

Abstract
The ability of cancer to evolve and adapt is a principal challenge to therapy in general, and to the paradigm of targeted therapy in particular. This ability is fueled by the co-existence of multiple, genetically heterogeneous subpopulations within the cancer cell population. Increasing evidence has supported the idea that these subpopulations are selected in a Darwinian fashion, by which the genetic landscape of the tumor is continuously reshaped. Massively parallel sequencing has enabled a recent surge in our ability to study this process, adding to previous efforts using cytogenetic methods and targeted sequencing. Altogether, these studies reveal the complex evolutionary trajectories occurring across individual hematological malignancies. They also suggest that while clonal evolution may contribute to resistance to therapy, treatment may also hasten the evolutionary process. New insights into this process challenge us to understand the impact of treatment on clonal evolution, and inspire the development of novel prognostic and therapeutic strategies.
Keywords: Cancer evolution, clonal heterogeneity, massively parallel sequencing
Introduction
The past decade has been a remarkable period of progress in the treatment of cancer in general, and hematologic malignancies in particular. Much of this progress has been based on exploiting knowledge of the genetic vulnerabilities of particular cancers so that they can be effectively targeted. For example, the impressive efficacy of tyrosine kinase inhibition (abrogating constitutive Abl kinase activity) for chronic myelogenous leukemia (CML) has unequivocally established the paradigm of targeted therapy for the treatment of malignant disease1. Likewise, understanding the role of APML-RARA in acute promyelocytic leukemia has led to a highly effective regimen with minimal toxicity that overcomes the effects of this gene fusion, and that does not include conventional chemotherapy2. Collectively, these examples suggest that the promise of precision medicine is finally coming to fruition in the treatment of blood malignancies.
At the same time, this revolution has also taught us important humbling lessons. Targeted cancer therapy, even when achieving highly effective responses, typically provides only short-lived relief. The malignant process often finds alternate routes to circumvent the roadblocks imposed on it by targeted monotherapy3–5. An instructive example is the case of Philadelphia chromosome positive B cell acute lymphoblastic leukemia (Ph+ B-ALL). The BCR-ABL1 oncogene is critical for the generation of Ph+ B-ALL, as shown by the high frequency of this lesion in ALL, its adverse prognostic impact6, as well as the strong in vitro transformative capacity of this driver7. The success of imatinib in the treatment of CML encouraged clinicians to attempt to inhibit the BCR-ABL1 oncogene in Ph+ B-ALL. Although a high response rate was observed (70% of patients)8, including in patients with refractory or relapsed disease9, the responses were uniformly short-lived with disease progression occurring within weeks. High failure rates were also seen with more potent, second generation, tyrosine kinase inhibitors such as dasatinib10, with the emergence of drug resistant clones.
Thus, even while the genomic revolution is rapidly expanding the list of potentially targetable genetic lesions, 11, the ability of cancer to adapt poses significant limitations to the therapeutic potential of both standard chemotherapy as well as targeted therapies. As reviewed herein, several lines of evidence lead to an increasing appreciation that the plasticity of cancer--its ability to adapt both to host defenses and to therapy--as an additional facet to consider in the selection and timing of cancer therapeutics.
Clonal heterogeneity, the engine of cancer plasticity
Genetic plasticity is defined as one of the enabling characteristics of cancer, in which the acquisition of the multiple cancer hallmarks depends on a succession of alterations in the genomes of neoplastic cells12. This plasticity results from ongoing accumulation of additional somatic mutations that are then positively selected. Cases of convergent evolution have been observed in which the same genetic target may sustain several different somatic mutations within the same tumor, yet affecting different subclones (e.g. the case of deletion BTG1 in ALL13). These findings strongly suggest that the lesions we detect at the level of large populations of cancer cells are the products of an astonishing amount of genetic “trial and error” that occurs in every cancerous process at the single cell level. This high degree of genetic variability provides a ready substrate for an evolutionary optimization process, as subclones compete over resources and adapt to external pressures such as cancer therapy. Cancer progression, therefore, is fundamentally a process of mutational diversification and clonal selection14.
The first experimental evidence supporting the idea that tumors are composed of heterogeneous subpopulations was obtained from mouse models of solid malignancies. These experiments showed that individual subclones possessed different phenotypic characteristics including varying metastatic potential15. Importantly, the link between heterogeneity and resistance to therapy was apparent even in other early experiments. For example, cell lines that exhibited a higher degree of phenotypic heterogeneity also acquired resistance to chemotherapy (methotrexate) at a higher rate compared with cell lines with lower phenotypic variability16.
Since cancer is a disease that results from the accumulation of genetic alterations17, a natural corollary of the above studies is that phenotypic evolution must stem from underlying genotypic evolution. This concept has been indeed confirmed over the past several decades with increasing technological sophistication, using approaches based on cytogenetics18,19, and Sanger sequencing20 (“first generation sequencing”). Mullighan et al, for example, in an elegant SNP-array analyses of pediatric pre-B cell ALL, demonstrated complex branched evolutionary growth associated with disease relapse11. This landmark study further showed how relapsed disease is genetically altered compared with disease at diagnosis. In chronic lymphocytic leukemia (CLL), clonal evolution was identified in up to 43% of patients using FISH or cytogenetic techniques, with frequent acquisition of the poor prognostic markers del(11q) and del(17p)21, and occurring at a higher rate in the poor prognosis group of IGHV unmutated cases 22.
Together, these experimental observations have demonstrated that the genetic makeup of hematologic malignancies is constantly reshaped during disease progression. Overall, they support the prescient ideas theorized by Nowell, who postulated that genetic instability would be expected to lead to enhanced heterogeneity with cancer progression23, resulting in diverse, genetically distinct, subpopulations within a neoplasm24. Thus, the selection process would be expected to promote the outgrowth of increasingly fit subclones, thereby continuously remodeling the fitness of the overall population.
If cancer plasticity is driven by clonal heterogeneity, it is important to consider the features that fuel the generation of clonal heterogeneity12. Genetic instability undoubtedly plays a key role in this process. The rate of acquisition of novel somatic mutations is likely closely tied to the diversification of the cancer population and therefore for enhancing its evolutionary potential, together with other features such as the population size25 (comprehensively review elsewhere26). A permissive genetic context that either inhibits DNA repair (e.g., BRCA mutations) or increases tolerance to novel mutations by removing critical checkpoints (e.g., TP53 or ATM mutations, enabling tolerance towards massive genomic damage27)28 is likely to increase the overall diversity of the tumor population. Adding to the complexity, different areas of the genome may have different rates of mutations acquisition29,30, which would need to be taken into account when inferring past rates of mutations from genomic information. A potentially provocative notion that arises from these data is whether genetic instability may be targeted as a measure to inhibit cancer evolution. For example, for hematological malignancies, the documented ongoing31 mutagenic32 activity of enzymes responsible for B and T cell receptor genetic modifications may be of particular interest.
