

Summary
In vitro, high-throughput screening (HTS) assays are seeing increasing use in toxicity testing. HTS assays can simultaneously test many chemicals, but have seen limited use in the regulatory arena, in part because of the need to undergo rigorous, time-consuming formal validation. Here we discuss streamlining the validation process, specifically for prioritization applications in which HTS assays are used to identify a high-concern subset of a collection of chemicals. The high-concern chemicals could then be tested sooner rather than later in standard guideline bioassays. The streamlined validation process would continue to ensure the reliability and relevance of assays for this application. We discuss the following practical guidelines: (1) follow current validation practice to the extent possible and practical; (2) make increased use of reference compounds to better demonstrate assay reliability and relevance; (3) deemphasize the need for cross-laboratory testing, and; (4) implement a web-based, transparent and expedited peer review process.
Keywords: Validation, in vitro, high-throughput screening
1 Introduction
Toxicity testing for human health effects is undergoing a paradigm shift from classical laboratory animal studies to in vitro assays that primarily use human cells and focus on assessing perturbations to key biological pathways (Ankley et al., 2010; Berg et al., 2010; Gohlke et al., 2009; Hamadeh et al., 2002; Hartung, 2009a; Takeuchi et al., 2006; Zhou et al., 2009; Ballatori et al., 2003; Nuwaysir et al., 1999; Reynolds, 2005; Dix et al., 2007; Judson et al., 2010; NRC, 2007; Collins et al., 2008; Doull et al., 2007; Singh et al., 2010; Stokes and Wind, 2010b; Stokes and Wind, 2010a; Bradbury et al., 2004). This shift is due to two major factors: 1) the recognition that current testing methods, which are costly, time consuming, and often use large numbers of animals without always providing correspondingly large benefits, are not adequate to manage the increasing backlog of largely untested chemicals; 2) the frequent inability of current in vivo tests to provide clear mechanistic insight into toxicity pathways, an advantage offered by the new types of in vitro assays that are able to directly probe human genes, cells, and tissues (NRC, 2007; Kavlock et al., 2009).
Currently, there are hundreds of in vitro high throughput screening (HTS) assays, many of which use human proteins or cells (primary cells or cell lines) and which are increasingly used in the toxicity testing of environmental chemicals and candidate pharmaceuticals. Before these HTS assays can be used for making regulatory decisions, however, there needs to be a formal process to appropriately evaluate their reliability, relevance and fitness for purpose. This is the rationale for test method validation, which is currently required by most regulatory bodies for assays used in making regulatory decisions on the safety of chemicals (ICCVAM, 2000, 1997, 2003; Birnbaum and Stokes, 2010; OECD, 2005). However, the current paradigm for validating new or revised tests for potential acceptance by regulatory agencies, while of high quality and ensuring that the use of the such tests would provide equivalent or better protection than current procedures, is time consuming, low throughput, and expensive. Thus, current processes used for test methods proposed for regulatory testing guidelines have not shown themselves to be capable of validating in a timely manner (less than one year) the many new HTS assays already in use in the research setting. Note however, that new validation approaches using the concept of performance standards have been proposed and used to more efficiently validate new innovative assays (Wind and Stokes, 2010). Hartung has discussed some of the rationale for and issues underlying current practice, especially in the context of the validation of alternative methods (Hartung, 2007). Leist et al. have further considered several important issues that specifically pertain to validation of in vitro assays for use in toxicity testing, which are particularly relevant to the current paper (Leist et al., 2010).
In general, HTS assays are relatively simple technologically. They can probe many specific key events (KE’s), such as a molecular initiating event (MIE), or an intermediate step associated with a pathway that can potentially lead to adverse health outcomes. KE’s (including MIE’s) are respectively defined in the context of toxicity pathways (NRC, 2007), modes of action (MOA) (Boobis et al., 2008; Meek et al., 2003; Seed et al., 2005; Sonich-Mullin et al., 2001) and adverse outcome pathways (AOP’s) (Ankley et al., 2010). The assays typically are focused on a particular target interaction or read-out, and measure endpoints such as the expression level or reporter signal of one or more genes, inhibition of enzymatic activity, or the binding of a chemical to a single receptor, as well as cellular phenotypes (e.g. changes in cell shape and size, cytotoxicity). Elucidating the toxicity MOA of chemicals by identifying and documenting the linkage from assay to KE to potential for adversity is a main objective in use of these assays. As a consequence, it is also key to evaluating the ultimate relevance of an HTS assay with respect to the information it provides.
While there is not a single accepted definition of HTS, for our purposes a working definition could be assays that are run in 96-well plates or higher; assays that are run in concentration-response format and yield a quantitative read-out at each concentration; and assays that (when run using cells) have simultaneous cytotoxicity measures.
Other significant advantages of HTS assays include the following. They scale to testing hundreds or thousands of chemicals at a time. The output of an assay is readily quantified, typically as a single response value for each concentration tested in each chemical replicate. One can repeatedly test in blinded fashion both reference and test chemicals, providing quantitative measures of reproducibility.
In this paper, we consider the use of HTS assays as tools for chemical prioritization as opposed to being replacements for regulatory guideline animal-based tests. Under the assumption that only a minority of chemicals will cause any specific adverse effect, it will be more health-protective and resource-efficient to use HTS assays to identify the chemicals most likely to cause particular adverse effects (and therefore to be positive in more expensive, low-throughput animal-based guideline bioassays) and to run these chemicals first in guideline bioassays that measure the effect identified as a potential concern. This entire process of identifying these first-in-line chemicals using HTS assays is what we mean by “prioritization”, and will be the focus of much of our discussion in this paper. (An important note is that a chemical that is “negative” in a prioritization assay will not necessarily be negative in the follow-on guideline test.) The ability of one or a collection of HTS assays to have reasonable sensitivity and specificity for identifying toxic chemicals is the basis for deciding the assays’ fitness for purpose, where in this paper the purpose is prioritization, rather than as a regulatory guideline test to generate data for definitive safety or hazard decisions.
A final implication of the comparative simplicity of the HTS assays is that it is relatively easy to implement new technologies and to develop new assays (e.g., new target; new readout for an old target; new, higher-throughput version of an existing assay). If newly introduced assays provide new or enhanced capabilities for mechanistic clarity in screening for potential toxicity, it is in the interest of public health to have them used as soon as possible in testing of potentially harmful chemicals.
The remainder of this paper will elaborate on these main points:
HTS assays provide a new capability for simultaneously testing the ability of thousands of chemicals to trigger intermediate biological or biochemical KE’s (as opposed to observable or apical endpoints) associated with toxicity pathways that can lead to adverse health outcomes.
The data from these assays can be used to prioritize which chemicals out of large sets of previously untested ones should be subject to further study sooner rather than later.
Before using these assays, even for prioritization, their relevance, reliability and fitness for purpose should be established and documented. In the present context, relevance is related to the ability to detect KE’s with documented links to adverse outcomes, and the ability to reproduce data and to respond appropriately to carefully selected reference compounds, either in a qualitative (e.g. positive/negative for effect) or quantitative (e.g. relative potency) manner. Fitness for purpose is more subjective since it is use-case dependent, but is typically established by characterizing the ability of an HTS assay to predict the outcome of guideline tests for which prioritization scores are being generated.
