

Abstract
Mutations in TP53 and RB1 have been shown to participate in the development of malignant brain tumors. Emerging evidence shows that mutations are involved in LGI1 in brain tumor progression. Herein we present data from the sequencing of a series of high- and low-grade gliomas with matched normal DNA. We report on 35 unique missense mutations in TP53, RB1 and LGI1 genes and use available information for each mutation in order to classify them as likely to be ‘driver’ or ‘passenger’ mutations. The identification of putatively deleterious mutations in LGI1 supports the notion that this locus may play a role in brain cancer development.
Keywords: TP53, RB1, LGI1, glioblastoma, brain tumors, mutation analysis
Introduction
The most common type of primary brain tumor in adults is glioma (MIM#137800), a tumor arising from glial cells (astrocytes, oligodendrocytes or both) (1). Most gliomas can be divided into three groups based on their histopathological appearance: astrocytic, oligodendroglial and mixed oligoastrocytic tumors (1). Although these tumor types make up the great majority of human gliomas, there are also a number of other subtypes (1). Brain tumors are highly heterogeneous with a wide morphological spectrum (2). Cytogenetic and molecular genetic studies have shown that tumor subtypes exist within larger histological brain tumor categories, such as astrocytoma, grade IV, also known as glioblastoma multiforme (GBM). GBM is the most commonly diagnosed type of malignant brain tumor in adults.
Two pathways for the progression of GBMs have been demonstrated: secondary and de novo (3-5). The two progression pathways show that these tumors have changes occurring in TP53 (MIM# 191170) and RB1 (MIM# 180200) and in other genes in these respective pathways (3-5).
Therefore, the evidence indicates that TP53 and RB1 play a key role in the molecular pathogenesis of brain tumors. Herein, we report on the somatic mutations in TP53 and RB1 found in a series of low- and high-grade gliomas. We analyze LGI1 whose locus is rearranged and down-regulated in malignant brain cancers (6). We also provide a tentative classification of mutations as that may confer a selective advantage to the cancer phenotypes, as ‘driver mutations’ or that accumulate in cancer cells but do not contribute to the cancer phenotype as ‘passenger mutations’.
Materials and methods
Subjects
Forty-three subjects with primary brain tumors were included in the study between May 2002 and July 2004 at the Karmanos Cancer Institute Neuro-oncology clinic and other institutions in the Detroit Metropolitan area, which included hospitals in the Detroit Medical Center system and the Henry Ford Health system. The tumor samples were re-read to ensure that a viable tumor was represented in the samples and to verify histological diagnosis. The DNA from blood or tumor samples was extracted according to established protocols. Therefore, matched tumor DNA (from tumor samples) and normal DNA (from blood samples) was available for each individual. Subsets of these samples were then used to investigate somatic and germline genetic changes in TP53, LGI1 and RB1.
TP53-sequencing analysis
Using a set of 20 matched tumor and normal DNA samples, single-strand conformation polymorphism (SSCP) assay and DNA sequencing were used to determine the frequency of TP53 mutations in exons 5-9 (accounting for 97% of mutations in tumor DNA). SSCP was performed for all 20 pairs for exons 5-7 and for 10 of these pairs for exons 5-9. Briefly, bidirectional oligonucleotide primers for the PCR amplification of exon 5-8 fragments were synthesized. The sequences for primers used are: 5′-gttcacttgt gccctga-3′ and 5′-agccctgtcgtctct-3′ for exon 5 (annealing temperature, 56°C); 5′-ctctgattcctcactg-3′ and 5′-ccagagac cccagttgcaaacc-3′ for exon 6 (annealing temperature, 52°C); 5′-tgcttgccacaggtct-3′ and 5′-acagcaggccagtgt-3′ for exon 7 (annealing temperature, 58°C); and 5′-aggacctgatttccttac-3′ and 5′-tctgaggcataactgc-3′ for exon 8 (annealing temperature, 55°C). PCR products were generated in a 30-μl reaction mixture including 50 ng of DNA, 20 μM dNTP, 10 mM Tris-HCl (pH 9.0), 1.5 mM MgCl2, 0.1% Triton X-100, 10 pmol of each primer, 1 unit Taq, (Perkin-Elmer Corp., Norwalk, CT) and 0.2 μCi [33P]-dCTP (DuPont New England Nuclear, Boston, MA). DNA with a known TP53 mutation was included as a positive control. The PCR reaction was carried out using 35 cycles (94°C for 30 sec, annealed for 30 sec at various temperatures as indicated above and 72°C for 1 min) on a Perkin-Elmer 9600 thermal cycler. PCR product (3 μl) was mixed with 2 μl of 0.1 N NaOH and then mixed with 5 μl of gel loading buffer solution from the United States Biochemical Corp. (Cleveland, OH) and heated at 94°C for 4 min. Gels were run at room temperature for 20 h and exposed for 16 h for the autoradiographic detection of bands. Direct sequencing of PCR fragments for the two DNA strands was performed with aberrant migration patterns on single-strand conformation polymorphism gel to determine the corresponding DNA sequences using a double-strand DNA cycle sequencing system from Life Technologies/Invitrogen, Inc. (Gaithersburg, MD). The IARC TP53 mutation database (http://www-p53.iarc.fr/) and Hollstein et al (7) were consulted to determine whether the mutations found in our samples had been reported previously.
