitors, our synthesis of isoflavanone derivatives was straightforward utilizing microwave irradiation and readily available hydroxyaldehydes and aromatic alkynes as starting materials. In addition, the most active compounds identified from this study showed favorable physicochemical properties which are promising for further development of this series of compounds.
In comparison to isoflavones, isoflavanone derivatives with functional groups such as methoxy, phenoxy, chloride, pyridyl, etc. demonstrated good inhibitory potencies against aromatase, revealing that the non-planarity configuration of the isoflavanone scaffold might play an important role in enzyme–ligand binding. Specifically, compound 2a bearing a methoxy group at C6 position stood out as the most potent inhibitor with a 100-fold enhancement in potency compared to model compound 1a. Our data also showed that the key structural features that enable isoflavanones bind to the aromatase active site with high affinity are as follows: (1) Isoflavanone rings A or B may participate in π–π stacking interactions with aromatase’s porphyrin ring or amino acid residues, such as Phe 221, Trp224 and Phe 134. (2) Methoxy groups could increase binding affinity by forming hydrogen bonds with aromatase residues, such as Met 374, Arg 192 and Ser 478. (3) Heterocyclic nitrogen or carbonyl oxygen atom of the inhibitors can coordinate to the heme iron atom and thereby enhance the binding interactions. The calculated physicochemical properties of these compounds indicated that it is possible to obtain more potent and selective aromatase inhibitors by appropriate modification on the isoflavanone scaffold. Compounds 2a, 2g and 3b showed not only high potency against aromatase but also favorable physicochemical properties, demonstrating that the isoflavanone scaffold can be further optimized for the development of new therapeutic agents for hormone-dependent breast cancer.
5. Experimental
5.1. Chemistry
Reagents and solvents were obtained from Aldrich and used without further purification unless otherwise noted. Toluene was freshly distilled from CaH2 prior to use. The microwave-assisted reactions were conducted on a single-mode Discover System from CEM Corporation. Power cooling was turned off manually during the reaction to ensure that the reaction temperature reached 200 °C. Thin-layer chromatography was performed using pre-coated silica gel F254 plates (Whatman). Column chromatography was performed using pre-packed RediSep Rf Silica columns on a CombiFlash Rf Flash Chromatography system (Teledyne Isco). NMR spectra were obtained on a Joel 500 MHz spectrometer. Chemical shifts were reported in parts per million (ppm) relative to the tetramethylsilane (TMS) signal at 0.00 ppm. Coupling constants, J, were reported in Hertz (Hz). The peak patterns were indicated as follows: s, singlet; d, doublet; t, triplet; dt, doublet of triplet; dd, doublet of doublet; m, multiplet; q, quartet. Analytical reverse-phase HPLC was carried out using a system consisting of a 1525 binary HPLC pump and 2996 photodiode array detector (Waters Corporation, Milford, MA). A Nova-Pak C18 column (4 μm, 3.9 × 150 mm), also from Waters, was used with a mobile phase of methanol and water (60:40, vol/vol) plus 0.25% acetic acid, flow rate 1.2 mL/min and UV detection wavelength at 250 nm. Control and data acquisition was done using the Empower 2 software (Waters Corporation, Milford, MA). Synthesized compounds were prepared in methanol to make 1.0 mg/mL stock solution and 10 μL solution was injected for HPLC test. The purity of all the compounds was assessed by HPLC under 254 nm. All final compounds were confirmed to be ≥95% purity by analysis of their peak area. Mass spectra were obtained on a Water TQD Tandem Quadrapole Mass Spectrometer, and data was collected in electrospray positive mode (ESI+). High resolution mass spectra were recorded on a Micromass Q-TOF 2 or a Thermo Scientific LTQ-FT™ mass spectrometer operating in electrospray (ES) mode. Calculation of important Physicochemical properties (logP, number of hydrogen bond donors and acceptors and polar surface area) was performed using Molinspiration Cheminformatics Software at URL http://www.molinspiration.com.
5.1.1. General procedure for isoflavanone formation
5.1.1.1. Method A (Thermal Process).
To a 5-mL micro reaction vial equipped with a stirring bar was added AuCN (0.03 mmol, 0.03 equiv, 6.6 mg), Bu3P(0.75 mmol, 0.75 equiv, 185.1 μL), aldehyde (1 mmol, 1 equiv), alkyne (3 mmol, 3 equiv) and 4 mL of freshly distilled toluene. The reaction mixture was stirred at 150 °C for 36 h. After cooling down to room temperature, the crude mixture was loaded directly on silica gel and was purified by Medium Performance Liquid Chromatography, eluding with an ethyl acetate/hexanes gradient to afford the desired products.
5.1.1.2. Method B (Microwave Irradiation Process)
The microwave-assisted reactions were conducted on a single-mode Discover System from CEM Corporation. To an oven-dried standard microwave reaction vial (capacity 10 mL) equipped with a stirring bar was added AuCN (0.05 mmol, 0.05 equiv, 11.0 mg), Bu3P(0.25 mmol, 0.25 equiv, 61.7 μL), aldehyde (1 mmol, 1 equiv), alkyne (3 mmol, 3 equiv) and 1 mL of freshly distilled toluene. The reaction vial was then sealed with a Teflon septum cap, and the sample was subjected to microwave irradiation at a power of 200 W for 10 min (hold time) at 200 °C. After being cooled down, the vial was opened, and the crude mixture was loaded directly on silica gel and was purified by Medium Performance Liquid Chromatography eluding with an ethyl acetate/hexanes gradient to afford the desired products.
5.1.1.3. 6-Methyl-3-phenylchroman-4-one (1b)
Synthesized from 2-hydroxy-5-methyl-benzaldehyde (1 mmol, 1 equiv, 136.15 mg) and phenylacetylene (3 mmol, 3 equiv, 330 μL) according to the general procedure for the synthesis of isoflavanone derivatives Method A (Thermal Process) described above. Light yellow solid. Rf = 0.39 (10% EtOAc/Hex). Yield, 26.4%. Purity, 96.5%. 1H NMR (CDCl3, 500 MHz, ppm): δ 7.76 (s, 1H, 5-H), 7.28 (m, 6H, Ar-H), 6.91 (d, J = 4.4 Hz, 1H, 8-H), 4.64 (m, 2H, 2-H), 3.96 (t, J = 7.1 Hz, 1H, 3-H), 2.32 (s, 3H, CH3). 13C NMR (CDCl3, 125 MHz, ppm): δ 192.5, 159.8, 137.3, 135.3, 131.2, 129.0, 128.71, 127.8, 127.4, 120.7, 117.8, 71.6, 52.5, 20.5. HRMS Calcd for C16H14O2Na [M+Na] 261.0891. Found: 261.0890.