It should be possible to develop a streamlined validation process to evaluate the relevance, reliability and fitness for purpose for HTS assays. This is largely because the data from the HTS assays generally provide quantitative, reproducible read-outs with a focused and mechanistically simple interpretation. These attributes should make evaluation of the performance of the HTS assays, and hence peer review and decisions on acceptance for use by regulatory bodies based on the scientific evidence, relatively straightforward.
It is unlikely that any single in vitro assay will ever yield the “perfect” result. Even mechanistically similar assays are expected to yield some degree of discordance due to the complexities of biological processes and assay-specific interference by some test chemicals. Hence multiple assays for critical targets and a weight of evidence approach is likely to be needed. Additionally, many environmental chemicals are likely to be of low potency, and hence subject to variation in hit calling from assay to assay.
Each of the above statements is consistent with current thinking about validation of tests for chemical toxicity. However, here we will propose modifications to current test method validation practice that are appropriate to, and can facilitate the use of HTS assays for prioritization. The two modifications that could have the largest impact on time and cost of validation pertain to cross-laboratory testing (or transferability requirements) and the peer review process. We will make a case for largely eliminating the requirement for cross-laboratory testing as part of the validation process for HTS assays for prioritization. In addition, because the output of HTS assays are for the most part easily interpreted, quantitative values, we will argue that the standard for regulatory acceptance should be commensurate with the focused biological interpretation of the assay and, therefore, be no more onerous than typical peer review of a scientific manuscript. Both of these propositions are perhaps controversial, so we discuss pros and cons of each.
Given the high burden of proof generally required of regulatory review and decisions applied to protecting public health, there is some reluctance in the regulatory community to even discuss alternative, more flexible validation approaches (Inside EPA, 2010). This is driven partially by the view that anything short of full, lengthy (multi-year), high-cost validation is an unacceptable compromise on quality. However, adhering to this strict standard effectively excludes the use of a large number of currently available HTS assays that provide the only practical approach to test thousands of previously untested chemicals. One option is to develop a new process that has fewer components than the full regulatory guideline study validation standards, and to call it something other than validation. We believe that this position has two problems. The first is that many statutes governing regulatory testing specifically stipulate that the assays used must be “validated” (see discussion below). The other problem is that users need to trust that the data yielded by these assays are reliable, relevant and fit for purpose, which is the very definition and goal of validation. This paper is not intended to be a definitive description of a new validation approach, nor is it a consensus statement that is endorsed by the authors or their institutions. Instead, it is meant to stimulate discussion and to propose a way forward towards developing a more streamlined validation process to accommodate and thereby facilitate the use of HTS assays in addressing some of the major shortfalls of existing testing approaches.
2 Use case: prioritization based on data from HTS assays
The focus of subsequent discussion will be on the use of HTS assays for prioritization rather than as replacements for regulatory test guidelines, so we begin by considering some issues relevant to this use case. One point sometimes made is that “prioritization” is not part of “regulation”, so that the tools used for prioritization do not need to be validated in the same way as those used for regulation. Regardless of whether this is true in the legal sense (see below), decisions are made in the prioritization process that ultimately can impact public and environmental health and affect regulatory decisions. Whether or not validation is required for prioritization, it is important to have confidence in the reliability, relevance and fitness for purpose of the tools being used for any purpose, including prioritization. Regulatory screening tests are in fact often used for decisions on whether further testing will or should be conducted, or if specific safety or hazard conclusions can be made without further testing (Stokes and Wind, 2010b; Stokes and Wind, 2010c, a). Data from assays that are validated are stronger than information from those that are not validated, and decisions are more defensible if informed by results from assays subject to some appropriate validation process. It is also possible that as validation data accrue, prioritization tools may be demonstrated to be sufficiently predictive so as to be used for definitive regulatory testing decisions.
Screening and prioritization (which are not always distinguished) are explicit components of the regulatory process within the United States. For example, the Endocrine Disruptor Screening Program (EDSP) of the U.S. Environmental Protection Agency (EPA) (U.S. EPA, 2007) uses a tiered testing approach in which less complex/expensive, but more sensitive and often less specific, tests form the first tier, and more complex/expensive and more definitive tests (definitive in terms of characterizing whether an adverse outcome was induced) form the second tier. Currently, chemicals are prioritized for inclusion in the EDSP Tier 1 battery (T1S) based on production volume, exposure potential or regulatory review schedule (i.e. for scheduled re-registration reviews for food-use pesticide active ingredients), but the EPA is moving towards the use of pathway-based in vitro assays for setting priorities of chemicals to be tested in T1S (U.S. EPA, 2011a). Compounds will be prioritized or selected for running in the T1S battery based on the results of HTS assays and in silico models.
The Toxic Substance Control Act (TSCA) of 1976 (TSCA, 1976) explicitly mentions screening: “The administrator shall coordinate … research … directed towards the development of rapid, reliable, and economical screening techniques for [toxic] effects of chemical substances…” [15 USC §2610 TSCA §10 (c)] (emphasis added). Under the Safe Drinking Water Act (U.S. EPA, 1996), the EPA is required to “identify and list unregulated contaminants which may require a national drinking water regulation in the future. … The EPA uses this list … to prioritize research and data collection efforts” (U.S. EPA, 2008) (emphasis added). These chemicals are entered into the Candidate Contaminant Lists (CCL) developed by the EPA Office of Water. Each of these laws requires the use of valid and scientifically supportable data in making regulatory decisions. In the European Union, although screening is not a specific requirement in chemicals legislation, REACH (Registration, Evaluation, Authorisation and Restriction of Chemical substances) (REACH, 2006) for example does make provision for identifying and managing chemicals of (very) high concern, while the Community Strategy for Endocrine Disrupters (COM, 1999) outlines actions to target chemicals that may have endocrine disrupting properties.
Whereas “screening” generally applies to all compounds of potential concern employing a variety of increasingly complex test methods, prioritization is critical because of the large size of the chemical landscape covered under these and other regulations – in the order of 100,000 unique substances (Judson et al., 2009). However, from an HTS perspective, this does not pose an insurmountable hurdle. Pharmaceutical companies routinely test libraries containing millions of compounds. Using this approach, it is possible to develop compound libraries consisting of thousands of chemicals of potential concern that could be tested repeatedly in any new assays that might be developed as a basis to evaluate new test performance. This process of repeated testing of a fixed library is illustrated in Figure 1 and discussed further in the Conclusions section. In subsequent discussion, we assume that such libraries are currently in development or will be developed. One validation-related requirement, which will not be discussed further here, is that these compound libraries undergo quality control procedures to assure that the chemicals being tested are what they purport to be, and are stable and sufficiently pure and soluble under the assay test conditions used.
Fig. 1.