RB1-sequencing analysis
Eight matched normal and tumor DNA samples from the original 20 matched samples were analyzed for RB1 mutations using custom Affymetrix resequencing chips (www.affymetrix.com). A multiple sequence alignment was generated using CLUSTAL (Vector NTI) with the following RB orthologs (accession number): Homo sapiens (NP_000312.1), Pan troglodytes (AAK32151.1), Canis familiaris (AAF67147.1), Mus musculus (NP_033055.1), Rattus norvegicus (P33568), Gallus gallus (NP_989750.1), Xenopus laevis (AAB23173.1), Oncorhynchus mykiss (AAD13390.1), Oryzias latipes (AAG21826.1), Fundulus heteroclitus (AAS80140.1), Caenorhabditis elegans (NP_491686.1), Drosophila melanogaster (CAA65661.1), Nicotiana benthamiana (AAU05979.1), Populus tremula (AAF61377.1), Oryza sativa (XP_483552.1) and Cocos nucifera (AAM77469.1). The corresponding amino acid was then identified in the orthologs. The flanking region was also analyzed for conservation. For residues located in regions for which there is a crystal structure (8,9), the amino acid location in the structure was determined (PDB: 1AD6 and 1N4M).
LGI1-sequencing analysis
The set of 8 matched normal and tumor DNA samples used for RB1 analysis was also analyzed for LGI1 mutations using custom Affymetrix resequencing chips. A multiple sequence alignment was generated using CLUSTAL (Vector NTI) with the following LGI1 orthologs (accession number): Homo sapiens (NP_005088), Mus musculus (NP_064674), Bos taurus (AAX08851) and Rattus norvegicus (NP_665712). The corresponding amino acid was then identified in the orthologs. The flanking region was also analyzed for conservation.
Results
Tumor mutations
We identified 38 missense and non-sense changes (38 unique) in 11 samples from brain tumors (Table I). Low-grade gliomas showed a limited number of mutations in the loci studied while the vast majority of high-grade gliomas displayed multiple mutations (Table I).
Table I.
Somatic mutations in brain cancer.
| IDa | Race/ethnicity | Type of tumorb | Gene | Exon | Genomicc | Predicted RNAd | Predicted protein |
|---|---|---|---|---|---|---|---|
| 72 | Caucasian | 9450 - Oligodendroglioma, NOS, LGG | TP53 | 6 | 12713G>T | 895G>T | S215I |
| 73 | Caucasian | 9451 - Oligodendroglioma, anaplastic, HGG | RB1 | 4 | 41314A>T | 555A>T | K130Il |
| 4 | 41329T>C | 570T>C | L135P | ||||
| 13 | 73189G>A | 1427G>A | D421N | ||||
| 17 | 77503C>T | 1695C>T | T510I | ||||
| 18 | 49221G>A | 1862G>A | D566N | ||||
| LGI1 | 4 | 19643A>G | 601A>G | N125S | |||
| 4 | 19697T>G | 655T>G | L144S | ||||
| 8 | 39800A>G | 1815A>G | N531D | ||||
| 78 | Caucasian | 9400 - Astrocytoma, NOS, HGG | RB1 | 2 | 3624A>G | 422A>G | G86R |
| 10 | 63759A>G | 1145A>G | K327E | ||||
| 12 | 69672A>G | 1335A>G | N390S | ||||
| 16 | 76409C>A | 1606C>A | N480K | ||||
| 20 | 155992A>G | 2201A>G | I679V | ||||
| LGI1 | 4 | 19631T>A | 589T>A | I122K | |||
| 6 | 34942A>G | 847A>G | K207R | ||||
| 6 | 34987T>C | 892T>C | I223T | ||||
| 8 | 39810A>T | 1825A>T | N534I | ||||
| 110 | Caucasian | 9440 - GBM, HGG | TP53 | 9 | 13797C>T | 1068C>T | R273C |
| 118 | Caucasian | 9440 - GBM, HGG | LGI1 | 4 | 19694A>G | 652A>G | H143R |
| 127 | Caucasian | 9400 - Astrocytoma, NOS, HGG | TP53 | 5 | 12524A>G | 787A>G | H179R |