Unraveling clonal complexity with massively parallel sequencing
While confirming the basic tenet of cancer as an evolutionary disease, the above described studies are inherently limited in their ability to decipher the true extent of genomic heterogeneity, given the limited amount of genetic lesions studied at any experiment and the limited sensitivity of experimental techniques that were then available to detect smaller subclones. Both of these limitations have been largely overcome with the advent of massively parallel sequencing (MPS). MPS of tumors has afforded an exponential increase in the ability to characterize the genetic landscape of cancer33. It has revealed a very high degree of intertumoral heterogeneity (i.e different genetic lesion affecting different tumors), with hundreds of different mutations affecting different tumors with a likely effect on fitness34,35. Moreover, this technology has also revealed a high level of intratumoral genetic heterogeneity (i.e., different genetic lesions affecting different subclones within an individual tumor), which also affects putative driver events36. In particular, the advent of MPS has allowed researchers to identify both subclonal somatic copy number alterations (SCNA) and subclonal somatic single nucleotide variations (SSNV) 37,38, which can be tracked over time to study tumor evolution 39,40. This ability to reconstruct the clonal structure is derived from an inherent property of MPS. It involves generating billions of independent sequencing reads, each derived from a single DNA molecule33. Thus, MPS data represents an informative random sample of individual DNA molecules contained within a tumor. At SSNV sites, the number of sequencing reads supporting the alternate and reference bases can be used to calculate a quantitative measurement of the variant allelic fraction (VAF).
In samples derived from diploid cancers such as AML that essentially lack SCNAs and are not contaminated with non-malignant cells, allelic fractions can be used to estimate SSNV clonality directly (in which any clonal SSNV should have a VAF of 0.5; while SSNVs in subclones will have lower VAFs). However, the vast majority of human cancers contain frequent SCNAs41, with many of them having undergone whole genome doubling during their evolution37. In addition, most tumor specimens contain a substantial fraction of normal cells37. Thus, in order to correctly infer SSNV clonality from MPS data, it is necessary to account for both local copy number occurring at SSNV sites and overall the tumor purity.
Recently, inference methods have been developed which attempt to account for these factors in order to estimate the actual cancer cell fraction (CCF) harboring a specific mutation 38,42 (Figure 1). Although moderate sequencing depth may result in considerable uncertainty in the CCF estimates of individual mutations, the fact that subclonal mutations are expected to co-occur in discrete subclonal cell populations has formed the basis of using clustering techniques to better resolve the subclonal structure of bulk tumor samples38,42.
Figure 1.
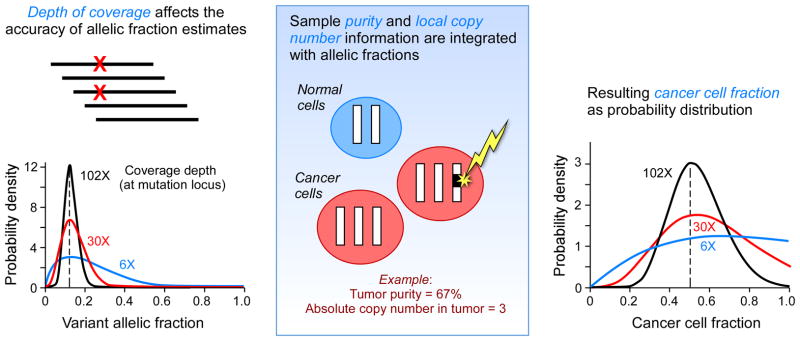
inferring the size of a subpopulation affected by somatic mutations from genomic data. Massively parallel sequencing provides an estimate of variant allelic fraction (VAF), which is calculated by counting the number of reads with the variant alleles and dividing it by the total number of reading from the specific location. The certainty of the estimate is a function of the depth of coverage, using binomial distribution (panel A). Subsequently, the VAF estimates are integrated with the purity and local copy number information (panel B) to yield cancer cell fractions (CCF, panel C). In the example provided a somatic mutation with a VAF of 0.125, a local copy number of 3 and a purity of 67% yields CCF estimates of 0.5.
Because DNA from all cells present in the bulk sample is mixed together prior to sequencing, information regarding which mutations co-occur in specific subclones is not readily accessible from analysis of a single sample. In general, analysis of bulk DNA from a single cancer sample cannot rule out the possibility that all subclones are nested inside one another in a linear phylogeny. One exception to this was reported in a study that applied deep whole genome sequencing (188X) of a single primary breast cancer sample, and could infer branched evolution based on co-occurrence of informative alleles on sequencing reads38. While this approach represents an elegant solution to the phylogenetic inference problem, the deep coverage needed over the whole genome renders it impractical for large studies using existing technology. We note that sequencing platforms capable of reliably producing longer reads will make this approach far more powerful.
An attractive approach to inferring phylogenetic structures from analysis of bulk tumor DNA is to sequence multiple specimens from the same individual’s cancer. Branched evolutionary relationships can be detected as clusters in which one subclone may increase in frequency while another sibling subclone may exhibit a concomitant decrease in frequency. This approach has been used to identify branched evolution in leukemia samples taken before and after treatment 39,40 as well as in solid tumors sampled at multiple anatomical locations36.
The surprising aspects of clonal complexity of hematologic malignancies
One of the first key lessons gleaned from genome-wide studies of hematologic cancers is that clonal evolution which follows a complex branched path (where multiple subpopulation co-exist in the same tumor and compete for ascendency) is at least as common as a more linear trajectory (in which progeny clones replace parent clones in full selective sweeps). A traditional linear model of successive clonal expansions43,44 could have be expected of hematologic malignancies, by virtue of the mobile nature of their cellular normal counterparts (compared to solid tissues that are often embedded in fixed tissue architecture). In theory, this feature could theoretically have led to a decreased level of clonal complexity since cancer cells can readily move across tissues and hence undergo more homogenous cellular mixing. This scenario is unlike solid tumor malignancies, in which the spatial compartments are formed. To the contrary, however, whole genome/exome investigation of clonal evolution in acute myeloid leukemia (AML)39,45, myelodysplastic syndrome (MDS)46, multiple myeloma (MM)47,48 and CLL40 have all consistently demonstrated not only a high degree of clonal heterogeneity and marked changes in the genetic makeup of the disease upon relapse, but also branching rather than linear as the predominant pattern of evolution (Table 1). A major implication of these findings is that the evolutionary process is expected to result from complex interactions amongst multiple highly diverse populations rather than a clear succession of selective sweeps. Clonal competition among co-existing subpopulations that harbor driver lesions49 thus shapes the eventual composition of the tumor such that multiple clonal variants are present at the same time 50,51.
Table 1.
Next-generation sequencing studies of clonal evolution in hematologic malignancies
| Disease | Methodology | Number of cases | Insights |
|---|---|---|---|
| AML39 | WGS, followed by targeted deep sequencing | 8 | Relapse after chemotherapy is associated with clonal evolution and acquisition of new mutations |
| Secondary AML46 | WGS, followed by targeted deep sequencing | 7 | Secondary AML clones are often evolved progeny of MDS clones |
| Multiple Myeloma47 | WES | 1 | Clonal shifts occur along the history of the disease |
| Multiple Myeloma118 | WES | 1 | Clonal shifts occur along the history of the disease |
| CLL76 | WGS, followed by targeted deep sequencing | 3 | Different patterns of evolution evident through cycles of therapy |
| CLL40 | WES | 149 (18 longitudinal samples) | Subclonal drivers can anticipate clonal evolution and impact outcome |
| Essential thrombocytosis115 | Single cell WES | 1 | ET is monoclonal in origin |
| Follicular Lymphoma119 | WES | 8 | Early and late drivers identified |
It is important to note that the published analyses to date have been limited to the detection of macroscopic clonal heterogeneity (clone size of greater than 1–10% of the entire cell population). This is because only clones that either represent a substantial proportion of the cancer cell mass or clones that become dominant at some point during the studied period are trackable using current methodologies. Emerging technologies capable of achieving deeper sequencing depth of bulk DNA52,53 or single cell genomic sequencing methods54, may enable the study of smaller subpopulations. Delineating the full extent of cancer heterogeneity down to the single-cell level will enable us to understand how the seemingly stochastic process of “trial and error” at the single cell level is integrated through selection to shape the genetic makeup of the tumor. It carries the potential to refine the dichotomy of driver versus passenger mutations, by quantifying the fitness contribution of each individual mutation to selection (manifested in varying clone sizes).