5.1.1.4. 7-Methyl-3-phenylchroman-4-one (1c)
Synthesized from 2-hydroxy-4-methylbenzaldehyde (1 mmol, 1 equiv, 138.9 mg) and phenylacetylene (3 mmol, 3 equiv, 330 μL) according to the general procedure for the synthesis of isoflavanone derivatives Method B (Microwave Irradiation Process) described above. Light yellow solid. Rf = 0.41 (10% EtOAc/Hex). Yield, 31.6%. Purity, 98.3%. 1H NMR (CDCl3, 500 MHz, ppm): δ 7.85 (d, J = 8.3 Hz, 1H, 5-H), 7.25–7.31 (m, 5H, Ar-H), 6.86 (d, J = 8.3 Hz, 1H, 6-H), 6.82 (s, 1H, 8-H), 4.64 (d, J = 9.2, 2H, 2-H), 3.96 (t, J = 7.8, 1H, 3-H), 2.37 (s, 3H, CH3). 13C NMR (CDCl3, 125 MHz, ppm): δ 191.9, 161.7, 147.7, 135.4, 128.9, 128.7, 127.8, 127.7, 123.1, 118.9, 117.9, 71.6, 52.3, 22.0. HRMS Calcd for C16H14O2Na+ [M+Na] 261.0891. Found: 261.0892.
5.1.1.5. 8-Methyl-3-phenylchroman-4-one (1d)
Synthesized from 2-hydroxy-3-methyl-benzaldehyde (1 mmol, 1 equiv, 120.0 μL) and phenylacetylene (3 mmol, 3 equiv, 330 μL) according to the general procedure for the synthesis of isoflavanone derivatives Method A (Thermal Process) described above. Yellow solid. Rf = 0.51 (10% EtOAc/Hex). Yield, 31.4%. Purity 98.8%. 1H NMR (CDCl3, 500 MHz, ppm): δ 7.84 (d, J = 7.3 Hz, 1H, 5-H), 7.28–7.40 (m, 6H, Ar-H), 6.96, (t, J = 7.5, 1H, 6-H), 4.68 (m, 2H, 2-H), 4.00 (dd, J = 9.1, 5.5 Hz, 1H, 3-H), 2.29 (s, 3H, CH3). 13C NMR (CDCl3, 125 MHz, ppm): δ 192.9, 159.8, 137.0, 135.3, 129.0, 128.8, 127.8, 127.2, 125.4, 121.1, 71.5, 52.3, 15.8. HRMS Calcd for C16H14O2Na [M+Na] 261.0891. Found: 261.0882.
5.1.1.6. 3-m-Tolylchroman-4-one (1e)
Synthesized from salicylaldehyde (1 mmol, 1 equiv, 105.0 μL) and 1-ethylyn-3-methylbenzene (3 mmol, 3 equiv, 387 μL) according to the general procedure for the synthesis of isoflavanone derivatives Method A (Thermal Process) described above. Light yellow solid. Rf = 0.42 (10% EtOAc/Hex). Yield, 38.0%. Purity, 99.2%. 1H NMR (CDCl3, 500 MHz, ppm): δ 7.97 (d, J = 9.6 Hz, 1H, 5-H), 7.50 (t, J = 7.8 Hz, 1H, 7-H), 7.24 (t, J = 7.8 Hz, 1H, 6-H), 7.01–7.13 (m, 5H, Ar-H), 4.66 (d, J = 6.8 Hz, 2H, 2-H), 3.97 (t, J = 7.3 Hz, 1H, 3-H), 2.34 (s, 3H, CH3). 13C NMR (CDCl3, 125 MHz, ppm): δ 192.7, 161.7, 138.6, 136.1, 135.0, 129.5, 128.9, 128.7, 127.9, 125.7, 121.7, 121.2, 117.7, 71.6, 52.4, 21.5. HRMS Calcd for C16H14O2Na [M+Na] 261.0891. Found: 261.0879.
5.1.1.7. 3-p-Tolylchroman-4-one (1f)39
Synthesized from salicylaldehyde (1 mmol, 1 equiv, 104.7 μL), and 4-ethynyltoluene (3 mmol, 3 equiv, 380.5 μL) according to the general procedure for the synthesis of isoflavanone derivatives Method A (Thermal Process) described above. Light yellow. Light yellow solid. Rf = 0.26 (10% EtOAc/Hex). Yield, 28.7%. Purity, 99.4%. 1H NMR (CDCl3, 500 MHz, ppm): δ 7.98 (d, J = 7.8 Hz, 1H, 5-H), 7.51 (t, J = 7.8 Hz, 1H, 7-H), 7.18 (m, 4H, Ar-H), 7.02–7.08 (m, 2H, 6-H, 8-H), 4.65 (d, J = 8.5 Hz, 2H, 2-H), 3.97 (t, J = 6.0 Hz, 1H, 3-H), 2.35 (s, 3H, CH3). 13C NMR (CDCl3, 125 MHz, ppm): δ 192.5, 179.7, 161.7, 137.6, 136.1, 132.0, 129.7, 128.5, 127.8, 121.6, 117.9, 71.6, 52.0, 21.2. HRMS Calcd for C16H14O2Na [M+Na] 261.0891. Found: 261.0889.
5.1.1.8. 2-Phenyl-2,3-dihydro-1H-benzo[f]chromen-1-one (1g)39
Synthesized from 2-hydroxy-1-naphthaldehyde (1 mmol, 1 equiv, 175.4 mg) and phenylacetylene (3 mmol, 3 equiv, 330 μL) according to the general procedure for the synthesis of isoflavanone derivatives Method B (Microwave Irradiation Process) described above. Light yellow solid. Rf = 0.42 (10% EtOAc/Hex). Yield, 17.1%. Purity, 96.1%. 1H NMR (CDCl3, 500 MHz, ppm): δ 9.48 (d, J = 8.7 Hz, 1H), 7.95 (d, J = 9.2, 1H), 7.76 (d, J = 7.8, 1H), 7.62 (t, J = 7.8, 1H), 7.43 (t, J = 7.6 Hz, 1H), 7.37–7.25 (m, 5H, Ar-H), 7.13 (d, J = 9.2 Hz, 1H), 4.80 (d, J = 7.4 Hz, 2H, 2-H), 4.07 (t, J = 6.9 Hz, 1H, 3-H). 13C NMR (CDCl3, 125 MHz, ppm): δ 193.3, 163.6, 137.7, 135.7, 131.9, 129.8, 129.4, 128.9, 128.6, 128.5, 127.8, 126.1, 125.0, 118.7, 112.6, 71.3, 52.9. HRMS Calcd for C19H14O2Na+ [M+Na] 297.0891. Found: 297.0888.
5.1.1.9. 3-(4-Phenoxyphenyl)chroman-4-one (1h)
Synthesized from salicylaldehyde (0.5 mmol, 1 equiv, 52.4 μL) and 1-ethynyl-4-phenoxybenzene (1.5 mmol, 3 equiv, 271 μL) according to the general procedure for the synthesis of isoflavanone derivatives Method A (Thermal Process) described above. Yellow solid. Rf = 0.38 (10% EtOAc/Hex). Yield, 22.0%. Purity, 98.2%. 1H NMR (CDCl3, 500 MHz, ppm): δ 7.97 (d, J = 6.8 Hz, 1H, 5-H), 7.51 (t, J = 7.8 Hz, 1H, 7-H), 7.34 (t, J = 8.0, 2H, Ar-H), 7.24 (d, J = 10.5, 2H, Ar-H), 6.98–7.06 (m, 7H, Ar-H), 4.65 (m, 2H, 2-H), 3.99 (dd, J = 8.7, 5.5 Hz, 1H, 3-H). 13C NMR (CDCl3, 125 MHz, ppm): δ 192.3, 161.7, 157.1, 157.0, 136.2, 130.0, 129.9, 129.7, 127.9, 123.6, 121.7, 121.1, 119.3, 119.1, 118.0, 71.6, 51.7. HRMS Calcd for C21H16O3Na [M+Na] 339.0997, found 339.1002.