| 189 | Caucasian | 9440 - GBM, HGG | TP53 | 5 | 12469G>A | 732G>A | A161T |
| 193 | Caucasian | 9440 - GBM, HGG | RB1 | 4 | 41322A>C | 563A>C | N133H |
| 13 | 73164A>C | 1402A>C | K412N | ||||
| 21 | 152566T>C | 2115T>C | F650S | ||||
| LGI1 | 4 | 19648A>G | 606A>G | K128E | |||
| LGI1 | 6 | 34982C>G | 887C>G | C221W | |||
| 8 | 39671C>T | 1686C>T | Q488X | ||||
| 8 | 39227A>G | 1242A>G | I340V | ||||
| 194 | African American | 9440 - Astrocytoma, GBM, HGG | RB1 | 13 | 73214A>G | 1452A>G | K429R |
| 205 | African American | 9401 - Anaplastic, HGG | TP53 | 5 | 12524A>G | 787A>G | H179R |
| 18 | 149243T>C | 1884T>C | I573T | ||||
| RB1 | 20 | 156037C>G | 2246C>G | L694V | |||
| 20 | 156032A>G | 2241A>G | Y692C | ||||
| 24 | 169609G>A | 2675G>A | E837K | ||||
| 25 | 173022C>T | 2778C>T | P871L | ||||
| 215 | African American | 9440 - GBM, HGG | RB1 | 19 | 152574A>G | 2123A>G | K652E |
| LGI1 | 4 | 19694A>G | 652A>G | H143R |
Patient identity.
ICD-O code with the associated name and further classified into LGG, low-grade glioma; HGG, high-grade glioma.
Nucleotide numbering according to the reference assembly: NC_000017 for TP53, NC_000013 for RB1 and NC_000010.9 for LGI1.
TP53 mutations
During the sequence analysis of exons 5-7 we identified 4 changes representing 3 unique missense variants while the analysis of exons 8-9 showed only 1 (Table II). The mutation found in an African-American sample, His179Arg, was the same as the one in a Caucasian sample. None of these variants were found in the normal matched controls. All five mutations have previously been reported (http://www-p53.iarc.fr/) and are expected to impact protein function.
Table II. Predicted functional consequence of somatic mutations in TP53.
| IDa | Predicted protein | Reported inb | Notes |
|---|---|---|---|
| 189 | A161T | 70 tumors | Part of the sequence-specific DNA-binding domain but not in any particular defined motif. |
| 127 205 |
H179R | 135 tumors | Part of motif III in the sequence-specific DNA-binding domain. |
| 72 | S215I | 21 tumors | Part of the sequence-specific DNA-binding domain but not in any particular defined motif. |
| 110 | R273C | 581 tumors 9 LF families |
Part of motif IV in the sequence-specific DNA-binding domain. This residue directly contacts DNA, and is one of the most frequently mutated in p53. |
Patient identity.
Number of times it has been found as a somatic or germline mutation (LF, Li-Fraumeni syndrome) and deposited at the IARC p53 mutation database (http://www-p53.iarc.fr/).
RB1 mutations
We obtained sequencing information from 8 brain tumor samples and compared it with normal samples. Brain tumor samples showed a large number of mutations many of which occurred in the intronic regions. We also found 20 missense alterations located throughout the coding region of the gene (Table III, Fig. 1A). Although it is difficult to infer causality or even contribution to the tumor phenotype, we have analyzed these missense alterations and found that some of them are likely to impact protein function and therefore play a role in tumor progression (Table III, Fig. 1A).
Table III. Predicted functional consequence of somatic mutations in RB1.