Epigenetic clonal heterogeneity
While genetic alteration has been the main focus of evolutionary dynamics in cancer thus far, epigenetic modifications are likely responsible to a large part of phenotypic differences55 that ultimately affect fitness. Similar to genetic alterations, epigenetic modifications are heritable and therefore subject to natural selection. The contribution of epigenetic modification to selection in cancer is probably substantial, since epigenetic alterations accumulate as the cell population evolves and diversifies at rates estimated to be orders of magnitude higher compared with somatic genetic alterations56. Indeed, a large degree of intra-tumor epigenetic heterogeneity was recently described in lymphoma using DNA methylation arrays57.
Genetic and epigenetic changes likely have complex bi-directional interactions and co-operate to mold the evolutionary landscape. This complex and bidirectional interplay between genetic and epigenetic features in cancer has been perhaps most deeply explored in the area of cancer stem cells. Specifically, early xenograft studies of ALL revealed leukemic repopulation that recapitulated the genetic heterogeneity of the patients’ original leukemia 58,59. Similar findings were also demonstrated in solid tumor malignancies60. Anderson et al. concluded that cancer stem cells-- an epigenetically uniform population-- are genetically diverse. On the other hand, even genetically uniform cell subpopulations have been reported to reveal profound epigenetic differences leading to differences in the phenotypes of survival capacity and pluripotency potential61,62.
Together, these observations prompt a model of cancer evolution in which epigenetic and genetic heterogeneity are integrated, thereby accounting for, epigenetic heterogeneity of genetically uniform populations, and genetic heterogeneity of epigenetically uniform populations. Such a model would designate cells with high self-renewal capacity (an attribute encoded in the epigenetic state) as the crucial units subjected to selection forces in genetic evolution. Hence, the appearance of a new somatic mutation within these specialized cells could lead to their clonal propagation. At the same time, such a model would also acknowledge the existence of a far less homogenous cancer stem cell population then previously considered with regard to various features including drug sensitivity 63.
It is important to consider that in cancer, the movement between different epigenetic states (along the spectrum of pluripotency to differentiation, for example) may be altered as well. In multicellular organisms, epigenetic transitions are tightly controlled through numerous regulatory mechanisms64. Neoplastic transformation can unhinge those mechanisms, reverting to a state more closely resembling unicellular organisms65, in which the fluid movement across diverse states can achieve high adaptivity by “trial and error.” Epigenetic heterogeneity, thus, can be a hedging strategy for enhanced survival65. Cancer progression, therefore, may be viewed as a scenario in which both genetic and epigenetic population structures become increasingly malleable, such that the lines between populations with different ‘stemness’ potential become more blurred. Within this framework, ‘stemness’ may exist as a functional phenotype, which can be manifested by any member of a malignant population given the appropriate endogenous and exogenous factors66. Thus, a high degree of interclonal competition would likely select for cells with the highest self-renewing capacity at the expense of more differentiated cells, as has been demonstrated in CML67. Therapy may also accelerate this process by providing a strong selection for cancer stem-cell survival and proliferation67,68. Acquired genetic alterations probably play an important role in this scenario as well. For example, the loss of TP53, often seen with disease progression21 provokes stem-cell-like transcriptional programs69,70. Other oncogenes may also afford leukemogenic potential to committed myeloid progenitors, again demonstrating that genetic lesions may enlarge the available pool of cells with stem-like features71.
In summary, integrating the stem cell hierarchy and the genetic phylogenetic tree yields a complex evolutionary picture that has only begun to be unraveled. In concert with genetic diversification and fitness optimization, a similar process very likely occurs at the epigenetic level. Cancer stem cells constitute a growing proportion within the cancer cell mass72,73 and have a greater plasticity in terms of bi-directional conversion from stem cells to more differentiated cells. Together, this leads to enhancement of the cellular substrate available for selection, with large, treatment resistant, and genetically heterogeneous cancer stem cell population.
How does clonal evolution contribute to resistance to therapy?
Relapsed malignancy shows an almost universal phenotypic evolution, resulting in a more aggressive and treatment-refractory phenotype74. We and others have shown that frequent genetic evolution underlies the phenotypic evolution11,40. Therefore, a central question in cancer treatment is what is the precise nature of the interaction of clonal evolution with cancer therapy. Initial studies highlighted the potential role of chemotherapy to induce novel mutagenesis75 and thereby to enhance the process of genetic diversification (Figure 2A). Although studies of WGS are inherently limited by the power to detect minute subclones within a sample, studies in acute myeloid malignancies have nonetheless suggested the novel mutagenesis may result from the genotoxic effects of chemotherapy, supported also by a changing spectrum of somatic single nucleotide alterations39,76. In contrast, in case of indolent blood malignancies such as CLL, evidence for the contribution of the chemotherapy’s mutagenizing effect is limited. Prior purine analog-based therapy was not associated with an increased total number of mutations in CLL77, and also was not associated with an altered mutational pattern40. Therefore, while chemotherapy-induced mutagenesis has the potential to contribute to further clonal diversification, other sources for generating evolutionary shifts appear to be at play, and likely involve pre-existing genetic variants or subclones78.
Figure 2. Three models of how cancer therapy may accelerate clonal evolution.

First, cancer therapy, particularly containing genotoxic agents, can induce novel mutagenesis (A). Second, therapy can accelerate clonal evolution by selecting a clone (here illustrated in red) containing a mutation that confers resistance to the therapeutic agent used (B). The resistance of the selected clone is reflected in the depiction of the cell population after cytoreduction, composed almost entirely of the resistant clone (in red). A third model postulates similar sensitivity to treatment of the different subpopulations, reflected in similar proportions before and after cytoreduction. The clearing niche alters the dynamic evolutionary landscape allowing a faster rise of a fitter clone.
How then does therapy induce evolution from pre-treatment genetic variation? Two explanations are considered, depending on the tumor kinetics, the efficacy of cell kill with treatment and other factors related to both the tumor type and the specific treatment strategy. The first is that resistant clones may be actively selected by therapy (Figure 2B). Examples for this model are numerous79–81, including MSH6 mismatch repair gene mutations in recurrent glioblastoma multiforme after treatment with temozolomide82,83, and the BCR-ABL T315I mutations in chronic myelogenous leukemia84,85. Indeed, this model of clonal evolution induced by the selective pressure of therapy maybe particularly relevant in the context of targeted therapy, as the therapy is often directed at a particular genetic context which may not be shared by all subclones. This relationship between therapy and genetic adaptation is likely to result in convergent evolution, in which a mutation that confers resistance will become highly prevalent in relapsed disease. Indeed, this process has been reported in relapsed T-cell ALL after treatment with nucleoside-analog chemotherapy drugs86.
An alternative process contributing to the emergence of continuously more aggressive clones may be entirely independent of differential sensitivity to therapy (Figure 2C). We recently observed a higher number of large subclones (>10% of cancer cells) in 149 CLL cases that were exposed to treatment prior to sampling compared to patients that received therapy after the sample was obtained. This finding of increased clonal diversity with treatment held true even after accounting for potential confounders such as longer follow up time40. We interpret this observation to result, at least in part, from the outgrowth of many diverse pre-existing minor but fit subclones 76,87. This latter interpretation is further supported by our observation of an increased frequency of subclonal driver events (presumably fitter) in treated relative to untreated patients. Overall, our data support the idea that CLL therapy, by markedly reducing disease bulk, may act as a classic evolutionary restriction point and reset interclonal dynamics88.