5.1.1.10. 3-(Biphenyl-4-yl)chroman-4-one (1i)39
Synthesized from salicylaldehyde (1 mmol, 0.5 equiv, 52.4 μL) and 4-ethynylbiphenyl (3 mmol, 1.5 equiv, 267.3 mg) according to the general procedure for the synthesis of isoflavanone derivatives Method A (Thermal Process) described above. Light yellow solid. Rf = 0.40 (10% EtOAc/Hex). Yield, 26.2%. Purity 97.0%. 1H NMR (CDCl3, 500 MHz, ppm): δ 8.00 (d, J = 7.8 Hz, 1H, 5-H), 7.25–7.60 (m, 10H, Ar-H), 7.03–7.09 (m, 2H, Ar-H), 4.71 (d, J = 6.4, 2H, 2-H), 4.06 (t, J = 7.3 Hz, 1H, 3-H). 13C NMR (CDCl3, 125 MHz, ppm): δ 192.4, 180.0, 161.7, 140.9, 140.8, 136.2, 134.1, 129.1, 128.9, 127.9, 127.7, 127.5, 127.2, 121.8, 118.0, 71.6, 52.1. HRMS Calcd for C21H16O2Na [M+Na] 323.1048. Found: 323.1038.
5.1.1.11. 3-(Phenanthren-9-yl)chroman-4-one (1j)
Synthesized from salicylaldehyde (0.5 mmol, 1 equiv, 52.4 μL) and 9-ethynylphenanthrene (1.5 mmol, 3 equiv, 303.4 mg) according to the general procedure for the synthesis of isoflavanone derivatives Method B (Microwave Irradiation Process) described above. Light yellow solid. Rf = 0.36 (10% EtOAc/Hex). Yield 16.2%. Purity, 98.2%. 1H NMR (CDCl3, 500 MHz, ppm): δ 8.77 (d, J = 8.3 Hz, 1H, Ar-H), 8.67 (d, J = 8.3 Hz, 1H, Ar-H), 8.05–8.10 (m, 2H, Ar-H), 7.80 (d, J = 8.3, 1H, Ar-H), 7.63–7.71 (m, 4H, Ar-H), 7.55–7.59 (m, 2H, Ar-H), 7.08–7.13 (m, 2H, Ar-H), 4.82–4.93 (m, 3H, 2-H and 3-H). 13C NMR (CDCl3, 125 MHz, ppm): δ 192.3, 161.7, 136.2, 131.3, 131.1, 130.6, 130.3, 129.9, 128.7, 128.0, 127.6, 127.1, 127.0, 126.9, 127.7, 124.1, 126.6, 122.6, 121.9, 124.5, 118.1, 71.5, 49.0. HRMS Calcd for C23H16O2Na [M+Na] 347.1048, found 347.1047.
5.1.1.12. 6-Methoxy-3-phenylchroman-4-one (2a)
Synthesized from 2-hydroxy-5-methoxy-benzaldehyde (1 mmol, 1 equiv, 123.7 μL) and phenylacetylene (3 mmol, 3 equiv, 330 μL) according to the general procedure for the synthesis of isoflavanone derivatives Method A (Thermal Process) described above. Light yellow solid. Rf = 0.28 (10% EtOAc/Hex). Yield, 30.4%. Purity, 98.2%. 1H NMR (CDCl3, 500 MHz, ppm): δ 7.26–7.38 (m, 6H, Ar-H), 7.11 (d, J = 4.6 Hz, 1H, 7-H), 6.95 (d, J = 4.6 Hz, 1H, 8-H), 4.64 (m, 2H, 2-H), 3.97 (d, J = 4.1 Hz, 1H, 3-H), 3.81 (s, 3H, OCH3). 13C NMR (CDCl3, 125 MHz, ppm): δ 192.2, 156.4, 154.3, 135.4, 129.0, 128.7, 127.9, 125.4, 120.9, 119.3, 108.1, 71.8, 55.9, 52.4. HRMS Calcd for C16H14O3Na [M+Na] 277.0841. Found: 277.0828.
5.1.1.13. 7-Methoxy-3-phenylchroman-4-one (2b)39
Synthesized from 2-hydroxy-4-methoxybenzaldehyde (1 mmol, 1 equiv, 159.2 mg) and phenylacetylene (3 mmol, 3 equiv, 330 μL) according to the general procedure for the synthesis of isoflavanone derivatives Method A (Thermal Process) described above. Light yellow solid. Rf = 0.22 (10% EtOAc/Hex). Yield, 37.1%. Purity, 97.7%. 1H NMR (CDCl3, 500 MHz, ppm): δ 7.90 (d, J = 8.8 Hz, 1H, 5-H), 7.25–7.34 (m, 5H, Ar-H), 6.61 (d, J = 8.7 Hz, 1H, 6-H), 6.45 (s, 1H, 8-H), 4.65 (m, 2H, 2-H), 3.92 (dd, J = 7.8, 5.5 Hz, 1H, 3-H), 3.85 (s, 3H, OCH3). 13C NMR (CDCl3, 125 MHz, ppm): δ 190.9, 166.2, 163.6, 135.5, 129.6, 128.9, 128.7, 127.8, 115.0, 110.3, 100.8, 72.0, 55.7, 52.0. HRMS Calcd for C16H14O3Na [M+Na] 277.0841. Found: 277.0830.
5.1.1.14. 8-Methoxy-3-phenylchroman-4-one (2c)
Synthesized from o-vanillin (1 mmol, 1 equiv, 166 mg), and phenylacetylene (3 mmol, 3 equiv, 330 μL) according to the general procedure for the synthesis of isoflavanone derivatives Method A (Thermal Process) described above. Yellow solid. Rf = 0.51 (10% EtOAc/Hex). Yield, 43.7%. Purity, 95.5%. 1H NMR (CDCl3, 500 MHz, ppm): δ 7.82 (d, J = 2.3 Hz, 1H, 5-H), 7.55 (d, J = 4.7 Hz, 1H, 7-H), 7.23–7.36 (m, 6H, Ar-H), 4.74–4.79 (m, 2H, 2-H), 4.00–4.03 (m, 1H, 3-H), 3.92 (s, 3H, OCH3). 13C NMR (CDCl3, 125 MHz, ppm): δ 190.3, 155.8, 135.6, 133.9, 129.1, 128.6, 128.2, 126.9, 125.8, 12.0, 60.5, 51.7, 14.3. MS Calcd for C16H15O3 [M+H] 255.29. Found: 255.04.