| IDa | Predicted protein | Allowed in orthologsb | Notes | Finalc |
|---|---|---|---|---|
| 78 | G86R | G, Y, D, E, S, F | The wide variety of different amino acids allowed at this position suggests that this change is not deleterious. | FB |
| 73 | K130I | K, R, D, Q, E, T | Although K is well conserved in mammalian species, the wide variety of different amino acids allowed at this position in non-mammalian species suggests that this change is not deleterious. | FB |
| 193 | N133H | N, D, R, T, G, Q, K | Although the wide variety of different amino acids allowed at this position suggests that this change is not deleterious, H is not within the range of variation. | UN |
| 73 | L135P | K, V, I, M, T | The amino acids found at this position in other orthologs have non-polar side chains as does proline, although a replacement for proline is not within the range of variation. | UN |
| 78 | K327E | K, Y, S, N, T | Although the wide variety of different amino acids allowed at this position suggests that this change is not deleterious, E is not within the range of variation. | UN |
| 78 | N390S | N, P, S, E, A | The wide variety of different amino acids allowed at this position suggests that this change is not deleterious. In addition, S is present in frogs as well as in some plant species. Located at an α helix at the start of domain A. | FB |
| 193 | K412N | K, E, G, T, D, R,Q | This position is highly divergent. However, N is a potential helix breaker. Located at an α helix in the A domain. | FD |
| 73 | D421N | D, H, S, I, T, E, M | This position is highly divergent. However, N is a potential helix breaker. Located at an α helix in the A domain. | FD |
| 73 | T510I | T, R, V, Y | Conservative change at a conserved position. Located in an unstructured loop in domain A. | FB |
| 194 | K429R | K, R, S, T | Conservative change in a position that is conserved in vertebrates with R present in fish (trout). Located at an α helix in domain A. | FB |
| 78 | N480K | N, D, E, A, S, K, V | The wide variety of different amino acids allowed at this position suggests that this change is not deleterious. In addition, K is present in Drosophila. Located at an α helix in domain A. An in-frame deletion of this residue has been reported in a family with low penetrance retinoblastoma (33). | UN |
| 73 | D566N | D, G, N | A conserved position. However, N is found in Drosophila. Located in an unstructured loop in domain A. | FB |
| 194 | I573T | I, V, L | Conservative change in a conserved position. The amino acids found at this position in other orthologs have non-polar side chains as does T, although it is not within the range of variation. Located in an α helix in domain B. | UN |
| 193 | F650S | F | Non-conservative change in a completely conserved position. Located at an α helix in the B domain. | FD |
| 215 | K652E | K, G, T, S, N, R | Despite the wide range of variation found at this position there is a high conservation in all animal species (K or R) and the change is from a positively to a negatively charged amino acid. Found in an α helix in domain B. | FD |
| 78 | I679V | I, V, Q S, N, R, W, T | The conservative nature of the change and the wide variety of different amino acids allowed at this position suggests that this change is not deleterious. In addition, V is present in frog and fish (Medaka). Located at an α helix in domain B. | FB |
| 194 | Y692C | Y, S, T | Y is conserved in all vertebrate species (except chicken) and C is not within the range of variation. Located in an unstructured loop in domain B. | UN |
| 205 | L694V | L, I | Highly conserved position in a relatively conserved region. This is a conservative change but V is not within the range of variation found. Located at an unstructured loop in domain B. | UN |
| 194 | E837K | E, T | Highly conserved position with E being found in all the orthologs (except T in C. elegans). Change from a negatively to a positively charged amino acid but not present in a recognizable domain. | UN |
| 194 | P871L | P, V, A, L, G | Although P is conserved in all vertebrate species, L can be found in Nicotiana. It is located in the cyclin/cdk docking site (KXLKXL). | FD |
Patient identity.
Amino acids that are found at the corresponding position in the multiple sequence alignment using the homologs as described in Materials and methods.
Tentative classification of whether the variant is likely to contribute or not to the tumor phenotype: FB, favor benign (or inconsequential, ‘passenger’); FD, favor deleterious (or consequential, ‘driver’) and UN, unknown or not enough information to propose a classification.
Figure 1.
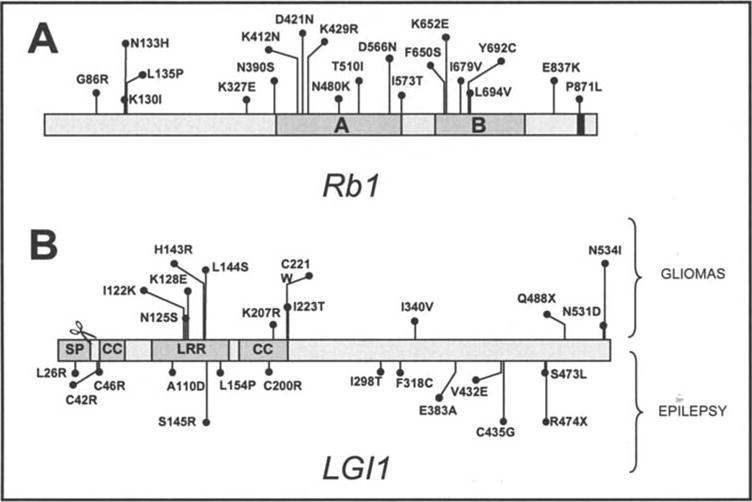
(A) The RB1 domain structure indicating conserved regions (A and B domains), cyclin/cdk docking site (black box) and the location of missense variants found in tumor samples. (B) The LGI1 domain structure indicating signal peptide (SP), cysteine-rich domains (CC) and the leucine-rich repeat (LRR). Mutations found mutated in the germline in epilepsy patients are shown at the bottom of the diagram. Mutations found in tumor samples (this study) are shown at the top of the diagram. The scissors show the cleavage site.