Within this conceptual framework, when subclones with high fitness already exist within a tumor population, treatment could favor the development of more-aggressive clones, potentially reducing post-relapse survival40. In this context, cytotoxic therapy would effectively remove the incumbent clone 89 -- acting like a ‘mass extinction’ event 89 -- and thereby shift the evolutionary landscape 90,91 in favor of one or more aggressive subclones 92. Thus, highly fit subclones likely benefit from treatment and exhibit rapid outgrowth 78. These data provides mechanistic support to the observation that the “watch and wait” strategy for CLL leads to superior clinical results93, as the earlier administration of chemotherapy may accelerate clonal evolution and the emergence of fitter clones with more aggressive disease phenotypes. This form of relationship between therapy and evolution maybe particularly important to CLL, since this cancer type is highly dependent on growth and survival signals provided by the local microenvironment94. This dependency may augment the importance of the role of interclonal competition in the evolutionary dynamics of CLL. Future in-depth studies would assist in confirming this model as well as whether it is generalizable to malignancies other than CLL, and in particular in other more indolent cancers.
Translating clonal evolution to the clinic
A major priority of precision cancer genomics is to use information on genetic lesions to define patient prognosis. In an illustrative example, Patel et al. could demonstrate that lesions such as internal tandem duplication of FLT3 (FLT3-ITD), partial tandem duplication in MLL (MLL-PTD), as well as mutations in ASXL1 and PHF6 associated with reduced overall survival in AML, while CEBPA and IDH2 mutations associated with improved overall survival. These associations were independent of established risk factors 95. Similar efforts have been carried out in other hematologic malignancies including CLL 77,96,97, and multiple myeloma98.
Across the blood malignancies, patients with apparently poor prognostic markers can nonetheless exhibit good survival, and vice versa88. The several studies reviewed above (Table 1) suggest that intratumoral clonal heterogeneity may be an important contributor to this complex picture. In aggregate, studies of clonal evolution have revealed cancers to be genetically heterogeneous in space and time 99. Hence, simply labeling an individual cancer as harboring a genetic lesion or not is not fully precise. From a practical standpoint, for a solid tumor mass, or even leukemia cells that are present in different tissue compartments (i.e. blood vs. marrow vs. lymph node), multiple samplings may be required to correctly assert the genetic landscape of an individual case (Figure 3).
Figure 3. Translating clonal heterogeneity insights to the clinic.

Possible prognostic and therapeutic implications of clonal heterogeneity are outlined. Image courtesy Broad Institute/Lauren Solomon; hourglass photo © iStockphoto/Dominik Pabis.
Finally, clonal heterogeneity in and of itself may impact clinical outcome. Our studies have shown that the presence of a strong subclonal driver event, but not a clonal driver, negatively impacts clinical outcome in CLL40. The link between clonal heterogeneity and specifically the presence of a subclonal driver to adverse clinical outcome, adds an additional dimension to the current efforts of linking discrete somatic mutations to outcome. In other words, it is not only the presence or absence of a mutation that should be considered but also the size of the subpopulation it affects.
From the therapeutic standpoint, studies of cancer genomics highlight the concept that cancer is not a single disease entity, but rather a collection of related disorders; hence, treatment should be targeted to the molecular subtype of disease. For example, high-dose daunorubicin, as compared with standard-dose daunorubicin, improves the rate of survival among patients with DNMT3A, NPM1 mutations or MLL translocations in AML but not among patients with wild-type DNMT3A, NPM1, and MLL95. The potential to integrate the available high throughput sequencing technologies (e.g., DNA-seq, RNA-seq and ChIP-seq) to provide a patient-specific genomic-epigenomic map may provide crucial prognostic perspective and inform therapeutic choices100. Furthermore, targeted treatments that are based on the presence of specific molecular lesions may greatly improve therapeutic response, as seen in, for example, FIP1L1-PDGFRA eosinophilia-associated myeloproliferative disorders101. These “actionable mutations” where a clinician matches a tumor mutation to a cancer drug, maybe either be missed given genetic heterogeneity in time and space, or alternatively might involve only a small subclone, which begs the question of the clinical efficacy were it to be solely targeted. For instance, synthetic lethal approaches were found to be highly effective in situations in which all cancer cells contain the targeted variation, as witnessed by the potent efficacy of PARP inhibition in tumors of BRCA germline carriers102.
These observations together raises the provocative question of whether it is preferable to target genetic variations that are found in the “trunk” compared to those found in “branches” of the evolutionary phylogenetic tree99. Intuitively, the former may be considered the superior approach. “Trunk” events, by definition, are mutations present in all the cells of the malignant process. Targeting of this event in theory carries the potential of a complete extinction of the entire population of malignant cells. Conversely, it is unclear whether the cell remains dependent on the specific “trunk” target after acquiring additional oncogenic events (a “branch” target), and therefore, how well they will be impacted by therapy directed against these founder targets. In solid malignancies, such as NSCLC, KRAS and EGFR mutations are rarely detected together; however, when they co-occur, targeting the “trunk”-type mutation (i.e. EGFR) is no longer effective103. Similarly, BRAF canonical mutations are discovered in benign colonic polyps and are therefore likely to be earlier, “trunk”-type events. However the response to BRAF targeting has been disappointing104. One may hypothesize that at least in more indolent malignancies, targeting “branch” mutations (“pruning”) may be an effective strategy, which could promote clonal equilibrium and hinder the selection of more aggressive phenotypes.
The differential effects of targeting “branch” vs. “trunk” lesions may be determined, in part, by the complex epistatic relationship between different genetic lesions within the same clonal population. Since new mutations do not occur in isolation but rather enter into an established genomic landscape, the existing gene network may have a profound effect on the fate of the cell, and determine whether the novel mutation will result in cell death or clonal expansion. For example, activation of many oncogenes together, including KRAS, can lead to a state of ‘oncogene-induced senescence’ 105. A similar relationship has been demonstrated for c-MYC induced apoptosis that is relieved in the context of BCL-2 over expression106. Hence, further study of the epistatic relationships in model systems as well as in clinical trials will help clarify in what context optimal effects will result from targeting the “trunk” event and when it is preferable to target the “branches”.
The broader evolutionary perspective allows us to view cancer as an ecology of different subpopulations in the context of their environment87. Intriguing data suggests that at least in some cases, complex co-dependency relationships between subpopulations may exist 107, in addition to competition. The understanding that disease is composed of diverse subpopulations is a challenge to our traditional schemes of clinical trials. A future in which both trunk and branch events are characterized, and in which no two cancers share the same genomic features, may be envisaged108. In this setting, performing large-scale clinical trials using present-day methodologies, in particular utilizing combinations of targeted agents, may prove highly challenging. The disease can no longer be defined as a single entity containing a uniform set of genetic abnormalities. Furthermore, the degree of genetic heterogeneity of a tumor is likely to be an important determinant of therapeutic outcome92,109.