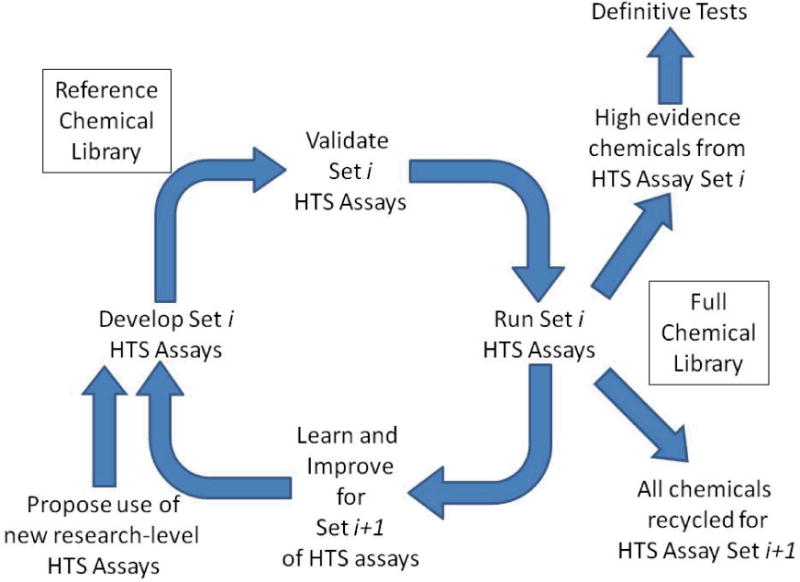
Conceptual model of a continuously improving battery of HTS assays to be used for prioritization. A library of chemicals of interest is identified. For each MOA of concern, in vitro assays that have been identified in a research setting are moved into an HTS platform suitable for screening large chemical libraries. These assays are then validated based on screening a set of reference chemicals. Validation depends on showing that the new assays are reliable (reproducible, giving good signal to background, and low well-to-well variation, etc.; and relevant, i.e. the results on the reference chemicals are in accord with what is known about their activity in the molecular pathway being probed with the assay, and with results in the in vivo definitive test). After validation, the full chemical library is tested. Based on what is learned from testing the library, and from related scientific studies, additional HTS assays for the MOA, or improved versions of the current assays will be introduced and those new assays, in turn, will be validated using the reference chemicals. Subsequently, the full chemical library will be rescreened. “High Evidence” chemicals, i.e. those whose activity in the KE of the MOA being probed is strongly supported by the assay data, will be recommended to be run in more definitive tests. An approach similar to this has been proposed for the U.S. EPA’s EDSP21 approach (U.S. EPA 2011a).
The scientific rationale for using in vitro HTS assays for prioritization is based on the idea that these assays probe key biological events in pathways that have been linked to or could lead to toxicity. This idea is well understood in the context of toxicity pathways (NRC, 2007), MOA (Meek et al., 2003; Seed et al., 2005; Sonich-Mullin et al., 2001) or AOP analysis (Ankley et al., 2010). Each of these paradigms includes the idea of a MIE, in which a chemical interacts directly with a target biomolecule(s). Whereas in vitro HTS assays do not, in general, allow one to follow all of the subsequent downstream processes described as part of the MOA, they can detect the necessary (initial) step(s). Each KE/MIE triggered by a chemical raises the likelihood that a chemical could produce an adverse outcome through the relevant pathway, factoring in issues such as ADME, local dosimetry, critical windows of sensitivity, genetic susceptibility, and confounding stressors.
3 Validation principles
The purpose of a validation process is to evaluate the reliability, relevance and fitness for purpose of an assay. The EPA, Food and Drug Administration (FDA), National Institute of Environmental Health Sciences (NIEHS), and other U.S. Federal agencies have developed definitions and principles for validation and regulatory acceptance of new, revised, and alternative methods (ICCVAM, 1997, 2003). To frame the discussion, we expand the definition of each of these concepts.
3.1 Reliability
To be reliable, an assay must be reproducible, e.g. it must produce qualitatively and quantitatively similar results over time, across lots and batches of reagents, and between different operators in the same laboratory. In the case where the assay is expected to be widely used, a demonstration of reproducibility across labs may also be desirable. ICCVAM defines reliability as the extent that a test or assay can be performed reproducibly within and among laboratories over time (ICCVAM, 1997). Reliability can also depend on the potency and efficacy of the compounds. If a compound has low potency or low efficacy, it may generate more variable results. An important point for toxicity testing is the goal of minimizing the false negative rate in order to ensure health protective testing. Note that one could argue for including the requirement that reference chemicals show the expected behavior in the assay under either “reliability” or “relevance”.
3.2 Relevance
Relevance describes the relationship of a test to the effect of interest and whether a test is meaningful and useful for a particular purpose (ICCVAM, 1997). To be relevant, an assay must probe some aspect of biology that will help assess the safety or hazard of a chemical, for instance by determining the ability of a chemical to trigger a KE in a toxicity pathway. Furthermore, a positive result in the assay should be indicative of perturbations to, or interactions with the target or pathway that the assay is designed to probe. Data on reference compounds with known activity in relation to a given target or pathway can be used to help assess the relevance of the assay. Relevance addresses the scientific basis of the test (does the assay measure interaction with a target that is linked to adverse outcomes through a pathway?) and the predictive capacity of the test (how well does a positive result in the prioritization assay predict a positive result in the more complex test whose outcome is the object of the prioritization?) (Hartung, 2007). See the fit for purpose discussion below. A relevant assay will have an acceptably low rate of false negative or false positive results i.e. chemicals that should interact with the target but give negative results in the assay, or chemicals that should not interact with the target but give positive results in the assay. In practice, relevant can be evaluated by repeated testing of positive and negative reference compounds, and by comparing the results against the expected behavior of these compounds.
3.3 Fitness for purpose
For a prioritization application, a positive or negative result in a single HTS assay does not have to directly predict a corresponding positive or negative result in the regulatory guideline bioassay for the corresponding apical endpoint. However, there should be sufficient positive and negative predictive power so that the prioritized chemicals are significantly enriched in positives when run in the guideline test, in comparison to the prevalence of positives in original population of chemicals. Additionally, it may be necessary to employ multiple assays (against the same or different targets) with orthogonal readouts to gain sufficient sensitivity and specificity for prioritization. As long as each individual assay is sufficiently reliable and relevant that it adds to the predictive power of the battery of assays, it can be said to be fit for purpose. We will focus our discussion on validating a single HTS assay at a time, but one should keep in mind that some HTS assays may be more useful when aggregated with related, complementary assays within a battery where deficiencies in one assay can be overcome by strengths of another.