5.1.1.15. 3-(2-Methoxyphenyl)chroman-4-one (2d)40
Synthesized from salicylaldehyde (1.5 mmol, 3 equiv, 59.3 μL) and 2-ethynylanisole (0.5 mmol, 1 equiv, 64.66 μL) according to the general procedure for the synthesis of isoflavanone derivatives Method B (Microwave Irradiation Process) described above. Yellow solid. Yield, 35.5%. Purity, 95.5%. Rf = 0.38 (10% EtOAc/Hex). 1H NMR (CDCl3, 500 MHz, ppm): δ 7.80 (d, J = 7.8 Hz, 1H, 5-H), 7.48–7.54 (m, 1H, 7-H), 7.29 (t, J = 7.9 Hz, 1H, 6-H), 6.92–7.12 (m, 5H, Ar-H), 4.66 (t, J = 11.5 1H, 2-H), 4.53 (dd, J = 11.0, 5.5 Hz, 1H, 3-H), 4.35–4.40 (m, 1H, 3-H), 3.78 (s, 3H, OCH3). 13C NMR (CDCl3, 125 MHz, ppm): δ 192.6, 161.9, 157.5, 135.7, 130.4, 129.2, 127.8, 123.6, 121.7, 121.5, 121.0, 117.9, 111.3, 70.8, 55.6, 48.6. HRMS Calcd for C16H14O3Na [M+Na] 277.0835. Found: 277.0835.
5.1.1.16. 3-(3-Methoxyphenyl)chroman-4-one (2e)
Synthesized from salicylaldehyde (1 mmol, 1 equiv, 104.7 μL) and 3-ethynylanisole (3 mmol, 3 equiv, 387.95 μL) according to the general procedure for the synthesis of isoflavanone derivatives Method B (Microwave Irradiation Process) described above. Yellow solid. Yield, 51.4%. Purity, 97.7%. Rf = 0.34 (10% EtOAc/Hex). 1H NMR (CDCl3, 500 MHz, ppm): δ 7.96 (d, J = 8.3 Hz, 1H, 5-H), 7.49 (t, J = 7.8 Hz, 1H, 7-H), 7.27 (t, J = 7.3 Hz, 1H, 6-H), 7.00–7.06 (m, 2H, Ar-H), 6.85 (t, J = 11.7 Hz, 3H, Ar-H), 4.67 (d, J = 6.87 Hz, 2H, 2-H), 3.96 (t, J = 7.1 Hz, 1H, 3-H), 3.78 (s, 3H, OCH3). 13C NMR (CDCl3, 125 MHz, ppm): δ 192.0, 161.7, 160.0, 136.5, 136.1, 130.0, 127.9, 121.7, 120.9, 118.0, 114.6, 113.2, 71.5, 55.3, 52.4. HRMS Calcd for C16H14O3Na [M+Na] 277.0835. Found: 277.0835.
5.1.1.17. 3-(4-Methoxyphenyl)chroman-4-one (2f)39
Synthesized from salicylaldehyde (1 mmol, 1 equiv, 104.7 μL), and 4-ethynylanisole (3 mmol, 3 equiv, 389 μL) according to the general procedure described above. Yellow solid. Rf = 0.14 (10% EtOAc/ Hex). Yield, 36.5%. Purity, 99.5%. 1H NMR (CDCl3, 500 MHz, ppm): δ 7.95 (d, J = 7.8 Hz, 1H, 5-H), 7.49 (t, J = 7.8 Hz, 1H, 7-H), 7.20 (d, J = 8.7 Hz, 2H, Ar-H), 6.87–7.04 (m, 4H, Ar-H), 4.62–4.65 (m, 2H, 2-H), 3.95 (dd, J = 8.7, 6.0 Hz, 1H, 3-H), 3.79 (s, 3H, OCH3). 13C NMR (CDCl3, 125 MHz, ppm): δ 192.3, 161.6, 159.2, 136.0, 129.7, 127.8, 127.0, 121.6, 121.1, 117.9, 114.4, 71.6, 55.3, 51.6. HRMS Calcd for C16H14O3Na [M+Na] 277.0841. Found: 277.0826.
5.1.1.18. 3-(3,5-Dimethoxyphenyl)chroman-4-one (2g)
Synthesized from salicylaldehyde (0.5 mmol, 1 equiv, 52.4 μL), and 1-ethynyl-3,5-dimethoxybenzene (1.5 mmol, 3 equiv, 243.5 mg) according to the general procedure for the synthesis of isoflavanone derivatives Method A (Thermal Process) described above. Yellow solid. Rf = 0.21 (10% EtOAc/Hex). Yield 24.6%. Purity, 99.3%. 1H NMR (CDCl3, 500 MHz, ppm): δ 7.95 (d, J = 7.8 Hz, 1H, 5-H), 7.49 (t, J = 7.8 Hz, 1H, 7-H), 6.99–7.05 (m, 2H, 6-H and 8-H), 6.44 (d, J = 1.85 Hz, 2H, Ar-H), 6.40 (s, 1H, Ar-H), 4.66 (d, J = 7.3 Hz, 2H, 2-H), 3.91 (t, J = 7.1 Hz, 1H, 3-H), 3.76 (s, 6H, OCH3). 13C NMR (CDCl3, 125 MHz, ppm): δ 197.2, 161.7, 161.1, 137.3, 136.0, 127.8, 121.6, 121.0, 117.9, 106.8, 99.76, 71.3, 55.3, 52.5. HRMS Calcd for C17H16O4Na [M+Na] 307.0946. Found: 307.0939.
5.1.1.19. 3-(Thiophen-3-yl)chroman-4-one (3a)
Synthesized from salicylaldehyde (1 mmol, 1 equiv, 104.7 μL) and 3-ethynylthiophene (3 mmol, 3 equiv, 295.5 μL) according to the general procedure for the synthesis of isoflavanone derivatives Method A (Thermal Process) described above. Light yellow oil. Rf = 0.26 (10% EtOAc/Hex). Yield, 28.7%. Purity, 99.8%. 1H NMR (CDCl3, 500 MHz, ppm): δ 7.98 (d, J = 7.8 Hz, 1H, 5-H), 7.50 (t, J = 6.1 Hz, 1H, 7-H), 7.01–7.18 (m, 5H, Ar-H), 4.65 (d, J = 6.9 Hz, 2H, 2-H), 3.96 (t, J = 7.3 Hz, 1H, 3-H). 13C NMR (CDCl3, 125 MHz, ppm): δ 192.5, 161.7, 137.5, 136.1, 132.0, 129.7, 128.5, 127.8, 121.6, 121.1, 117.9, 71.6, 60.5, 52.0, 21.2. HRMS Calcd for C13H10O2SNa [M+Na] 253.0299. Found: 253.0291.
5.1.1.20. 3-(Pyridin-3-yl)chroman-4-one (3b)
Synthesized from salicylaldehyde (0.5 mmol, 1 equiv, 52.4 μL) and 3-ethylnylpyridine (3 mmol, 1.5 equiv, 267.3 mg) according to the general procedure for the synthesis of isoflavanone derivatives Method A (Thermal Process) described above. Light yellow solid. Rf = 0.22 (40% EtOAc/Hex). Yield, 28.0%. Purity 99.7%. 1H NMR (CDCl3, 500 MHz, ppm): δ 8.55 (s, 2H, Pyr-H), 7.93 (d, J = 2.4, 1H, 5-H), 7.58 (d, J = 7.8, 1H, Pyr-H), 7.51 (t, J = 7.3, 1H, 7-H), 7.25–7.28 (m, 1H, 6-H), 6.99–7.06 (m, 2H, Ar-H and Pyr-H), 4.62–4.68 (m, 2H, 2-H), 4.02 (dd, J = 9.1, 5.0 Hz, 1H, 3-H). 13C NMR (CDCl3, 125 MHz, ppm): δ 191.1, 161.6, 150.0, 149.2, 136.5, 136.2, 130.9, 127.8, 123.8, 122.0, 120.8, 118.0, 71.0, 50.0. HRMS Calcd for C14H11NO2Na [M+Na] 248.0687. Found: 248.0682.