A better understanding is needed of the impact of therapy on the evolutionary landscape, possibly through the use of the more applicable whole-exome sequencing technologies to study large cohorts on patients37. Researchers may consider incorporating approaches such as WES that identify at least larger subpopulations (greater than 1–10% of cancer mass), and characterize their evolution in ancillary studies of prospective clinical trials. Such information may inform us regarding the adaptive processes responsible for treatment failure as well as eventually spark the development of novel therapeutic paradigms. For one, it has been proposed that alternative approaches could potentially maintain interclonal equilibrium at the expense of trying to maximize cell kill110. This approach supports preventing the elimination of therapy-sensitive clones, as they (theoretically) could continue to suppress the growth of therapy-resistant clones in a competitive manner, and thereby maintain an equilibrium state. A second approach that requires further consideration is the idea of limiting the underlying diversification that serves as the substrate for clonal evolution before the full expression of the genetic or the epigenetic heterogeneity in cancer is evident. Finally, the therapeutic challenge posed by a continuously adapting and reshaping malignant process provides strong rationale to support the pursuit of immunity-based therapies, since this approach may effectively pit one complex adaptive process against another. There is already limited evidence that allogeneic hematopoietic stem cell transplant (a non-specific example of a immune based therapy), imposes evolutionary pressures on the tumor which are distinct from other therapeutic modalities (leading for example to loss of donor-recipient mismatched HLA alleles111,112, or multiple cytogenetic abnormalities113). These alterations demonstrate that the leukemic cell population is being molded by a powerful immune response, and hence to the efficacy of the immune based therapy. The process of co-evolution of the cancer cells and the immune response in the setting of effective immunotherapy is an area of great interest for future study.
Conclusions and future directions
Understanding the evolutionary capacity of cancer is emerging as a key element in developing improved therapeutic strategies in the era of precision medicine, as it presents one of the most formidable obstacles to the successful application of targeted therapy.
As aforementioned, the intensive application of high-throughput genomic platforms has enabled rapid progress in our understanding of the process of clonal evolution in hematologic malignancies. Collectively, these studies have provided several core insights, including first, that clonal heterogeneity is common in malignancy both at the genetic and the epigenetic level; second, that clonal evolution is frequently observed in relation to therapy, leading to emergence of more aggressive and resistant disease; and finally, that the process of clonal evolution is linked to adverse clinical outcomes.
While these recent studies point to the key role played by clonal evolution in cancer progression, current perspectives of cancer as an evolutionary problem are fairly limited. Even as knowledge about germline and acquired genetic lesions associated with cancers has grown exponentially, we still know only little about the background rate of heterogeneity – which is the substrate of evolution -- and possess only a rudimentary understanding of how the epigenetic program affects this substrate. In this respect, developing methodologies to integrate data from complementary -- genetic and epigenetic-- high throughput platforms is key, both at the cell population and at the single cell level. Moreover, the dynamics of interactions between clones – whether they compete or are co-depend upon each other -- has not been elucidated. Additionally, the examination of key mechanistic question relating to clonal evolution using genomic tools (e.g., different types of selective pressure, interaction with microenvironment niches and interactions between multiple genetic lesions within the same cell) has yet to be accomplished. Thus, the ability to foresee the evolutionary trajectory of any individual cancer is presently still in its infancy. Improving this capacity to predict how cancer will evolve with treatment carries a significant potential to allow us to anticipate and tailor treatment to the likely future trajectory (so-called ‘anticipation-based chemotherapy’)114.
Ongoing technological developments are now generating tools ideally suited for the study of these questions. Recently, proof-of-principle studies of single-cell sequencing have been conducted which have catalogued the point mutations in protein-coding regions 115,116. In the not too distant future, single-cell sequencing will allow the detailed study of genetic heterogeneity that provide the backdrop against which evolution at the subpopulation level occurs. The application of single cell RNA-seq117 to the study of hematological malignancies would enable the study of the heterogeneous transcriptional changes and signaling networks that stem from heterogeneous somatic genetic alterations. In addition to single cell examination, novel methodologies are capable of deconvoluting subpopulations from bulk material37, and may be helpful in delineating the basic underlying principles of evolution in in vivo and in vitro models, with deeper sequencing providing both higher sensitivity for smaller subclones as well as more precise estimates of their size. The ability to use WES as an alternative approach to WGS40 as well as the projected downtrend of sequencing costs, will enable multi-sampling in time and space of hematologic malignancies, clarifying the nature of spatial heterogeneity in blood malignancies as well as questions regarding the nature repopulation of the ecological niche upon relapse. It may also allow the study of these questions in large clinical trials. These efforts can potentially answer questions of fundamental importance to the clinical application of these insights, namely what is the prognostic significance of the size of the subclone that harbors a genetic marker, when should we target branch or trunk lesions, and how to integrate this knowledge in combinatorial therapies.
Acknowledgments
DAL acknowledges support by the American Society of Hematology (Research Award for Fellows-in-Training), and the American Cancer Society. C.J.W. acknowledges support from the Blavatnik Family Foundation, the Lymphoma Research Foundation, NHLBI (1RO1HL103532-01; 1RO1HL116452-01) and NCI (1R01CA155010-01A1) and is a recipient of a Leukemia Lymphoma Translational Research Program Award and an AACR SU2C Innovative Research Grant. We thank all members of the Broad Institute’s Biological Samples and Genome Sequencing Platforms, as well as the Cancer Genome Analysis group who made this work possible (NHGRI-U54HG003067).
Glossary of abbreviations
- ALL
acute lymphoblastic leukemia
- AML
acute myeloid leukemia
- CCF
cancer cell fraction (the proportion of cancer cells harboring a specific genetic alteration of the entire cancer cell population sampled)
- CLL
chronic lymphocytic leukemia
- CML
chronic myelogenous leukemia
- FISH
Flourescent in situ hybridization
- IGHV status
Ig heavy variable region. Mutated typically defined as <98% homology with reference genome, signifying a past process of affinity maturation through somatic hypermutation
- LOH
loss of heterozygocity
- MDS
myelodysplastic syndrome
- MM
Multiple Myeloma
- MPS
Massively parallel sequencing
- sCNA
somatic copy number alteration
- SNP
single nucleotide polymorphism
- sSNV
somatic single nucleotide variant
- VAF
variant allele frequency (the proportion of reads that contain a genetic variant of the entire of sequenced reads for a given locus)
Footnotes
Conflict of interests
The authors declare no conflict of interests.