LGI1 variants
We obtained sequencing information from 8 brain tumors samples and compared it with normal samples. We also screened 24 additional normal tissue DNAs. Normal samples revealed two recurring changes in the LGI1 gene: i) a silent mutation (g.34976T>C, c.881T>C and p.Phe219Phe) that has previously been reported (refSNP ID 1111820), found in 31 out of 32 samples and ii) a missense variant (g.39285T>C, c.1301T>C and p.Ile359Thr) that has not been reported, found in 8 out of 32 samples. It is unclear whether the latter variant may have any impact on protein function (Table IV and Fig. 1B).
Table IV.
Predicted functional consequence of somatic mutations in LGI1.
| IDa | Predicted protein | Allowed in orthologsb | Allowed in paralogsc | Notes | Finald |
|---|---|---|---|---|---|
| 78 | I122K | I | I | Highly conserved in paralogs. Hydrophobic residues can be found in all leucine repeat-rich proteins with L present in all except in this second repeat of LGI1 (I). This change from a hydrophobic to a positively charged amino acid at this position is likely to impact function. | FD |
| 73 | N125S | N | N | Located in the leucine-rich repeat. N is completely conserved in LGI1 orthologs, paralogs, and other leucine repeat-rich proteins (Slit, Toll, Osteomodulin, Tartan, Biglycan, ALS, lumican, PGPIX and Chondroadherin) (6) suggesting an important function. | FD |
| 194 | K128E | K | E, W, G | Conserved in orthologs but highly variable stretch in paralogs. Part of the leucine repeat-rich region but this position is highly variable in other LRR proteins, with D, Q, T, S, E and R being found. | UN |
| 118215 | H143R | H | H | Highly conserved in paralogs and is part of the leucine repeat-rich region but this position is highly variable in other LRR proteins, with K, Y, S, D, E and Q being found. | FB |
| 73 | L144S | L | L | Highly conserved in paralogs. Hydrophobic residues can be found in all leucine repeat-rich proteins with L present in all except PGPIX (T) and Lumican (I) (6). Located at the splicing junction. | FD |
| 78 | K207R | K | K, Q, S | Conservative change in a region that is variable in the paralogs. Located in the cysteine cluster of the LRR flanking region but position is highly variable. | FB |
| 193 | C221W | C | C | Conserved C in LRR proteins located at the cysteine cluster of the LRR flanking region, probably important for function. | FD |
| 78 | I223T | I | I, T, A | Replacement is allowed in paralogs and is a relatively conservative change. | FB |
| 193 | I340V | I | I, L | Conservative change in a conserved position in orthologs and paralogs. | FB |
| 194 | Q488X | n/a | n/a | Stop codon that truncates the extreme C-terminus. Notably, frameshifts on codons 440 and 547 have been shown to be deleterious in autosomal-dominant partial epilepsy with auditory features, suggesting that the extreme C-terminus is important for function (28,29). | FD |
| 73 | N531D | N | N, D, G, A | Conserved in orthologs but highly variable stretch in paralogs which may suggest a role in conferring specificity. | UN |
| 78 | N534I | N | N,D, Q, R | Conserved in orthologs but highly variable stretch in paralogs which may suggest a role in conferring specificity. |
Patient identity.
Amino acids that are found at the corresponding position in the multiple sequence alignment using the homologs as described in Materials and methods.
Amino acids that are found at the corresponding position in paralogs LGI1-4 (34), (accession numbers for LGI2-LGI4: AF467954-AF467956).
Tentative classification of whether the variant is likely to contribute or not to the tumor phenotype: FB, favor benign (or inconsequential, ‘passenger’); FD, favor deleterious (or consequential, ‘driver’) and UN, unknown or not enough information to propose a classification.
Additionally, brain tumor samples revealed 11 and 1 missense and nonsense tumor-specific mutations, respectively (Tables I and IV). Unfortunately, due to the limited number of orthologs, the known conservation analysis is not comprehensive. Importantly, a majority of the mutations (9/13) are located at the N-terminal region (Fig. 1B). Several of them (8/13) are located in defined domains that appear to be important for the function of LGI1 such as the leucine-rich repeats (LRR) and the cysteine clusters that flank the LRR, with one mutation targeting one of the conserved cysteines (C221W) (Fig. 1B).
Discussion
In this study, we performed a sequence analysis of matched normal and tumor DNA derived from brain tumors in order to identify mutations in TP53, RB1 and LGI1. We chose to concentrate on these genes due to established, as is the case for TP53 and RB1, or emerging, as is the case for LGI1, roles in the development of brain tumors.