5.1.1.21. 6-Fluoro-3-phenylchroman-4-one (4a)
Synthesized from 5-fluorosalicylaldehyde (0.5 mmol, 1 equiv, 70.1 mg) and phenylacetylene (1.5 mmol, 3 equiv, 164.7 μL) according to the general procedure for the synthesis of isoflavanone derivatives Method B described above. Light yellow solid. Rf = 0.44 (10% EtOAc/Hex). Yield, 21.7%. Purity, 99.5%. 1H NMR (CDCl3, 500 MHz, ppm): δ 7.60 (d, J = 8.3 Hz, 1H, 5-H), 7.38–7.20 (m, 6H, Ar-H), 7.00 (dd, J = 9.2, 4.1 Hz, 1H, 8-H), 4.65 (d, J = 6.4 Hz, 2H, 2-H), 3.99 (t, J = 7.3 Hz, 1H, 3-H). 13C NMR (CDCl3, 125 MHz, ppm): δ 191.5, 158.2, 156.4, 134.7, 129.0, 128.6, 128.0, 123.7, 121.5, 119.7, 112.7, 71.7, 52.2. HRMS Calcd for C15H11FO2 [M+Na] 265.0641. Found: 265.0631.
5.1.1.22. 6-Chloro-3-phenylchroman-4-one (4b)39
Synthesized from 5-chlorosalicylaldehyde (1 mmol, 1 equiv, 156.6 mg and phenylacetylene (3 mmol, 3 equiv, 330 μL) according to the general procedure for the synthesis of isoflavanone derivatives Method A (Thermal Process) described above. Light yellow solid. Rf = 0.44 (10% EtOAc/Hex). Yield 32.8%. Purity, 98.8%. 1H NMR (CDCl3, 500 MHz, ppm): δ 7.91 (d, J = 2.8 Hz, 1H, 5-H), δ 7.43 (dd, J = 9.0, 6.0 Hz, 1H, 7-H), 7.30–7.38 (m, 3H, Ar-H), 7.26 (d, J = 6.0 Hz, 2H, Ar-H), 6.98 (d, J = 9.2 Hz, 1H, 8-H), 4.67 (d, J = 3.7 Hz, 2H, 2-H), 3.99 (t, J = 7.1 Hz, 1H, 3-H). 13C NMR (CDCl3, 125 MHz, ppm): δ 190.9, 160.0, 135.9, 134.5, 129.0, 128.6, 128.0, 127.3, 127.1, 121.9, 119.7, 71.6, 52.0. HRMS Calcd for C15H11O2ClNa [M+Na] 281.0345. Found: 281.0349.
5.1.1.23. 6-Bromo-3-phenylchroman-4-one (4c)
Synthesized from 5-bromosalicylaldehyde (1 mmol, 1 equiv, 201 mg) and phenylacetylene (3 mmol, 3 equiv, 330 μL) according to the general procedure for the synthesis of isoflavanone derivatives Method B (Microwave Irradiation Process) described above. Light yellow solid. Yield, 31.8%. Purity, 97.4%. Rf = 0.46 (10% EtOAc/Hex). 1H NMR (CDCl3, 500 MHz, ppm): δ 8.06 (d, J = 2.8 Hz, 1H, 5-H), 7.56 (dd, J = 8.7, 2.3 Hz, 1H, 7-H), 7.25–7.44 (m, 5H, Ar-H), 6.92 (d, J = 8.7 Hz, 1H, 8-H), 4.66 (d, J = 7.8 Hz, 2H, 2-H), 3.99 (t, J = 3.9 Hz, 1H, 3-H). 13C NMR (CDCl3, 125 MHz, ppm): δ 191.1, 160.6, 138.8, 134.5, 130.2, 129.1, 128.6, 128.1, 122.3, 120.1, 114.4, 71.6, 52.0. HRMS Calcd for C15H11O2BrNa+ [M+Na] 324.9840. Found: 324.9833.
5.1.1.24. 6-tert-Butyl-3-phenylchroman-4-one (4d)
Synthesized from 5-tert-butyl-2-hydroxy-benzaldehyde (0.5 mmol, 1 equiv, 85.8 μL), and phenylacetylene (1.5 mmol, 3 equiv, 164.7 μL) according to the general procedure for the synthesis of isoflavanone derivatives Method B (Microwave Irradiation Process) described above. A yellow oil product was formed. Light yellow solid. Yield, 14.4%. Purity, 94.4%. Rf = 0.49 (10% EtOAc/Hex). 1H NMR (CDCl3, 500 MHz, ppm): δ 7.95 (t, J = 2.1 Hz, 1H, 5-H), 7.57 (d, J = 8.7 Hz, 1H, 7-H), 7.35–7.28 (m, 5H, Ar-H), 6.96 (d, J = 8.7 Hz, 1H, 8-H), 4.65 (d, J = 5.95 Hz, 2H, 2-H), 3.97 (t, J = 6.9 Hz, 1H, 3-H), 1.31 (s, 9H, C(CH3)3). 13C NMR (CDCl3, 125 MHz, ppm): δ 192.1, 159.7, 144.6, 135.4, 133.9, 128.9, 128.7, 127.8, 123.7, 117.6, 71.6, 52.5, 34.5, 31.4, 22.7, 14.2. HRMS Calcd for C19H20O2Na+ [M+Na] 303.13555. Found: 303.13560.
5.2. Aromatase activity assay
Inhibitory potencies of compounds were determined according to an established procedure using a commercially available aromatase test kit from BD Gentest.33 This fluorescence-based assay measures the rate at which recombinant human aromatase (baculovirus/insect cell-expressed) converts the substrate 7-methoxy-trifluoromethylcoumarin (MFC) into a fluorescent product (λex = 409 nm, λem = 530 nm) in a NADPH regenerating system. Briefly, concentrated stock solutions of test compounds were prepared in acetonitrile. 100 μL samples containing serial dilutions of test compounds (dilution factor of 3 between samples) and cofactor mixture (0.4 U/mL glucose-6-phosphate dehydrogenase; 16.2 μM NADP+; 825 μMMgCl2; 825 μM glucose-6-phosphate; 50 μM citrate buffer, pH 7.5) were prepared in a 96 well plate. After incubating the plate for 10 min at 37 °C, 100 μL of an aromatase/P450 reductase/substrate solution (105 lg protein/mL enzyme; 50 μM MFC; 20 mM phosphate buffer, pH 7.4) were added to each well. The plate was covered and incubated for 30 min at 37 °C. 75 μL of 0.5 M Tris base were then added to stop the reaction and the fluorescence of the formed de-methylated MFC was measured with a plate reader (SpectraMax Gemini, Molecular Devices).