References
- 1.Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–7. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 2.Lo-Coco F, Avvisati G, Vignetti M, Thiede C, Orlando SM, Iacobelli S, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med. 2013;369:111–21. doi: 10.1056/NEJMoa1300874. [DOI] [PubMed] [Google Scholar]
- 3.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 4.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–19. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arkenau HT, Kefford R, Long GV. Targeting BRAF for patients with melanoma. Br J Cancer. 2011;104:392–8. doi: 10.1038/sj.bjc.6606030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Westbrook CA, Hooberman AL, Spino C, Dodge RK, Larson RA, Davey F, et al. Clinical significance of the BCR-ABL fusion gene in adult acute lymphoblastic leukemia: a Cancer and Leukemia Group B Study (8762) Blood. 1992;80:2983–90. [PubMed] [Google Scholar]
- 7.Lugo TG, Pendergast AM, Muller AJ, Witte ON. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science. 1990;247:1079–82. doi: 10.1126/science.2408149. [DOI] [PubMed] [Google Scholar]
- 8.Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344:1038–42. doi: 10.1056/NEJM200104053441402. [DOI] [PubMed] [Google Scholar]
- 9.Ottmann OG, Druker BJ, Sawyers CL, Goldman JM, Reiffers J, Silver RT, et al. A phase 2 study of imatinib in patients with relapsed or refractory Philadelphia chromosome-positive acute lymphoid leukemias. Blood. 2002;100:1965–71. doi: 10.1182/blood-2001-12-0181. [DOI] [PubMed] [Google Scholar]
- 10.Foa R, Vitale A, Vignetti M, Meloni G, Guarini A, De Propris MS, et al. Dasatinib as first-line treatment for adult patients with Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood. 2011;118:6521–8. doi: 10.1182/blood-2011-05-351403. [DOI] [PubMed] [Google Scholar]
- 11.Mullighan CG, Phillips LA, Su X, Ma J, Miller CB, Shurtleff SA, et al. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. 2008;322:1377–80. doi: 10.1126/science.1164266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 13.Waanders E, Scheijen B, van der Meer LT, van Reijmersdal SV, van Emst L, Kroeze Y, et al. The origin and nature of tightly clustered BTG1 deletions in precursor B-cell acute lymphoblastic leukemia support a model of multiclonal evolution. PLoS Genet. 2012;8:e1002533. doi: 10.1371/journal.pgen.1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Merlo LM, Pepper JW, Reid BJ, Maley CC. Cancer as an evolutionary and ecological process. Nat Rev Cancer. 2006;6:924–35. doi: 10.1038/nrc2013. [DOI] [PubMed] [Google Scholar]
- 15.Harris JF, Chambers AF, Hill RP, Ling V. Metastatic variants are generated spontaneously at a high rate in mouse KHT tumor. Proc Natl Acad Sci U S A. 1982;79:5547–51. doi: 10.1073/pnas.79.18.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cillo C, Dick JE, Ling V, Hill RP. Generation of drug-resistant variants in metastatic B16 mouse melanoma cell lines. Cancer Res. 1987;47:2604–8. [PubMed] [Google Scholar]
- 17.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–67. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 18.Zhang L, Znoyko I, Costa LJ, Conlin LK, Daber RD, Self SE, et al. Clonal diversity analysis using SNP microarray: a new prognostic tool for chronic lymphocytic leukemia. Cancer Genet. 2011;204:654–65. doi: 10.1016/j.cancergen.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 19.Calissano C, Damle R, Hayes G, Murphy E, Hellerstein M, Moreno C, et al. In vivo intraclonal and interclonal kinetic heterogeneity in B-cell chronic lymphocytic leukemia. Blood. 2009;114:4832–42. doi: 10.1182/blood-2009-05-219634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bagnara D, Callea V, Stelitano C, Morabito F, Fabris S, Neri A, et al. IgV gene intraclonal diversification and clonal evolution in B-cell chronic lymphocytic leukaemia. Br J Haematol. 2006;133:50–8. doi: 10.1111/j.1365-2141.2005.05974.x. [DOI] [PubMed] [Google Scholar]
- 21.Stilgenbauer S, Sander S, Bullinger L, Benner A, Leupolt E, Winkler D, et al. Clonal evolution in chronic lymphocytic leukemia: acquisition of high-risk genomic aberrations associated with unmutated VH, resistance to therapy, and short survival. Haematologica. 2007;92:1242–5. doi: 10.3324/haematol.10720. [DOI] [PubMed] [Google Scholar]
- 22.Gunnarsson R, Mansouri L, Isaksson A, Goransson H, Cahill N, Jansson M, et al. Array-based genomic screening at diagnosis and during follow-up in chronic lymphocytic leukemia. Haematologica. 2011;96:1161–9. doi: 10.3324/haematol.2010.039768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–8. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 24.Fidler IJ. Tumor heterogeneity and the biology of cancer invasion and metastasis. Cancer Res. 1978;38:2651–60. [PubMed] [Google Scholar]
- 25.Cairo MS, Jordan CT, Maley CC, Chao C, Melnick A, Armstrong SA, et al. NCI first International Workshop on the biology, prevention, and treatment of relapse after allogeneic hematopoietic stem cell transplantation: report from the committee on the biological considerations of hematological relapse following allogeneic stem cell transplantation unrelated to graft-versus-tumor effects: state of the science. Biol Blood Marrow Transplant. 2010;16:709–28. doi: 10.1016/j.bbmt.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Loeb LA. Human cancers express mutator phenotypes: origin, consequences and targeting. Nat Rev Cancer. 2011;11:450–7. doi: 10.1038/nrc3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bassaganyas L, Bea S, Escaramis G, Tornador C, Salaverria I, Zapata L, et al. Sporadic and reversible chromothripsis in chronic lymphocytic leukemia revealed by longitudinal genomic analysis. Leukemia. 2013 doi: 10.1038/leu.2013.127. Epublication: 2013/04/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat Rev Cancer. 2012;12:587–98. doi: 10.1038/nrc3342. [DOI] [PubMed] [Google Scholar]
- 29.Schwartz M, Zlotorynski E, Kerem B. The molecular basis of common and rare fragile sites. Cancer Lett. 2006;232:13–26. doi: 10.1016/j.canlet.2005.07.039. [DOI] [PubMed] [Google Scholar]
- 30.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–8. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gawad C, Pepin F, Carlton VE, Klinger M, Logan AC, Miklos DB, et al. Massive evolution of the immunoglobulin heavy chain locus in children with B precursor acute lymphoblastic leukemia. Blood. 2012;120:4407–17. doi: 10.1182/blood-2012-05-429811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mahowald GK, Baron JM, Sleckman BP. Collateral damage from antigen receptor gene diversification. 2008;135:1009–12. doi: 10.1016/j.cell.2008.11.024. [DOI] [PubMed] [Google Scholar]
- 33.Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second-generation sequencing. Nat Rev Genet. 2010;11:685–96. doi: 10.1038/nrg2841. [DOI] [PubMed] [Google Scholar]
- 34.Pleasance E, Cheetham R, Stephens P, McBride D, Humphray S, Greenman C, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191–6. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, et al. A census of human cancer genes. Nat Rev Cancer. 2004;4:177–83. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–21. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nik-Zainal S, Van Loo P, Wedge DC, Alexandrov LB, Greenman CD, Lau KW, et al. The life history of 21 breast cancers. Cell. 2012;149:994–1007. doi: 10.1016/j.cell.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–10. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Landau D, Carter S, Stojanov P, McKenna A, Stevenson K, Lawrence M, et al. Evolution and Impact of Subclonal Mutations in Chronic Lymphocytic Leukemia. Cell. 2013;152:714–26. doi: 10.1016/j.cell.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486:395–9. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maley CC, Galipeau PC, Li X, Sanchez CA, Paulson TG, Reid BJ. Selectively advantageous mutations and hitchhikers in neoplasms: p16 lesions are selected in Barrett’s esophagus. Cancer Res. 2004;64:3414–27. doi: 10.1158/0008-5472.CAN-03-3249. [DOI] [PubMed] [Google Scholar]