Current validation principles and practice were developed to insure the quality of guideline tests and to provide confidence to all stakeholders of the reliability and relevance of the resulting data (ICCVAM, 1997; OECD, 2005). Therefore, to the extent possible, it is important to adhere to these well-accepted practices in any alternative, streamlined validation framework, and only deviate where there is a clear net benefit. Accordingly, we will discuss in some detail current practices and our proposed variants. We base our discussion on guidance for the validation process developed by the Organization for Economic Co-operation and Development (OECD), which was developed using guidance and principles developed by the Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM), the European Centre for the Validation of Alternative Methods (ECVAM), and other input from OECD member countries (OECD, 2005). The OECD states that the purpose of validation is “… to determine the performance characteristics, usefulness, and limitations of a test method that is under consideration for use in a regulatory context, and to determine the extent that the results from the test can be used for hazard identification, and to support risk assessments or other health and safety decisions” (OECD, 2005).
The ICCVAM regulatory acceptance criteria (ICCVAM, 1997, 2003) and OECD guidance provide acceptance criteria that should be met for regulatory adoption of a new test (OECD, 2005). Some of the OECD criteria include:
The test has been sufficiently validated
The test provides at least as much scientifically credible information as an existing test while using fewer animals …
The test improves the safety assessments for man and the environment
We will spend most of the subsequent discussion on point #1, but it is worth considering the other two points, albeit briefly. In vitro, target or pathway-based HTS assays provide information not readily available from existing animal-based tests, and do not make use of whole animals, thus addressing point #2. Further, because these assays are high-throughput and low cost, we can examine large numbers of chemicals simultaneously and in multiple assays. For practical reasons, many of these chemicals will likely never be evaluated in standard animal-based assays, but by generating new data on existing chemicals for which little to no data may exist, these assays directly address points #2 and #3. Assuming the data are of high quality, generating new, biologically relevant information always has the potential to improve safety assessments relative to those informed by little or no data.
The OECD validation guidance document (OECD, 2005) endeavors to provide guidance for developing data and information to address the validation criteria developed by ICCVAM (ICCVAM, 1997, 2003; Stokes, 2007) and the modules in the framework developed by ECVAM (Hartung et al., 2004). The overall validation process traditionally proceeds through 5 stages described in the ICCVAM, OECD, and ECVAM documents:
Development
Pre-validation
Validation
Peer Review
Regulatory Acceptance
During development and pre-validation, the assay is characterized and initial optimization is carried out, typically using a set of known reference compounds specific to the assay and its ostensible target, and in HTS mode, perhaps with respect to a larger test library. Precisely because of the ability to simultaneously test many chemicals in HTS mode, there will likely not be a strong demarcation between development and pre-validation steps. For HTS assays, guidelines similar to those available at the National Institutes of Health (NIH) Chemical Genomics Center (NCGC)website2 can be used for the development and pre-validation steps. These guidelines are intended to ensure robust statistical performance and include considerations such as evaluating the assay signal window, well-to-well variation in the plates, ideal assay operational conditions (such as compound treatment time and cell density in the plate), day-to-day reproducibility (such as assessed by AC50-correlation of the positive controls across the plates, and consistent signal to background window. AC50 is the concentration at which assay activity is at 50% of maximum.). For in vitro assays, one could define a list of variables that must be analyzed and documented as part of the assay characterization and validation process. Current validation guidelines requirethat tests be conducted under GLP (Good Laboratory Practice) guidelines. Among other things, these stipulate that a testing laboratory demonstrate the purity and stability of the chemical to be tested (Cooper-Hannan et al., 1999).
The validation process described by ICCVAM, OECD, and ECVAM consists of well-defined stages that typically generate the information needed to evaluate the validity of a test method (ICCVAM, 1997, 2003; Stokes, 2007; OECD, 2005). The validation step (#3) itself is modular, as outlined in Table 1, below (Hartung et al., 2004). The points raised in the rest of this paper will generally place the validation requirements for our use-case into the context of these modules.
Table 1.
The validation modules (Hartung et al. 2004)
|
3.4 Test definition
As can be seen from the outline above, this validation module deals mostly with description of the test itself, including what the test is designed to measure (addressing issues of relevance) as well as test protocols. For HTS assays, these principles can be followed very closely. Test protocols should be carefully documented in Standard Operating Procedures (SOPs). The endpoint (e.g., KE) being tested and its mechanistic basis should be clearly stated (e.g., binding to a target protein is aKEin a documented toxicity pathway). When HTS assays are being validated individually, there is no “model;” instead, the HTS assay readout is a simple quantitative value such as percent inhibition/activation or fold-change in expression, relative to the negative control. The testing is done in concentration-response mode and a potency value such as the AC50 calculated. It is important, however, to document the statistical analysis procedures for data processing steps such as background subtraction, normalization, curve fitting, and hit calling. It is also important to document known limitations of the assay; these are often well understood based on the particular target or assay class. For instance, assays using fluorescent readouts can give unreliable results for compounds that are themselves fluorescent (e.g., azo dyes). With cell-based assays, simultaneous cytotoxicity measurements are usually needed because cytotoxicity can confound the target-specific readout. Defining the media used is also critical, for instance because the available free concentration of the test compound will be a function of serum protein and lipid composition of the media. In addition, many assays are run in cell-free conditions or in cells that do not have metabolic capacity, so in these cases, only effects of the parent compound will be measured. Leist et al. further discuss issues related to the appropriate level of description required in a validation package for an in vitro assay (Leist et al., 2010).
The ICCVAM and OECD guidance makes several recommendations regarding reference chemicals, including that they be representative of the range of responses and effects that the test is capable of measuring (ICCVAM, 2003). In addition, they should:
Have produced consistent results and potency ranking order in relevant reference tests
Reflect the accuracy of the reference test
Have well defined chemical structure and purity
Are readily available
Are not excessively hazardous
An important consequence of the high-throughput nature of the assays is that during validation and testing, large numbers of reference chemicals that span a diverse range of features and properties can be used. If available, one can use multiple strong, moderate, weak, and negative reference compounds for the target, as well as compounds that are known to interfere with assays in a variety of ways that could lead to false positive or false negative results. Furthermore, because many compounds are run simultaneously during actual testing, it is usually possible to run some or all of the reference chemicals concurrently with the test compounds to enable real-time quality control in a way that is not possible with standard one-chemical-at-a-time tests. This provides the ability to better judge assay performance and applicability domain than is the case for low-throughput assays. The issue of applicability domain is considered in more detail below.
One confounding issue with selecting the reference compounds and defining the expected behavior in a new assay is occasional disagreement within the literature as to whether a specific chemical is truly active against a given target. This discordance may be due to use of different species, cell types, or in vitro vs. in vivo conditions. Particularly for less potent chemicals, reports of activity are often discordant. This can be an issue for chemicals that act as partial agonists or antagonists or exhibit different pharmacological behaviors in different tissues (e.g. chemicals interacting with alpha and beta estrogen receptors). Including such chemicals can still be useful, but caution must be exercised in interpretation of the results. This issue of chemicals that give ambiguous or variant results in different versions of tests that ostensibly measure interactions with the same target is not unique to HTS and can be of use in evaluating the assay.
3.5 Within-laboratory variability
There are many known sources of variability within HTS in vitro assays. Some important ones are lot-to-lot reagent variation, stability across batches of cells (especially when primary cells are used), multiple tip variation within the instrument, and tip carry-over in the compound-transferring step. However, none of these are unique to the high-throughput assays described here, so variability characterization should be handled as with any other in vitro assay used for regulatory purposes, for instance those used in genotoxicity testing or in the EDSP.