TP53 and RB1 mutations are a common event in gliomas of all types (10-15). TP53 is mutated in 40-70% of grade II and III astrocytomas and 30-40% of GBMs (16) but less frequently in oligodendrogliomas (∼5%) and mixed gliomas (∼30%) (17). In a study of almost 200 astrocytomas of grades II-IV, 76% of GBMs, 72% of anaplastic astrocytomas and 67% of grade II astrocytomas were found to have a disruption in the TP53 pathway, while 67% of GBMs, 13% of anaplastic astrocytomas and 0% of grade II astrocytomas were found to have a disruption in the RB1 pathway (11). The critical role of TP53 in gliomagenesis has also been confirmed in numerous pre-clinical models (18,19). Mutations, allelic loss at 13q or homozygous deletion of RB1 is found in ∼30% of GBMs and a smaller proportion of anaplastic astrocytomas (11-13).
Our analysis identified 4 unique missense mutations in TP53 (Table II), all of which had previously been reported as somatic mutations in tumors (http://www-p53.iarc.fr/). One such mutation, Arg273Cys, has also been found to mutate in the germline in 9 Li-Fraumeni syndrome families indicating that it is clearly an inactivating mutation. All mutations detected in TP53 are located in the DNA binding region, a region of the protein that is less tolerant to mutations (20). Accordingly, they are predicted to be deleterious by structure-based (21), AGVGD (22) and SIFT (23) prediction methods. Functional assays in yeast also indicate that all mutations are non-functional, with the one exception being Ala161Thr which is partially non-functional (20). We conclude that the TP53 mutations found in our study are ‘driver mutations’ and are likely to contribute to brain tumor progression.
Twenty mutations were identified in RB1 (Table III, Fig. 1A), none of which had been reported to the Rb mutation database (http://rb1-lsdb.d-lohmann.de/) or could be found in the literature. Since these mutations have yet to be characterized it is difficult to know whether they contribute to the tumor phenotype. Six variants are found in the N-terminal region, but there is no information regarding protein motifs in this region. The N-terminal mutations which are part of the three conserved N-terminal sub-domains (aa195-235, aa270-289 and aa317-343) (24) appear to be inconsequential as judged by their position and relative lack of conservation in other orthologs. Twelve mutations localize to the A and B domains many of which are in secondary structures and are likely to impact the RB1 function, although it is unknown to which extent (Table III). Finally, two mutations are found in the extreme C-terminus in relatively well-conserved residue positions, one of them in the cyclin/cdk docking site. Their impact on protein function is unclear. We conclude that although most mutations found in RB1 are likely to be ‘passengers’, some amino acid changes that may impact protein function are likely to contribute to the tumor phenotype.
LGI1 has been implicated in the development of brain tumors, although it is still debated whether the evidence support a role as a bona fide tumor suppressor in brain tumors (6,25-27). Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features (28-30). Herein, we report on 12 novel variants in LGI1. Notably, the identified mutations differ from those that have been detected in patients with epilepsy (Table IV, Fig. 1B). Although it is difficult to infer causality or even contribution to the tumor phenotype we have analyzed these missense alterations and found that some of them are likely to impact protein function and therefore play a role in tumor progression (Tables I-IV). However, previous studies did not show mutations in LGI1 in glioblastomas, suggesting that they are not a common event (31,32). Further studies will be needed to determine the role of LGI1 in gliomagenesis.
Acknowledgments
This work was supported by the American Brain Tumor Association (JSBS) and by grants CA92309 and CA101954 from the National Institutes of Health (ANAM; AES). This work has also been supported in part by the Molecular Imaging and the Molecular Biology cores at the H.L. Moffitt Cancer Center.