- 44.Bozic I, Antal T, Ohtsuki H, Carter H, Kim D, Chen S, et al. Accumulation of driver and passenger mutations during tumor progression. Proc Natl Acad Sci U S A. 2010;107:18545–50. doi: 10.1073/pnas.1010978107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264–78. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Walter MJ, Shen D, Ding L, Shao J, Koboldt DC, Chen K, et al. Clonal architecture of secondary acute myeloid leukemia. N Engl J Med. 2012;366:1090–8. doi: 10.1056/NEJMoa1106968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Egan JB, Shi CX, Tembe W, Christoforides A, Kurdoglu A, Sinari S, et al. Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, evolution, and clonal tides. Blood. 2012;120:1060–6. doi: 10.1182/blood-2012-01-405977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Keats JJ, Chesi M, Egan JB, Garbitt VM, Palmer SE, Braggio E, et al. Clonal competition with alternating dominance in multiple myeloma. Blood. 2012;120:1067–76. doi: 10.1182/blood-2012-01-405985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Snuderl M, Fazlollahi L, Le LP, Nitta M, Zhelyazkova BH, Davidson CJ, et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell. 2011;20:810–7. doi: 10.1016/j.ccr.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 50.Ene CI, Fine HA. Many tumors in one: a daunting therapeutic prospect. Cancer Cell. 2011;20:695–7. doi: 10.1016/j.ccr.2011.11.018. [DOI] [PubMed] [Google Scholar]
- 51.Quentmeier H, Amini RM, Berglund M, Dirks WG, Ehrentraut S, Geffers R, et al. U-2932: two clones in one cell line, a tool for the study of clonal evolution. Leukemia. 2013;27:1155–64. doi: 10.1038/leu.2012.358. [DOI] [PubMed] [Google Scholar]
- 52.Campbell PJ, Pleasance ED, Stephens PJ, Dicks E, Rance R, Goodhead I, et al. Subclonal phylogenetic structures in cancer revealed by ultra-deep sequencing. Proc Natl Acad Sci U S A. 2008;105:13081–6. doi: 10.1073/pnas.0801523105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smith AE, Mohamedali AM, Kulasekararaj A, Lim Z, Gaken J, Lea NC, et al. Next-generation sequencing of the TET2 gene in 355 MDS and CMML patients reveals low-abundance mutant clones with early origins, but indicates no definite prognostic value. Blood. 2010;116:3923–32. doi: 10.1182/blood-2010-03-274704. [DOI] [PubMed] [Google Scholar]
- 54.Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90–4. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14:204–20. doi: 10.1038/nrg3354. [DOI] [PubMed] [Google Scholar]
- 56.Siegmund KD, Marjoram P, Woo YJ, Tavare S, Shibata D. Inferring clonal expansion and cancer stem cell dynamics from DNA methylation patterns in colorectal cancers. Proc Natl Acad Sci U S A. 2009;106:4828–33. doi: 10.1073/pnas.0810276106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.De S, Shaknovich R, Riester M, Elemento O, Geng H, Kormaksson M, et al. Aberration in DNA methylation in B-cell lymphomas has a complex origin and increases with disease severity. PLoS Genet. 2013;9:e1003137. doi: 10.1371/journal.pgen.1003137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Anderson K, Lutz C, van Delft FW, Bateman CM, Guo Y, Colman SM, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011;469:356–61. doi: 10.1038/nature09650. [DOI] [PubMed] [Google Scholar]
- 59.Clappier E, Gerby B, Sigaux F, Delord M, Touzri F, Hernandez L, et al. Clonal selection in xenografted human T cell acute lymphoblastic leukemia recapitulates gain of malignancy at relapse. J Exp Med. 2011;208:653–61. doi: 10.1084/jem.20110105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Piccirillo SG, Combi R, Cajola L, Patrizi A, Redaelli S, Bentivegna A, et al. Distinct pools of cancer stem-like cells coexist within human glioblastomas and display different tumorigenicity and independent genomic evolution. Oncogene. 2009;28:1807–11. doi: 10.1038/onc.2009.27. [DOI] [PubMed] [Google Scholar]
- 61.Kreso A, O’Brien CA, van Galen P, Gan OI, Notta F, Brown AM, et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science. 2013;339:543–8. doi: 10.1126/science.1227670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Spencer SL, Gaudet S, Albeck JG, Burke JM, Sorger PK. Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature. 2009;459:428–32. doi: 10.1038/nature08012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Greaves M. Cancer stem cells: back to Darwin? Semin Cancer Biol. 2010;20:65–70. doi: 10.1016/j.semcancer.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 64.Arias AM, Hayward P. Filtering transcriptional noise during development: concepts and mechanisms. Nat Rev Genet. 2006;7:34–44. doi: 10.1038/nrg1750. [DOI] [PubMed] [Google Scholar]
- 65.Balazsi G, van Oudenaarden A, Collins JJ. Cellular decision making and biological noise: from microbes to mammals. Cell. 2011;144:910–25. doi: 10.1016/j.cell.2011.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat Med. 2009;15:1010–2. doi: 10.1038/nm0909-1010. [DOI] [PubMed] [Google Scholar]
- 67.Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351:657–67. doi: 10.1056/NEJMoa040258. [DOI] [PubMed] [Google Scholar]
- 68.Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138:645–59. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mizuno H, Spike BT, Wahl GM, Levine AJ. Inactivation of p53 in breast cancers correlates with stem cell transcriptional signatures. Proc Natl Acad Sci U S A. 2010;107:22745–50. doi: 10.1073/pnas.1017001108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cicalese A, Bonizzi G, Pasi CE, Faretta M, Ronzoni S, Giulini B, et al. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell. 2009;138:1083–95. doi: 10.1016/j.cell.2009.06.048. [DOI] [PubMed] [Google Scholar]
- 71.Huntly BJ, Shigematsu H, Deguchi K, Lee BH, Mizuno S, Duclos N, et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell. 2004;6:587–96. doi: 10.1016/j.ccr.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 72.Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature. 2008;456:593–8. doi: 10.1038/nature07567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pece S, Tosoni D, Confalonieri S, Mazzarol G, Vecchi M, Ronzoni S, et al. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. 2010;140:62–73. doi: 10.1016/j.cell.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 74.Fielding AK, Richards SM, Chopra R, Lazarus HM, Litzow MR, Buck G, et al. Outcome of 609 adults after relapse of acute lymphoblastic leukemia (ALL); an MRC UKALL12/ECOG 2993 study. Blood. 2007;109:944–50. doi: 10.1182/blood-2006-05-018192. [DOI] [PubMed] [Google Scholar]
- 75.Parsons D, Jones S, Zhang X, Lin J, Leary R, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–12. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schuh A, Becq J, Humphray S, Alexa A, Burns A, Clifford R, et al. Monitoring chronic lymphocytic leukemia progression by whole genome sequencing reveals heterogeneous clonal evolution patterns. Blood. 2012;120:4191–6. doi: 10.1182/blood-2012-05-433540. [DOI] [PubMed] [Google Scholar]
- 77.Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med. 2011;365:2497–506. doi: 10.1056/NEJMoa1109016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306–13. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Maheswaran S, Sequist LV, Nagrath S, Ulkus L, Brannigan B, Collura CV, et al. Detection of mutations in EGFR in circulating lung-cancer cells. N Engl J Med. 2008;359:366–77. doi: 10.1056/NEJMoa0800668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111–5. doi: 10.1038/nature06548. [DOI] [PubMed] [Google Scholar]
- 81.Inukai M, Toyooka S, Ito S, Asano H, Ichihara S, Soh J, et al. Presence of epidermal growth factor receptor gene T790M mutation as a minor clone in non-small cell lung cancer. Cancer Res. 2006;66:7854–8. doi: 10.1158/0008-5472.CAN-06-1951. [DOI] [PubMed] [Google Scholar]