The U.S. cross-agency Tox21 project provides an extreme example of testing within-lab variability for HTS assays [Tice, et al. in preparation]. For this project, the NCGC is using their ultra high-throughput robotics system to test a large library of environmental and consumer-use chemicals and drugs in 1536-well format, using a battery of toxicologically-relevant HTS assays. A library of approximately 10,000 test samples is being screened, of which more than 1,000 are separately sourced (same chemical purchased independently from different sources or different lot/batches). In addition, each 1536-well plate being assayed includes the same duplicate set of 88 chemicals, derived from a single stock solution for each chemical. Finally, all plates will be run in concentration-response format in triplicate in each of the assays. The library also contains multiple reference chemicals selected for a variety of targets being tested. All of these data will provide ample statistics for assessing chemical lot-to-lot variability, plate positional effects, and assay reproducibility within and across plates, across runs, and across time. This illustrates the unique ability to have robust measurements of assay reliability for HTS assays during both validation and production testing.
3.6 Transferability and between-laboratory variability
It is for these validation steps that we consider the potential for significant changes to current practice. Running tests during validation in multiple laboratories serves two purposes. Firstly, it is often the case that no single laboratory has the capacity to handle all of the world’s testing needs, or there are other commercial or practical reasons for routing testing orders to multiple laboratories. Hence, it is important to know that the results of a test will be consistent across independent laboratories (i.e., that the assay can be transferred successfully to multiple testing facilities). Secondly, by demonstrating that a test can be run in one or more independent laboratories and give the same result (within tolerances), one verifies that the protocols are adequately described and that there are no subtle (and perhaps unknown) features of the assay that have not been considered and documented. Much of the focus in establishing transferability and reproducibility of in vitro assays is related to the particulars of the cell model since differences between laboratories often indicate weaknesses in cell culturing protocols (e.g. documentation or practice). Clearly, if one is to move away from required cross-lab testing, this issue must be dealt with in a satisfactory way.
The case for not requiring cross-laboratory testing as part of the validation process for HTS assays used in prioritization for our proposed use case(s) can be stated briefly as follows:
Most of the assays to be used in our envisioned prioritization applications can be run for all chemicals of interest in a single laboratory, meaning that, from a purely practical standpoint, there is no need to have multiple laboratories demonstrate competency in running the assay.
An extensive number of reference chemicals (blinded to laboratory personnel for most assays) will be used both during the validation process and concurrently during testing. All the test compounds in the wells will also be blinded inall the assays during screening using a robotic system. This provides significantly more quality assurance and control over the process than is the case in most guideline tests. (How large this reference chemical set needs to be is an issue that will require significant discussion.)
Some laboratories (e.g., NCGC) use very expensive, customized robotics equipment, such that no other laboratory is available that could readily duplicate their exact protocol.
Due to the rapid pace of technological development of HTS assays in the commercial realm, some of the tests we envision using are proprietary, and so for legal and business reasons, replication in other laboratories is unlikely to occur.
Items #1 and #2 are practical reasons why one might not need to do cross-laboratory testing, whereas items #3 and #4 are practical reasons why one might not be able to do cross-laboratory testing.
Addressing point #1, an important aspect of the prioritization approach is that the assays are all run in HTS mode. Although there is no formal definition, an assay is considered high-throughput if hundreds of chemicals can be run in a minimum of 96 or 384 well format, and up to 1536 well format, within a limited period of time, usually days to weeks. Therefore, a single laboratory can test hundreds to thousands of chemicals in a few months. At the higher end, the NCGC is able to simultaneously test a library of 10,000 chemicals in triplicate at 15 concentrations in a single week, using their quantitative HTS (qHTS) platform (Inglese et al., 2006; Shukla et al., 2010). This high-throughput capability requires the use of a customized and expensive robotic infrastructure that is not readily replicated in other laboratories (see discussion of point #3 below).
Point #2 is supported by the fact that, for at least the first set of assays being considered for prioritization applications at the EPA, there is an extensive literature on both reference chemicals and other assays against the same targets, such as the estrogen receptor (ER). As an extreme example, we have compiled literature on in vitro ER assays (including from the FDA Endocrine Disruptor Knowledgebase (Ding et al., 2010)) and have found ~100 publications detailing results for ~800 chemicals. This literature was also surveyed by the National Toxicology Program (NTP) Interagency Center for the Evaluation of Alternative Toxicological Methods (NICEATM) while developing a reference chemical set for validation of ER assays; they identified 78 possible ER reference compounds (eventually reduced to a definitive set of 35) with associated indications of relative strength in transactivation assays (ICCVAM, 2011). For validation of low-throughput assays, it has been infeasible or impractical to run such a large set of chemicals multiple times. However, running a large set of reference chemicals, such as this, during HTS assay development and validation would pose no particular challenges. Hence, a protocol could be developed whereby a large number of strong, moderate and weak positive, along with true negative chemicals (based on clear and consistent data from reports in the literature), are run in a new HTS assay and the results are compared to reports in the literature, not only for similar assays, but also for assays testing other modes of activity (binding, transactivation, proliferation, co-factor recruitment).
This evaluation process would provide much more information on the behavior of the new HTS assay than is available for any of the current low-throughput assays, and would concurrently improve our knowledge of reliability, relevance, and domain of applicability. This information would be sufficiently robust to obviate the need for direct cross-laboratory testing for the new protocol while at the same time providing a sound basis for conducting a cross-laboratory study of a manual version of the assay if there was a desire to make it widely available. In addition to running the reference chemicals during validation, they could also be run concurrently with the test chemicals during production testing. This would allow for a level of ongoing quality control that is not possible with any low-throughput assay. In summary, we argue for a compromise in which no cross-laboratory testing is required during validation, but in- and post-validation testing of many reference chemicals is required. An argument can be made that this strategy is superior in some respects to the current situation for validation of low throughput assays in which only a few reference chemicals are evaluated during validation, albeit in multiple laboratories, whereas few or none are evaluated during production testing. This particular line of reasoning, of course, fails for assays that test targets or pathways for which there is no extensive literature background on chemicals and assays, and no well characterized set of reference compounds.
We next address issue #3, having to do with the one-of-a-kind nature of some candidate testing laboratories, and use the NCGC as an example. They have implemented a complex and expensive robotic system capable of processing up to 300,000 chemicals at a time in concentration-response mode in 1536-well plates. Typically, they start with a published, precursor test that has been run in small format plates (often 24 or 96-well) and then optimize the assay to run in their qHTS format. The optimization process frequently involves changing parameters, such as cell number, reagent volumes, incubation times and number of handling steps. Typically, the readout is the same type as was used in the precursor assay. The goal of the optimization process is to achieve the same or better assay performance (in terms of signal-to-noise, variability, etc.) as the precursor low-throughput assay. In some cases, the precursor assay has itself been subject to validation, including cross-lab testing in the lower-throughput format. Hence, the issue is whether the 1536-well modified protocol assay can be considered the same as the precursor assay for validation purposes. If this proposition were accepted, then the case could be made that cross-laboratory testing had already been completed (low-throughput to high-throughput). If the proposition is rejected, then there are two possible recourses. In the first case, the assay validation package could be accepted as is, based on extensive use of reference chemicals and comparison to published assays against the same target, as just described. A careful review of the completeness of the SOP would also be required, as a matter of course. A second approach would be to take the high-throughput protocol exactly as specified, and to run it in low-throughput mode with a limited number of chemicals using as close to the same protocol as possible, including plate format, cell number per well, media concentrations, etc. This is analogous to the requirement by most journals that microarray data be replicated by an independent technique.