References
- 1.Kleihues P, Cavenee WK, editors. WHO Classification of Tumours: Pathology and Genetics of Tumours of Nervous System. IARC Press; Lyon: 2000. [Google Scholar]
- 2.Coons SW, Johnson PC, Scheithauer BW, Yates AJ, Pearl DK. Improving diagnostic accuracy and interobserver concordance in the classification and grading of primary gliomas. Cancer. 1997;79:1381–1393. doi: 10.1002/(sici)1097-0142(19970401)79:7<1381::aid-cncr16>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 3.Kleihues P, Ohgaki H. Phenotype vs genotype in the evolution of astrocytic brain tumors. Toxicol Pathol. 2000;28:164–170. doi: 10.1177/019262330002800121. [DOI] [PubMed] [Google Scholar]
- 4.Nozaki M, Tada M, Kobayashi H, Zhang CL, Sawamura Y, Abe H, Ishii N, Van Meir EG. Roles of the functional loss of p53 and other genes in astrocytoma tumorigenesis and progression. Neurooncol. 1999;1:124–137. doi: 10.1093/neuonc/1.2.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lang FF, Miller DC, Koslow M, Newcomb EW. Pathways leading to glioblastoma multiforme: a molecular analysis of genetic alterations in 65 astrocytic tumors. J Neurosurg. 1994;81:427–436. doi: 10.3171/jns.1994.81.3.0427. [DOI] [PubMed] [Google Scholar]
- 6.Chernova OB, Somerville RP, Cowell JK. A novel gene, LGI1, from 10q24 is rearranged and downregulated in malignant brain tumors. Oncogene. 1998;17:2873–2881. doi: 10.1038/sj.onc.1202481. [DOI] [PubMed] [Google Scholar]
- 7.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 8.Xiao B, Spencer J, Clements A, Ali-Khan N, Mittnacht S, Broceno C, Burghammer M, Perrakis A, Marmorstein R, Gamblin SJ. Crystal structure of the retinoblastoma tumor suppressor protein bound to E2F and the molecular basis of its regulation. Proc Natl Acad Sci USA. 2003;100:2363–2368. doi: 10.1073/pnas.0436813100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee C, Chang JH, Lee HS, Cho Y. Structural basis for the recognition of the E2F transactivation domain by the retinoblastoma tumor suppressor. Genes Dev. 2002;16:3199–3212. doi: 10.1101/gad.1046102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Collins VP. Brain tumours: classification and genes. J Neurol Neurosurg Psychiatry. 2002;75:ii2–ii11. doi: 10.1136/jnnp.2004.040337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ichimura K, Bolin MB, Goike HM, Schmidt EE, Moshref A, Collins VP. Deregulation of the p14ARF/MDM2/p53 pathway is a prerequisite for human astrocytic gliomas with G1-S transition control gene abnormalities. Cancer Res. 2000;60:417–424. [PubMed] [Google Scholar]
- 12.Ichimura K, Schmidt EE, Goike HM, Collins VP. Human glioblastomas with no alterations of the CDKN2A (p16INK4A, MTS1) and CDK4 genes have frequent mutations of the retinoblastoma gene. Oncogene. 1996;13:1065–1072. [PubMed] [Google Scholar]
- 13.Ueki K, Ono Y, Henson JW, Efird JT, von DA, Louis DN. CDKN2/p16 or RB alterations occur in the majority of glioblastomas and are inversely correlated. Cancer Res. 1996;56:150–153. [PubMed] [Google Scholar]
- 14.Lee SH, Kim JH, Rhee CH, Kang YS, Lee JH, Hong SI, Choi KS. Loss of heterozygosity on chromosome 10, 13q(Rb), 17p, and p53 gene mutations in human brain gliomas. J Korean Med Sci. 1995;10:442–448. doi: 10.3346/jkms.1995.10.6.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watanabe T, Yokoo H, Yokoo M, Yonekawa Y, Kleihues P, Ohgaki H. Concurrent inactivation of RB1 and TP53 pathways in anaplastic oligodendrogliomas. J Neuropathol Exp Neurol. 2001;60:1181–1189. doi: 10.1093/jnen/60.12.1181. [DOI] [PubMed] [Google Scholar]
- 16.Ichimura K, Ohgaki H, Kleihues P, Collins VP. Molecular pathogenesis of astrocytic tumours. J Neurooncol. 2004;70:137–160. doi: 10.1007/s11060-004-2747-2. [DOI] [PubMed] [Google Scholar]
- 17.von Deimling A, Fimmers R, Schmidt MC, Bender B, Fassbender F, Nagel J, Jahnke R, Kaskel P, Duerr EM, Koopmann J, Maintz D, Steinbeck S, Wick W, Platten M, Muller DJ, Przkora R, Waha A, Blumcke B, Wellenreuther R, Meyer-Puttlitz B, Schmidt O, Mollenhauer J, Poustka A, Stangl AP, Lenartz D, et al. Comprehensive allelotype and genetic analysis of 466 human nervous system tumors. J Neuropathol Exp Neurol. 2000;59:544–558. doi: 10.1093/jnen/59.6.544. [DOI] [PubMed] [Google Scholar]
- 18.Zhu Y, Parada LF. The molecular and genetic basis of neurological tumours. Nat Rev Cancer. 2002;2:616–626. doi: 10.1038/nrc866. [DOI] [PubMed] [Google Scholar]