- 82.Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med. 2011;17:313–9. doi: 10.1038/nm.2304. [DOI] [PubMed] [Google Scholar]
- 83.Yip S, Miao J, Cahill DP, Iafrate AJ, Aldape K, Nutt CL, et al. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin Cancer Res. 2009;15:4622–9. doi: 10.1158/1078-0432.CCR-08-3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2:117–25. doi: 10.1016/s1535-6108(02)00096-x. [DOI] [PubMed] [Google Scholar]
- 85.Roche-Lestienne C, Soenen-Cornu V, Grardel-Duflos N, Lai JL, Philippe N, Facon T, et al. Several types of mutations of the Abl gene can be found in chronic myeloid leukemia patients resistant to STI571, and they can pre-exist to the onset of treatment. Blood. 2002;100:1014–8. doi: 10.1182/blood.v100.3.1014. [DOI] [PubMed] [Google Scholar]
- 86.Tzoneva G, Perez-Garcia A, Carpenter Z, Khiabanian H, Tosello V, Allegretta M, et al. Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed ALL. Nat Med. 2013;19:368–71. doi: 10.1038/nm.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wu CJ. CLL clonal heterogeneity: an ecology of competing subpopulations. Blood. 2012;120:4117–8. doi: 10.1182/blood-2012-09-452805. [DOI] [PubMed] [Google Scholar]
- 88.Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer. 2012;12:335–48. doi: 10.1038/nrc3257. [DOI] [PubMed] [Google Scholar]
- 89.Jablonski D. Lessons from the past: evolutionary impacts of mass extinctions. Proc Natl Acad Sci U S A. 2001;98:5393–8. doi: 10.1073/pnas.101092598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nowak MA, Sigmund K. Evolutionary dynamics of biological games. Science. 2004;303:793–9. doi: 10.1126/science.1093411. [DOI] [PubMed] [Google Scholar]
- 91.Vincent TL, Gatenby RA. An evolutionary model for initiation, promotion, and progression in carcinogenesis. Int J Oncol. 2008;32:729–37. [PubMed] [Google Scholar]
- 92.Maley CC, Galipeau PC, Finley JC, Wongsurawat VJ, Li X, Sanchez CA, et al. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat Genet. 2006;38:468–73. doi: 10.1038/ng1768. [DOI] [PubMed] [Google Scholar]
- 93.CLL Trialists Collaborative Group. Chemotherapeutic options in chronic lymphocytic leukemia: a meta-analysis of the randomized trials. J Natl Cancer Inst. 1999;91:861–8. doi: 10.1093/jnci/91.10.861. [DOI] [PubMed] [Google Scholar]
- 94.Burger JA, Ghia P, Rosenwald A, Caligaris-Cappio F. The microenvironment in mature B-cell malignancies: a target for new treatment strategies. Blood. 2009;114:3367–75. doi: 10.1182/blood-2009-06-225326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366:1079–89. doi: 10.1056/NEJMoa1112304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Döhner H, Stilgenbauer S, Benner A, Leupolt E, Kröber A, Bullinger L, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343:1910–6. doi: 10.1056/NEJM200012283432602. [DOI] [PubMed] [Google Scholar]
- 97.Rossi D, Rasi S, Spina V, Bruscaggin A, Monti S, Ciardullo C, et al. Integrated mutational and cytogenetic analysis identifies new prognostic subgroups in chronic lymphocytic leukemia. Blood. 2013;121:1403–12. doi: 10.1182/blood-2012-09-458265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Avet-Loiseau H, Li C, Magrangeas F, Gouraud W, Charbonnel C, Harousseau JL, et al. Prognostic significance of copy-number alterations in multiple myeloma. J Clin Oncol. 2009;27:4585–90. doi: 10.1200/JCO.2008.20.6136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Swanton C. Intratumor heterogeneity: evolution through space and time. Cancer Res. 2012;72:4875–82. doi: 10.1158/0008-5472.CAN-12-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–74. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pardanani A, Tefferi A. Imatinib therapy for hypereosinophilic syndrome and eosinophilia-associated myeloproliferative disorders. Leuk Res. 2004;28 (Suppl 1):S47–52. doi: 10.1016/j.leukres.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 102.Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–34. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 103.Zhu CQ, da Cunha Santos G, Ding K, Sakurada A, Cutz JC, Liu N, et al. Role of KRAS and EGFR as biomarkers of response to erlotinib in National Cancer Institute of Canada Clinical Trials Group Study BR. 21. J Clin Oncol. 2008;26:4268–75. doi: 10.1200/JCO.2007.14.8924. [DOI] [PubMed] [Google Scholar]
- 104.Yang H, Higgins B, Kolinsky K, Packman K, Bradley WD, Lee RJ, et al. Antitumor activity of BRAF inhibitor vemurafenib in preclinical models of BRAF-mutant colorectal cancer. Cancer Res. 2012;72:779–89. doi: 10.1158/0008-5472.CAN-11-2941. [DOI] [PubMed] [Google Scholar]
- 105.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 106.Bissonnette RP, Echeverri F, Mahboubi A, Green DR. Apoptotic cell death induced by c-myc is inhibited by bcl-2. Nature. 1992;359:552–4. doi: 10.1038/359552a0. [DOI] [PubMed] [Google Scholar]
- 107.Inda MM, Bonavia R, Mukasa A, Narita Y, Sah DW, Vandenberg S, et al. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev. 2010;24:1731–45. doi: 10.1101/gad.1890510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gillies RJ, Verduzco D, Gatenby RA. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat Rev Cancer. 2012;12:487–93. doi: 10.1038/nrc3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Merlo LM, Maley CC. The role of genetic diversity in cancer. J Clin Invest. 2010;120:401–3. doi: 10.1172/JCI42088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gatenby RA, Silva AS, Gillies RJ, Frieden BR. Adaptive therapy. Cancer Res. 2009;69:4894–903. doi: 10.1158/0008-5472.CAN-08-3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Waterhouse M, Pfeifer D, Pantic M, Emmerich F, Bertz H, Finke J. Genome-wide profiling in AML patients relapsing after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2011;17:1450–9. e1. doi: 10.1016/j.bbmt.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 112.Stolzel F, Hackmann K, Kuithan F, Mohr B, Fussel M, Oelschlagel U, et al. Clonal evolution including partial loss of human leukocyte antigen genes favoring extramedullary acute myeloid leukemia relapse after matched related allogeneic hematopoietic stem cell transplantation. Transplantation. 2012;93:744–9. doi: 10.1097/TP.0b013e3182481113. [DOI] [PubMed] [Google Scholar]
- 113.Bacher U, Haferlach T, Alpermann T, Zenger M, Kroger N, Beelen DW, et al. Comparison of cytogenetic clonal evolution patterns following allogeneic hematopoietic transplantation versus conventional treatment in patients at relapse of AML. Biol Blood Marrow Transplant. 2010;16:1649–57. doi: 10.1016/j.bbmt.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 114.Puente XS, Lopez-Otin C. The evolutionary biography of chronic lymphocytic leukemia. Nat Genet. 2013;45:229–31. doi: 10.1038/ng.2556. [DOI] [PubMed] [Google Scholar]
- 115.Hou Y, Song L, Zhu P, Zhang B, Tao Y, Xu X, et al. Single-cell exome sequencing and monoclonal evolution of a JAK2-negative myeloproliferative neoplasm. Cell. 2012;148:873–85. doi: 10.1016/j.cell.2012.02.028. [DOI] [PubMed] [Google Scholar]
- 116.Xu X, Hou Y, Yin X, Bao L, Tang A, Song L, et al. Single-cell exome sequencing reveals single-nucleotide mutation characteristics of a kidney tumor. Cell. 2012;148:886–95. doi: 10.1016/j.cell.2012.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shalek AK, Satija R, Adiconis X, Gertner RS, Gaublomme JT, Raychowdhury R, et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature. 2013;498:236–40. doi: 10.1038/nature12172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Weston-Bell N, Gibson J, John M, Ennis S, Pfeifer S, Cezard T, et al. Exome sequencing in tracking clonal evolution in multiple myeloma following therapy. Leukemia. 2013;27:1188–91. doi: 10.1038/leu.2012.287. [DOI] [PubMed] [Google Scholar]
- 119.Green MR, Gentles AJ, Nair RV, Irish JM, Kihira S, Liu CL, et al. Hierarchy in somatic mutations arising during genomic evolution and progression of follicular lymphoma. Blood. 2013;121:1604–11. doi: 10.1182/blood-2012-09-457283. [DOI] [PMC free article] [PubMed] [Google Scholar]