Elaborating on point #4, the EPA ToxCast program (Dix et al., 2007; Judson et al., 2010) is making extensive use of unique, proprietary assays developed by companies supporting the pharmaceutical industry. Intellectual property considerations restrict the commercial use of these assays to those who have licensed them, or to those who pay for testing services from the assay owner or licensee. As a policy, OECD will not develop guidelines for patented assays or for assays that have proprietary components, to avoid a monopoly situation, except in cases where (i)the value of the information derived from the assay is perceived as high, (ii) there is no equivalent assay in the public domain, and (iii) the preceding validation study has established performance standards that can be used by others to develop a similar method. Whatever the origin of the assay however, our modified HTS validation approach is applicable for both “me too” assays and those that are first-in-class, that explore some new mechanism or readout.
ICCVAM developed guidelines for performance standards that could be used to document the basis for the acceptance of test methods with proprietary components, so that such methods could be adopted by EPA and other regulatory authorities (ICCVAM, 2003; Stokes et al., 2006; Wind and Stokes, 2010; Stokes, 2007). OECD test guidelines have now been adopted that are based on a proprietary method and that incorporate performance standards (Wind and Stokes, 2010). Linge and Hartung have also discussed some issues surrounding the validation of proprietary tests in the context of OECD and ECVAM guidelines (Linge and Hartung, 2007). Firstly, the European Commission supports the development and commercialization of proprietary methods for obvious economic reasons. Several proprietary tests have been submitted as alternative tests in the area of eye irritation and skin corrosivity. Interestingly, these are “black box” assays for which detailed protocols were not public, whereas for the assays used in ToxCast, most details of the protocols have been published. One concern about proprietary tests is that if one of those assays constituted a sole test for some purpose, and the company went out of business, that the corresponding testing program would come to a halt. Secondly, again if there were a single commercial test for some application, the owner of that test would have a monopoly, with the corresponding limits and threats that implies. In our proposed prioritization application, tests would often be used in a battery, so the disappearance of a single test would not precipitate a crisis, nor would the owner of a single test have any particular power to disrupt the overall testing program. Even if a proprietary test was considered to provide some unique capability, presumably the performance standard would be sufficiently described so as to be replicated in some fashion. An interesting point made by Linge and Hartung is that the life of a patent is “only” 20 years, so that once the lengthy development and validation process is completed, there may not be many years of monopoly control remaining. In contrast, one of our goals is to provide a quicker route to validation, so tests could have a longer period of patent protection while being used for commercial testing. Ultimately however, the shift towards the design of validation studies that deliver generic performance standards for classes of assays, rather than validating single methods, will both mitigate the risk of a unique assay becoming unavailable and will help facilitate the efficient and cost effective development of similar assays that deliver equivalently reliable and relevant information but which exploit a variety of techniques and technologies.
Recently ECVAM has demonstrated the first practical steps in how HTS approaches combined with performance standards can actually be used to support the validation of in vitro assays that lend themselves to either manual or automated implementation. The motivation is to use HTS upstream of validation to identify promising assays and, where possible, to use HTS within a validation study to generate data on large sets (10s to 100s) of reference chemicals to explore the predictive capacity and applicability domain of an assay. An initial case study (Bouhifd et al., 2012) centered on a well known cytotoxicity assay (uptake of neutral red dye by mouse fibroblasts cells after 48h exposure to a test chemical), which is the basis of a recently adopted OECD guidance on how to estimate starting doses for acute oral systemic toxicity testing in rodents (OECD, 2010). It was demonstrated how the performance standards developed during the original validation study (manual protocol) could be used to implement an automated version of the assay that delivered data of an equivalent or higher quality but with higher throughput. A subsequent study has dealt with the automation of another important class of assay, namely a transcriptional reporter-gene assay, using a protocol based on BG1Luc4E2 cells (Rogers and Denison, 2000)that is the subject of a draft OECD test guideline to identify ER agonists and antagonists in vitro. The comprehensive performance standards, defined during an inter-lab validation study and based on 35 reference chemicals, were used to verify an automated version of the assay and demonstrate how the modified experimental design (e.g. titration series across plates rather than within a plate) could still satisfy important acceptance criteria laid out in the manual protocol. Since this exercise (manuscript in preparation) demonstrates that the manual and automated versions of this class of assay can deliver essentially the same results, the expectation is that historic data generated manually for an assay can be combined with HTS data generated on a single automation platform to provide a comprehensive evaluation of assay performance.
3.7 Predictive capacity (accuracy)
In the context of our use case, we define the predictive capacity or accuracy of each assay as its ability to correctly determine whether or not a chemical can perturb the target or pathway that the assay is designed to probe. This is most directly measured by the performance of the assay against a set of reference compounds whose ability to perturb the pathway is well documented. This approach raises an important point concerning the ability to compare different implementations of the same basic assay.
To illustrate this point, consider the case of ER assays. Multiple different assay formats are available, including cell-free binding assays; coactivator recruitment assays; reporter gene assays using full length and chimeric ER; proliferation assays; variants of these assays run using ER from human, rat, mouse and other species; variants of these assays run in different cell lines or primary cells; assays run in agonist and antagonist mode; and, finally, choices of different assay technologies for each of the assay formats. We argue that there is no single perfect assay and no unique “right” answer for testing a set of chemicals across these assay types. For the ER example, any assay should show a clear response for known ER actives (e.g., 17β-estradiol or Bisphenol A), and should show no response for known inactives (e.g. atrazine). However, it is recognized that each assay format has its own set of susceptibilities to both false positive and false negative results. For example, reporter gene assays using luciferase are prone to false positive results by indirect effects on protein stability (Auld et al., 2008). Fluorescence-based assays can be interfered with by compounds with fluorescent emission in the same range as the assay signal (Simeonov et al., 2008) or by quenching of the excitation or emission wavelengths. In practice, it is difficult to control for each of the many possible modes of interference. Thus, a multimodal approach, in which multiple orthogonal assays (i.e. assays that test the same pathway but use different technologies) probing the same target or related targets associated with the same pathway, are employed to ensure a minimal false negative rate.
Beyond assay interference issues, there are weak actives that may be positive in one assay but not in another, and these help to define the relative sensitivity of the assays. Differences in sensitivity may be due to technical factors or to differences in the fundamental biology related to the use of different cell types and cell-clone-specific stable cell lines.