Fluorescence intensities, which were proportional to the amount of reaction product generated by aromatase, were graphed as a function of inhibitor concentration and then fit to a 3-parameter logistic function. Inhibitory potencies were expressed in terms of the IC50 value, the inhibitor concentration necessary to reduce the enzyme activity by half. Each experiment was performed at least in triplicate.
In cases in which the intrinsic fluorescence of a test compound overlapped with the fluorescence of the reaction product, the substrate MFC was substituted by dibenzylfluorescein (DBF) which was converted by aromatase to fluorescein (λex = 485 nm; λem = 530 nm).41 The protocol was the same as the one above, except for a longer incubation time (2 h) and the use of a different stop reagent (2 M NaOH).
5.3. Molecular modeling
The coordinates of the X-ray crystal structure of the aromatase/androstenedione complex (PDB code 3EQM) were downloaded from the Protein Databank (http://www.rcsb.org) and imported into the modeling program SYBYL (version 8.0; Tripos, St. Louis, MO). All non-protein components were deleted and hydrogen atoms were added to the protein structure. Partial charges were assigned according to the Amber library and the positions of the added hydrogen atoms were optimized by molecular mechanics minimization that kept the positions of the heavy atoms static.42 Energy minimization was performed with the Powell method in combination with the Amber7 FF99 force field, a distance-dependent dielectric constant of 4, and a convergence criterion of 0.05 kcal/(mol Å). The molecular structures of inhibitors were also prepared in SYBYL and the conformational energy of each structure was minimized by molecular mechanics (MMFF94s force field, MMFF94 charges, and distance-dependent dielectric constant of 4) using the conjugate gradient method and a termination criterion of 0.01 kcal/(mol Å).
Inhibitor structures were computationally docked into the enzyme’s binding site using the program GOLD (version 5.0.1.; CCDC, Cambridge, UK). GOLD operates with a genetic search algorithm and allows for complete ligand and partial binding site flexibility.43 All four scoring functions (GoldScore, ChemScore, ASP, and Chem-PLP) were tested and various sizes and centers of the docking sphere were evaluated. The settings for the genetic algorithm runs were kept at their default values (population size: 100, selection pressure: 1.1; number of operations: 100,000, the number of islands: 5, niche size: 2, probability for migration, mutation, and crossover: 10%, 95%, and 95%, respectively). For each ligand, 30 runs were performed under identical conditions.
Acknowledgments
This project was supported by Grants from the National Center for Research Resources (5P20RR016481-12) and the National Institute of General Medical Sciences (8 P20 GM103436-12) from the National Institutes of Health, the Center for Integrated Science and Mathematics (CINSAM) and Northern Kentucky University Office of Research, Grants and Contracts Seed Grant. We thank Dr. Stuart Oehrle for the assistance with LC/MS, Dr. Grant Edwards and Dr. Heather Bullen for their help with HPLC and Dr. Isabelle Lagadic for the assistance with Microwave Reactor. We also thank Dr. Peter Crooks for his advice regarding the preparation of the manuscript.
Abbreviations
- Phe 134
phenylalanine 134
- Arg 192
arginine
- Trp
tryptophan 224 phenylalanine 224
- Asp 309
aspartic acid 309
- Met 374
methionine 374
- Ser 478
serine 478
- CYP19
cytochrome P450 aromatase
- AIs
aromatase inhibitors
- SERMs
selective estrogen receptor modulators
- NSAIs
non-steroidal aromatase inhibitors
- NMR
nuclear magnetic resonance
- MS
mass spectrometry
- HPLC
high performance liquid chromatography
References and notes
- 1.American Cancer Society . Cancer Facts & Figures 2010. American Cancer Society; Atlanta: 2010. pp. 3–9. [Google Scholar]
- 2.Brueggemeier RW, Hackett JC, Diaz-Cruz ES. Endocr. Rev. 2005;26:331. doi: 10.1210/er.2004-0015. [DOI] [PubMed] [Google Scholar]
- 3.Margolese RG, Fisher B, Hortobagyi GN, Bloomer WD. Cancer Med. 2000:1. [Google Scholar]
- 4.Maughan Karen L, Lutterbie Mark A, Ham Peter S. Am. Fam. Physician. 2010;81:1339. [PubMed] [Google Scholar]
- 5.Buzdar AU. Endocr. Relat. Cancer. 1999;6:219. doi: 10.1677/erc.0.0060219. [DOI] [PubMed] [Google Scholar]
- 6.(a) Muftuoglu Y, Mustata G. Bioorg. Med. Chem. Lett. 2010;20:3050. doi: 10.1016/j.bmcl.2010.03.113. [DOI] [PubMed] [Google Scholar]; (b) Ghosh D, Griswold J, Erman M, Pangborn W. Nature. 2009;457:219. doi: 10.1038/nature07614. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gobbi S, Cavalli A, Bisi A, Recanatini M. Curr. Top. Med. Chem. (Sharjah, United Arab Emirates) 2008;8:869. doi: 10.2174/156802608784911590. [DOI] [PubMed] [Google Scholar]; (d) Recanatini M, Cavalli A, Valenti P. Med. Res. Rev. 2002;22:282. doi: 10.1002/med.10010. [DOI] [PubMed] [Google Scholar]; (e) Benson JR, Ravisekar O. Curr. Cancer Ther. Rev. 2007;3:67. [Google Scholar]; (f) Wang M, Lacy G, Gao M, Miller KD, Sledge GW, Zheng QH. Bioorg. Med. Chem. Lett. 2007;17:332. doi: 10.1016/j.bmcl.2006.10.065. [DOI] [PubMed] [Google Scholar]