- 19.Zhu Y, Guignard F, Zhao D, Liu L, Burns DK, Mason RP, Messing A, Parada LF. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell. 2005;8:119–130. doi: 10.1016/j.ccr.2005.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kato S, Han SY, Liu W, Otsuka K, Shibata H, Kanamaru R, Ishioka C. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci USA. 2003;100:8424–8429. doi: 10.1073/pnas.1431692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mathe E, Olivier M, Kato S, Ishioka C, Vaisman I, Hainaut P. Predicting the transactivation activity of p53 missense mutants using a four-body potential score derived from Delaunay tessellations. Hum Mutat. 2006;27:163–172. doi: 10.1002/humu.20284. [DOI] [PubMed] [Google Scholar]
- 22.Tavtigian SV, Deffenbaugh AM, Yin L, Judkins T, Scholl T, Samollow PB, de Silva D, Zharkikh A, Thomas A. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J Med Genet. 2006;43:295–305. doi: 10.1136/jmg.2005.033878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ng PC, Henikoff S. Accounting for human polymorphisms predicted to affect protein function. Genome Res. 2002;12:436–446. doi: 10.1101/gr.212802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goodrich DW. How the other half lives, the amino-terminal domain of the retinoblastoma tumor suppressor protein. J Cell Physiol. 2003;197:169–180. doi: 10.1002/jcp.10358. [DOI] [PubMed] [Google Scholar]
- 25.Kunapuli P, Kasyapa CS, Hawthorn L, Cowell JK. LGI1, a putative tumor metastasis suppressor gene, controls in vitro invasiveness and expression of matrix metalloproteinases in glioma cells through the ERK1/2 pathway. J Biol Chem. 2004;279:23151–23157. doi: 10.1074/jbc.M314192200. [DOI] [PubMed] [Google Scholar]
- 26.Piepoli T, Jakupoglu C, Gu W, Lualdi E, Suarez-Merino B, Poliani PL, Cattaneo MG, Ortino B, Goplen D, Wang J, Mola R, Inverardi F, Frassoni C, Bjerkvig R, Steinlein O, Vicentini LM, Brustle O, Finocchiaro G. Expression studies in gliomas and glial cells do not support a tumor suppressor role for LGI1. Neurooncol. 2006;8:96–108. doi: 10.1215/15228517-2005-006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gu W, Brodtkorb E, Piepoli T, Finocchiaro G, Steinlein OK. LGI1: a gene involved in epileptogenesis and glioma progression? Neurogenetics. 2005;6:59–66. doi: 10.1007/s10048-005-0216-5. [DOI] [PubMed] [Google Scholar]
- 28.Kalachikov S, Evgrafov O, Ross B, Winawer M, Barker-Cummings C, Martinelli BF, Choi C, Morozov P, Das K, Teplitskaya E, Yu A, Cayanis E, Penchaszadeh G, Kottmann AH, Pedley TA, Hauser WA, Ottman R, Gilliam TC. Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nat Genet. 2002;30:335–341. doi: 10.1038/ng832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morante-Redolat JM, Gorostidi-Pagola A, Piquer-Sirerol S, Saenz A, Poza JJ, Galan J, Gesk S, Sarafidou T, Mautner VF, Binelli S, Staub E, Hinzmann B, French L, Prud'homme JF, Passarelli D, Scannapieco P, Tassinari CA, Avanzini G, Marti-Masso JF, Kluwe L, Deloukas P, Moschonas NK, Michelucci R, Siebert R, Nobile C, et al. Mutations in the LGI1/Epitempin gene on 10q24 cause autosomal dominant lateral temporal epilepsy. Hum Mol Genet. 2002;11:1119–1128. doi: 10.1093/hmg/11.9.1119. [DOI] [PubMed] [Google Scholar]
- 30.Gu W, Brodtkorb E, Steinlein OK. LGI1 is mutated in familial temporal lobe epilepsy characterized by aphasic seizures. Ann Neurol. 2002;52:364–367. doi: 10.1002/ana.10280. [DOI] [PubMed] [Google Scholar]
- 31.Krex D, Hauses M, Appelt H, Mohr B, Ehninger G, Schackert HK, Schackert G. Physical and functional characterization of the human LGI1 gene and its possible role in glioma development. Acta Neuropathol. 2002;103:255–266. doi: 10.1007/s004010100463. [DOI] [PubMed] [Google Scholar]
- 32.Somerville RP, Chernova O, Liu S, Shoshan Y, Cowell JK. Identification of the promoter, genomic structure, and mouse ortholog of LGI1. Mamm Genome. 2000;11:622–627. doi: 10.1007/s0033500101280. [DOI] [PubMed] [Google Scholar]
- 33.Lohmann DR, Brandt B, Hopping W, Passarge E, Horsthemke B. Distinct RB1 gene mutations with low penetrance in hereditary retinoblastoma. Hum Genet. 1994;94:349–354. doi: 10.1007/BF00201591. [DOI] [PubMed] [Google Scholar]
- 34.Gu W, Wevers A, Schroder H, Grzeschik KH, Derst C, Brodtkorb E, de Vos R, Steinlein OK. The LGI1 gene involved in lateral temporal lobe epilepsy belongs to a new subfamily of leucine-rich repeat proteins. FEBS Lett. 2002;519:71–76. doi: 10.1016/s0014-5793(02)02713-8. [DOI] [PubMed] [Google Scholar]