3.8 Domain of applicability
Domain of applicability is a concept originating in the Quantitative Structure-Activity Relationship (QSAR) field that has rough parallels in assay validation (Jaworska et al., 2005). The applicability domain of a QSAR model has been defined as follows by ECVAM (Netzeva et al., 2005) and the OECD (OECD, 2004): “The applicability domain of a (Q)SAR model is the response and chemical structure space in which the model makes predictions with a given reliability”. QSAR models are “trained” and parameterized using a set of chemicals with known activities relative to the endpoint being modeled, and model performance is evaluated against some validation set, usually consisting of chemicals external to the training set. Although, in principle, the model could make predictions for any chemical structure for which model parameters can be computed, the reliability of prediction for a chemical whose model parameters are “outside” of the training and validation structure domain is not well characterized. Therefore, the development and validation model parameter space (or some variant, thereof) is typically designated as the domain of applicability of the model, and the conservative recommendation is that one should not trust predictions on chemicals whose parameters fall outside of this domain.
For assays (in vitro or in vivo), development and validation are also typically carried out on a limited set of chemicals, and there may be reasons to question the reliability of test results for chemicals significantly dissimilar to those in the development and validation sets. This concept of assay domain of applicability has not been often examined in standard assays, because the number of chemicals tested during validation has typically been too small. However, for HTS assays, even during the development and validation stage, one typically tests many (up to thousands) of chemicals, so domain of applicability may be more carefully considered here. One important influence on chemical-assay data reliability has to do with whether a chemical (or its close structural analogs) can be successfully evaluated in a particular test system, i.e. does the assay result accurately reflect the target or pathway interaction of the administered chemical or its biotransformation product (the latter in the case of metabolically competent assays). For example, chemicals would likely have to be soluble in an aqueous buffer and be relatively non-volatile in order to be tested in most HTS formats. As already mentioned, chemicals with light emission/absorption activity in the fluorescence detection region of the assay (such as dyes) could produce assay interference and false positives. Similarly, semi-volatile chemicals could potentially contaminate surrounding plate wells and produce false positives/negatives, and reactive chemicals could decompose upon exposure to air or water and produce false positives or negatives. All such chemicals, which in principle could be identified based on molecular structure features or physicochemical properties (as in QSAR approaches), could be considered to fall outside the domain of applicability of an HTS assay operated under a set of standard protocols. Additionally, as is the case for QSAR models, the structural and property dimensions of the chemicals in the test library define the range of historical application of the assay. Hence, chemicals having properties or features that differ significantly from previously tested chemicals could be considered to be outside the domain of “past experience” of the assay, which could trigger increased scrutiny of the results for these chemicals.
3.9 Performance standards
Performance standards are principally associated with documenting the aspects of a validated test that need to be included in a subsequent “me too” test (i.e., assays that are mechanistically and functional similar to the original, validated assay) (ICCVAM, 2003; Stokes et al., 2006; Stokes, 2007; Wind and Stokes, 2010). These include essential test method components and procedures, a minimal set of reference compounds and required accuracy and reliability values that the follow-on test would have to meet. Documenting performance standards for HTS assays would be no different than for low-throughput assays, so the OECD procedures could be followed as written. In the section above on predictive capacity, we discussed different versions of a basic assay, but with significant differences in protocol. As an example, consider two versions of a basic reporter gene assay, both using the same cell line and reporter gene construct, but one being run manually in 24 well plates, and the other being run in 1536 well plates using a robotic system. As discussed above, it is not clear whether the second assay is a “me too” assay that only needs to meet performance standards developed during validation of the former, or whether it is a wholly new assay that would require complete validation. One could certainly argue that the underlying assay similarities, both functional and operational, are sufficiently compelling to warrant the more limited performance standard requirement.
3.10 Peer review
Independent scientific peer review is considered as an essential step for a new test method prior to regulatory acceptance. ICCVAM and OECD guidelines provide detailed processes for conducting peer review of proposed assays (ICCVAM, 1997; Stokes, 2007; OECD, 2005). The formality of the peer review process and the overall validation process are related to the desire to be as rigorous and impartial as possible and to avoid (even unintentional) bias in validation studies. Typically, independent validation of a new test method involves the appointment of a working group comprising external experts and/or members of a validation body (e.g. ICCVAM, ECVAM Scientific Advisory Committee, etc.). Once the validation study report is completed, then this report is subjected to a highly transparent and independent scientific peer review by a panel of experts who do not have a financial or other conflict of interest with the test method or outcome of the review (ICCVAM, 1997; Sailstad et al., 2001; Stokes, 2007). These panels meet in public session, and all materials considered by the panels are also made available to the public for review and comment. The opportunity for comments by public stakeholders is also provided during the meetings of the peer review panel.
We believe that the peer review stage is one place where the overall validation process can be significantly streamlined for HTS assays, while at the same time increasing transparency. This is because the outputs of HTS assays are easily interpreted, quantitative read-outs of mechanistically simple interactions. As a result, objective evaluation criteria can be easily formulated, and the performance against these can be measured automatically. This makes judging performance more of a quantitative and statistical task rather than one requiring significant expert judgment. As previously discussed, for each assay, there would be an extensive set of reference chemicals, to the extent supported by the literature and existing knowledge, and the evaluation of assay performance would be based on the data generated for these chemicals. One also needs to have guidance on the minimal information that must be supplied about the conduct of the assay, for instance similar to the MIAME (Minimum Information About a Microarray Experiment) standards for gene arrays (Brazma et al., 2001). There are a wide range of proposed “Minimal Information” standards from which a standard appropriate for HTS assay validation could be constructed.3 For HTS assays, the newly developed BioAssay Ontology (BAO) (Schurer et al., 2011; Visser et al., 2011) could provide a framework for standard descriptions of assays for use in our proposed process, and could help guide minimal sets of information to be required as part of the assay description. The goal of the peer review would then be to assess objective criteria such as: Did the reference compounds yield the expected positive or negative responses? Are the efficacy and potency values in line with expectations? How well did the assay perform across time and reagent batches, and across chemical replicates? Is the assay protocol documented well enough that another group could replicate the assay in their lab given the appropriate resources?
Regarding the selection of reference chemicals, an important use of an outside expert group would be in the selection and publication of acceptable reference chemical sets for each assay target, similar to the NICEATM effort in relation to ER assays (ICCVAM, 2011). A peer reviewer requires information on the assay protocols and quality procedures in the laboratories, literature or other historical data on the reference compounds, and data generated during the testing phases (including concentration-response curves and analysis of replicates). For the ToxCast and Tox21 projects, all of this information is captured electronically in a single database. This type of database could be enhanced to manage all of the required validation information, and all of this information (except for some potential proprietary information) could be made public online. Because all of the data would be in a common format, it would simplify and make practical the peer-review of any number of assays. Any group wishing to propose a new assay for use in a regulatory prioritization application would then have immediate access to all existing validation information on similar assays, and could submit their validation package into the central system to be queued up for subsequent peer review. (Recall that there is still no consensus on whether “prioritization” is a regulatory activity.)