- 7.(a) Furr BJA. Milestones in Drug Therapy. Aromatase Inhibitors. 2nd Birkhauser; 2006. p. 182. [Google Scholar]; (b) Herold CI, Blackwell KL. Clin. Breast Cancer. 2008;8:50. doi: 10.3816/CBC.2008.n.003. [DOI] [PubMed] [Google Scholar]; (c) Nabholtz JM, Buzdar A, Pollak M, Harwin W, Burton G, Mangalik A, Steinberg M, Webster A, Von Euler M. J. Clin. Oncol. 2000;18:3758. doi: 10.1200/JCO.2000.18.22.3758. [DOI] [PubMed] [Google Scholar]
- 8.(a) Barker S. Curr. Opin. Investig. Drugs (Thomson Sci.) 2006;7:1085. [PubMed] [Google Scholar]; (b) Miller WR, Bartlett J, Brodie AM, Brueggemeier RW, di Salle E, Lonning PE, Llombart A, Maass N, Maudelonde T, Sasano H, Goss PE. Oncologist. 2008;13:829. doi: 10.1634/theoncologist.2008-0055. [DOI] [PubMed] [Google Scholar]
- 9.Bhatnagar AS. Breast Cancer Res. Treat. 2008;112:385. [Google Scholar]
- 10.Plourde PV, Dyroff M, Dukes M. Breast Cancer Res. Treat. 1994;30:103. doi: 10.1007/BF00682745. [DOI] [PubMed] [Google Scholar]
- 11.(a) Le Borgne M, Marchand P, Delevoye-Seiller B, Robert J-M, Le Baut G, Hartmann RW, Palzer M. Bioorg. Med. Chem. Lett. 1999;9:333. doi: 10.1016/s0960-894x(98)00737-9. [DOI] [PubMed] [Google Scholar]; (b) Marchand P. Bioorg. Med. Chem. Lett. 2003;13:1553. doi: 10.1016/s0960-894x(03)00182-3. [DOI] [PubMed] [Google Scholar]; (c) Leze MP, Le Borgne M, Pinson P, Palusczak A, Duflos M, Le Baut G, Hartmann RW. Bioorg. Med. Chem. Lett. 2006;16:1134. doi: 10.1016/j.bmcl.2005.11.099. [DOI] [PubMed] [Google Scholar]; (d) Wood PM, Woo LWL, Labrosse J-R, Trusselle MN, Abbate S, Longhi G, Castiglioni E, Lebon F, Purohit A, Reed MJ, Potter BVL. J. Med. Chem. 2008;51:4226. doi: 10.1021/jm800168s. [DOI] [PubMed] [Google Scholar]
- 12.(a) Paoletta S, Steventon GB, Wildeboer D, Ehrman TM, Hylands PJ, Barlow DJ. Bioorg. Med. Chem. 2008;16:8466. doi: 10.1016/j.bmc.2008.08.034. [DOI] [PubMed] [Google Scholar]; (b) Brueggemeier RW, Richards JA, Joomprabutra S, Bhat AS, Whetstone JL. J. Steroid Biochem. Mol. Biol. 2001;79:75. doi: 10.1016/s0960-0760(01)00127-3. [DOI] [PubMed] [Google Scholar]; (c) Sun B, Hoshino J, Jermihov K, Marler L, Pezzuto JM, Mesecar AD, Cushman M. Bioorg. Med. Chem. 2010;18:5352. doi: 10.1016/j.bmc.2010.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Karkola S, Waehaelae K. Mol. Cell. Endocrinol. 2009;301:235. doi: 10.1016/j.mce.2008.10.003. [DOI] [PubMed] [Google Scholar]; (e) Adlercreutz H, Bannwart C, Wahala K, Makela T, Brunow G, Hase T, Arosemena PJ, Kellis JT, Jr., Vickery LE. J. Steroid Biochem. Mol. Biol. 1993;44:147. doi: 10.1016/0960-0760(93)90022-o. [DOI] [PubMed] [Google Scholar]
- 13.(a) Efferth T, Koch E. Curr. Drug Targets. 2011;12:122. doi: 10.2174/138945011793591626. [DOI] [PubMed] [Google Scholar]; (b) Balunas MJ, Su B, Brueggemeier RW, Kinghorn AD. Anticancer Agents Med. Chem. 2008;8:646. [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Yuan Q, Liu Z, Xiong C, Wu L, Wang J, Ruan J. Bioorg. Med. Chem. Lett. 2011;21:3427. doi: 10.1016/j.bmcl.2011.03.108. [DOI] [PubMed] [Google Scholar]; (b) Sakamoto T, Horiguchi H, Oguma E, Kayama F. J. Nutr. Biochem. 2010;21:856. doi: 10.1016/j.jnutbio.2009.06.010. [DOI] [PubMed] [Google Scholar]; (c) Balunas Marcy J, Su B, Brueggemeier Robert W, Kinghorn AD. J. Nat. Prod. 2008;71:1161. doi: 10.1021/np8000255. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Balunas MJ, Kinghorn AD. Planta Med. 2010;76:1087. doi: 10.1055/s-0030-1250169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caporuscio F, Rastelli G, Imbriano C, Del Rio A. J. Med. Chem. 2011;54:4006. doi: 10.1021/jm2000689. [DOI] [PubMed] [Google Scholar]
- 16.van Meeuwen JA, Korthagen N, de Jong PC, Piersma AH, van den Berg M. Toxicol. Appl. Pharmacol. 2007;221:372. doi: 10.1016/j.taap.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 17.Campbell DR, Kurzer MS. J. Steroid Biochem. Mol. Biol. 1993;46:381. doi: 10.1016/0960-0760(93)90228-o. [DOI] [PubMed] [Google Scholar]
- 18.Yao N, Song A, Wang X, Dixon S, Lam KS. J. Comb. Chem. 2007;9:668. doi: 10.1021/cc070009y. [DOI] [PubMed] [Google Scholar]
- 19.(a) Quintin J, Buisson D, Thoret S, Cresteil T, Lewin G. Bioorg. Med. Chem. Lett. 2009;19:3502. doi: 10.1016/j.bmcl.2009.05.008. [DOI] [PubMed] [Google Scholar]; (b) Blank VC, Poli C, Marder M, Roguin LP. Bioorg. Med. Chem. Lett. 2004;14:133. doi: 10.1016/j.bmcl.2003.10.029. [DOI] [PubMed] [Google Scholar]
- 20.(a) Pouget C, Lauthier F, Simon A, Fagnere C, Basly J-P, Delage C, Chulia A-J. Bioorg. Med. Chem. Lett. 2001;11:3095. doi: 10.1016/s0960-894x(01)00617-5. [DOI] [PubMed] [Google Scholar]; (b) Ibrahim AR, Abul-Hajj YJ. J. Steroid Biochem. Mol. Biol. 1990;37:257. doi: 10.1016/0960-0760(90)90335-i. [DOI] [PubMed] [Google Scholar]
- 21.Yahiaoui S, Fagnere C, Pouget C, Buxeraud J, Chulia AJ. Bioorg. Med. Chem. 2008;16:1474. doi: 10.1016/j.bmc.2007.10.057. [DOI] [PubMed] [Google Scholar]
- 22.Gobbi S, Cavalli A, Rampa A, Belluti F, Piazzi L, Paluszcak A, Hartmann RW, Recanatini M, Bisi A. J. Med. Chem. 2006;49:4777. doi: 10.1021/jm060186y. [DOI] [PubMed] [Google Scholar]
- 23.Kao Y-C, Zhou C, Sherman M, Laughton CA, Chen S. Environ. Health Perspect. 1998;106:85. doi: 10.1289/ehp.9810685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim Y-W, Hackett JC, Brueggemeier RW. J. Med. Chem. 2004;47:4032. doi: 10.1021/jm0306024. [DOI] [PubMed] [Google Scholar]
- 25.(a) Su B, Hackett JC, Diaz-Cruz ES, Kim YW, Brueggemeier RW. Bioorg. Med. Chem. 2005;13:6571. doi: 10.1016/j.bmc.2005.07.038. [DOI] [PubMed] [Google Scholar]; (b) Hackett JC, Kim YW, Su B, Brueggemeier RW. Bioorg. Med. Chem. 2005;13:4063. doi: 10.1016/j.bmc.2005.03.050. [DOI] [PubMed] [Google Scholar]
- 26.(a) Sugimoto T, Itagaki K, Irie K. Bioorg. Med. Chem. 2008;16:650. doi: 10.1016/j.bmc.2007.10.045. [DOI] [PubMed] [Google Scholar]; (b) Endringer DC, Guimaraes KG, Kondratyuk TP, Pezzuto JM, Braga FC. J. Nat. Prod. 2008;71:1082. doi: 10.1021/np800098f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.(a) Cavalli A, Recanatini M. J. Med. Chem. 2002;45:251. doi: 10.1021/jm015567k. [DOI] [PubMed] [Google Scholar]; (b) Stefanachi A, Favia AD, Nicolotti O, Leonetti F, Pisani L, Catto M, Zimmer C, Hartmann RW, Carotti A. J. Med. Chem. 2011;54:1613. doi: 10.1021/jm101120u. [DOI] [PubMed] [Google Scholar]
- 28.Auvray P, Sourdaine P, Moslemi S, Seralini GE, Sonnet P, Enguehard C, Guillon J, Dallemagne P, Bureau R, Rault S. J. Steroid Biochem. Mol. Biol. 1999;70:59. doi: 10.1016/s0960-0760(99)00093-x. [DOI] [PubMed] [Google Scholar]
- 29.(a) Leonetti F, Favia A, Rao A, Aliano R, Paluszcak A, Hartmann RW, Carotti A. J. Med. Chem. 2004;47:6792. doi: 10.1021/jm049535j. [DOI] [PubMed] [Google Scholar]; (b) Yahiaoui S, Pouget C, Fagnere C, Champavier Y, Habrioux G, Chulia AJ. Bioorg. Med. Chem. Lett. 2004;14:5215. doi: 10.1016/j.bmcl.2004.07.090. [DOI] [PubMed] [Google Scholar]
- 30.(a) Gobbi S, Zimmer C, Belluti F, Rampa A, Hartmann RW, Recanatini M, Bisi A. J. Med. Chem. 2010;53:5347. doi: 10.1021/jm100319h. [DOI] [PubMed] [Google Scholar]; (b) Gobbi S, Cavalli A, Negri M, Schewe KE, Belluti F, Piazzi L, Hartmann RW, Recanatini M, Bisi A. J. Med. Chem. 2007;50:3420. doi: 10.1021/jm0702938. [DOI] [PubMed] [Google Scholar]; (c) Recanatini M, Bisi A, Cavalli A, Belluti F, Gobbi S, Rampa A, Valenti P, Palzer M, Palusczak A, Hartmann RW. J. Med. Chem. 2001;44:672. doi: 10.1021/jm000955s. [DOI] [PubMed] [Google Scholar]; (d) Cavalli A, Bisi A, Bertucci C, Rosini C, Paluszcak A, Gobbi S, Giorgio E, Rampa A, Belluti F, Piazzi L, Valenti P, Hartmann RW, Recanatini M. J. Med. Chem. 2005;48:7282. doi: 10.1021/jm058042r. [DOI] [PubMed] [Google Scholar]; (e) Yao N, Chen CY, Wu CY, Motonishi K, Kung HJ, Lam KS. J. Med. Chem. 2011;54:4339. doi: 10.1021/jm101440r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.(a) Kumar S, Deshpande S, Chandra V, Kitchlu S, Dwivedi A, Nayak VL, Konwar R, Prabhakar YS, Sahu DP. Bioorg. Med. Chem. 2009;17:6832. doi: 10.1016/j.bmc.2009.08.034. [DOI] [PubMed] [Google Scholar]; (b) Park JY, Ullapu PR, Choo H, Lee JK, Min S-J, Pae AN, Kim Y, Baek D-J, Cho YS. Eur. J. Org. Chem. 2008;2008:5461. [Google Scholar]; (c) Vasquez-Martinez Y, Ohri RV, Kenyon V, Holman TR, Sepulveda-Boza S. Bioorg. Med. Chem. 2007;15:7408. doi: 10.1016/j.bmc.2007.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Valenti P, Belluti F, Rampa A, Bisi A. Synth. Commun. 1999;29:3895. [Google Scholar]; (e) Goto H, Terao Y, Akai S. Chem. Pharm. Bull. 2009;57:346. doi: 10.1248/cpb.57.346. [DOI] [PubMed] [Google Scholar]
- 32.(a) Skouta R, Li C-J. Tetrahedron Lett. 2007;48:8343. [Google Scholar]; (b) Skouta R, Li C-J. Angew. Chem. Int. Ed. 2007;46:1117. doi: 10.1002/anie.200603495. [DOI] [PubMed] [Google Scholar]
- 33.(a) Maiti A, Reddy PVN, Sturdy M, Marler L, Pegan SD, Mesecar AD, Pezzuto JM, Cushman M. J. Med. Chem. 2009;52:1873. doi: 10.1021/jm801335z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Stresser DM, Turner SD, McNamara J, Stocker P, Miller VP, Crespi CL, Patten CJ. Anal. Biochem. 2000;284:427. doi: 10.1006/abio.2000.4729. [DOI] [PubMed] [Google Scholar]
- 34.Negrerie M, Kruglik SG, Lambry JC, Vos MH, Martin JL, Franzen S. J. Biol. Chem. 2006;281:10389. doi: 10.1074/jbc.M513375200. [DOI] [PubMed] [Google Scholar]
- 35.(a) Teles JH, Brode S, Chabanas M. Angew. Chem. Int. Ed. 1998;37:1415. doi: 10.1002/(SICI)1521-3773(19980605)37:10<1415::AID-ANIE1415>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]; (b) Dash C, Shaikh MM, Butcher RJ, Ghosh P. Inorg. Chem. 2010;49:4972. doi: 10.1021/ic100087d. [DOI] [PubMed] [Google Scholar]; (c) Chiou JYZ, Luo SC, You WC, Bhattacharyya A, Vasam CS, Huang CH, Lin IJB. Eur. J. Inorg. Chem. 2009;2009:1950. [Google Scholar]
- 36.(a) Ganesan A. Curr. Opin. Chem. Biol. 2008;12:306. doi: 10.1016/j.cbpa.2008.03.016. [DOI] [PubMed] [Google Scholar]; (b) Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv. Drug Delivery Rev. 2001;46:3. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 37.Ertl P, Rohde B, Selzer P. J. Med. Chem. 2000;43:3714. doi: 10.1021/jm000942e. [DOI] [PubMed] [Google Scholar]
- 38.Molinspiration Cheminformatics Available from: < http://www.molinspiration.com> (accessed 11/8/2011)
- 39.Skouta R, Li CJ. Angew. Chem. 2007;46:1117. doi: 10.1002/anie.200603495. [DOI] [PubMed] [Google Scholar]
- 40.Bellina F, Masini T, Rossi R. Eur. J. Org. Chem. 2010;2010:1339. [Google Scholar]
- 41.Stresser DM, Blanchard AP, Turner SD, Erve JC, Dandeneau AA, Miller VP, Crespi CL. Drug Metab. Dispos. 2000;28:1440. [PubMed] [Google Scholar]
- 42.Paula S, Ball WJ., Jr. Proteins. 2004;56:595. doi: 10.1002/prot.20105. [DOI] [PubMed] [Google Scholar]
- 43.(a) Jones G, Willett P, Glen RC. J. Mol. Biol. 1995;245:43. doi: 10.1016/s0022-2836(95)80037-9. [DOI] [PubMed] [Google Scholar]; (b) Jones G, Willett P, Glen RC, Leach AR, Taylor R. J. Mol. Biol. 1997;267:727. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]