This rapid and continuous preparation of validation documentation would facilitate the continuous improvement of assays to be used in regulatory prioritization. Other advantages of such an online system include capturing electronic records of all validation data and past review documentation, and allowing reviewers to access all information remotely. Further peer review could proceed on a continuing basis.
Despite this clear-cut scenario, it is important to stress that peer review should not be set up as a pass-fail test, but should be used to provide valuable feedback. The process should encourage outside experts to offer insight and advice on the construction of the assay and its performance, and assay developers should be encouraged to incorporate suggestions for improvements. Involving an expert peer review group early, even in the case of straightforward single endpoint assays, can help achieve the best performing assay sooner than would occur otherwise. Peer review should be a constructive process that aims to highlight the strengths of the method and to identify limitations that end-users and regulators should keep in mind when basing decisions on data generated using the method.
Finally, an important issue that must be addressed is who will manage the peer review process. Under our proposal, there needs to be centralized databases holding validation data, and for the sake of efficiency, some organization needs to coordinate this as well as other tasks such as organizing peer reviews (recruiting reviewers, publishing guidelines, etc.), publishing results, etc. Organizations that could potentially play this role are the U.S. EPA, ICCVAM, ECVAM, and the Japanese Center for the Validation of Alternative Methods (JaCVAM).
3.11 Regulatory acceptance
Our purpose in developing and implementing a validation process is to provide regulatory scientists the information they need to decide if an HTS assay, or battery of assays, is reliable, relevant and fit for purpose. The primary goal of the validation process is regulatory acceptance so that data generated with the assay can be used to help assess the safety of chemicals. The analysis presented in this paper is driven by the specific need to provide the U.S. EPA and NTP with acceptable tools for prioritization applications in cases where we have large numbers of untested chemicals and limited mandate to require, or insufficient resources to carry out further testing. The most mature plans are for the U.S. EPA’s Endocrine Disruptor Screening Program (EDSP), where HTS assays for endocrine pathways (estrogen, androgen, thyroid, and steroidogenesis) will be used in prioritizing which of the thousands of chemicals subject to EDSP should have Tier 1 test orders issued first (U.S. EPA, 2011b).
Clearly, it is in all stakeholders’ interest to insure the relevance and reliability of the assays and the transparency of the process for generating assay data. The OECD Guidance Document 34 recommends validation and peer review for assays that will ultimately be incorporated into a Test Guideline (TG), recommended by the Working Group of the National Coordinators of the Test Guidelines Programme (WNT). The recommendations of the WNT are subsequently considered by the Joint Meeting of the Chemicals Committee and the Working Party on Chemicals, Pesticides and Biotechnology and if found acceptable, and are then subsequently accepted by all OECD members under the Mutual Acceptance of Data (MAD) agreement. In the European Union, such TGs are often taken up in legislation (where relevant), for example in the Test Methods Regulation. They can then be referenced in the information requirements for regulatory submissions/registrations, under REACH, for example.
4 Conclusions
At its core, validation is about doing good science. For an HTS assay (or collection of related assays) to be considered “valid” for a particular use and purpose, it needs to have a sound rationale, provide explainable and reproducible results, and be documented in a way that a scientist can understand the results and potentially repeat them. We have presented an analysis of how HTS in vitro assays could mostly conform to standard validation practice, including some issues that are specific to this type of assay, and some suggested changes to standard practice. The goal is to develop a validation procedure that is as streamlined (fast and inexpensive) as possible, while still providing the information that regulators need in terms of relevance, reliability and fitness for purpose. ICCVAM also seeks to streamline the validation process and updates its guidances periodically to achieve this (Schechtman et al., 2006). Flexibility is also important, and is reflected in the introduction for the ICCVAM interagency validation criteria (ICCVAM, 1997): “For a new or revised test method to be considered validated for regulatory risk assessment purposes, it should generally meet the following criteria (the extent to which these criteria are met will vary with the method and its proposed use). However, there needs to be flexibility in assessing a method given its purpose and the supporting database.” (ICCVAM, 1997). Similarly, the OECD validation guidelines state that “Scientific rigor is always required. … However, the level of assurance that is appropriate for a specific purpose and type of test varies and should be assessed on a case-by-case basis” (OECD, 2005).
We have specifically focused on the use of HTS assays for prioritization, rather than replacement of existing in vivo assays. In this use case, the assays are intended to provide data on KE’s in toxicity pathways, which is a level of biological organization that is less complex than that typically evaluated in standard, animal-based toxicity assays. Whereas these biological activities are thought to underlie certain adverse effects, there is no one-to-one matching with adverse outcomes in animals. Therefore, the goal here is not to recapitulate in vivo results. Instead, it is to provide a comprehensive enough set of data to suggest the possibility of toxicity via a particular set of mechanisms, or to suggest the lack of such a possibility. As assays spanning more potential mechanisms of action are implemented in HTS format, more of the universe of mechanisms that could underlie toxicity will be covered. At some point in the future, as such HTS coverage increases, we may reach the point where such assays can be used within a systems biology or modeling framework to quantitatively predict in vivo toxicity.
In line with our stated use case of prioritization, we have proposed two potential changes to standard assay validation practice that could significantly streamline the acceptance criteria for new HTS technologies, namely elimination of the mandatory requirement to do cross-laboratory testing, and the development of a straightforward on-line peer review process, which offers not only greater efficiency, but also additional transparency relative to the current approach when the test methods are not proposed for regulatory guidelines, but rather for prioritization. Although both of these recommendations might be considered controversial, due to their departure from current validation practice, we believe that both merit serious consideration given the significant advantages offered by HTS assays.
As stated in the introduction, this paper is not intended to be a prescription for a new process, but instead to offer some suggestions and to start a conversation about the possibility of developing a streamlined validation practice for use of HTS assays as prioritization tools. To that end, we offer a set of questions that need to be addressed:
Do assays used for prioritization require validation, i.e. will regulators accept their use for prioritization without formal demonstration of relevance, reliability and fitness for purpose?
Are HTS versions of existing assays, where there are at least some technical changes in the underlying protocol, really new assays?
Is it an acceptable tradeoff to require testing of greater numbers of reference chemicals in HTS assays, more than used in traditional assays, in exchange for not requiring cross-laboratory testing during validation?
Is this tradeoff more acceptable in a prioritization context, in which the assay is not replacing an existing validated test?
Can the peer review of HTS assays proposed for use in prioritization be adequately streamlined to largely a review of protocols and quantitative results, thus enabling at least a semi-automated review process?
Assuming that some level of transparency is maintained and a set of performance standards can be achieved, is there any compelling reason to treat proprietary assays differently than non-proprietary ones in the validation process?
Given that some proposed changes to the validation process rely on having an adequately large number of reference chemicals, how many reference chemicals is enough? For how many targets are we likely to have this large-enough set of well-characterized reference chemicals?
Given that the ultimate user of the test result is a regulator, how does the current or new validation process help that person understand the best use of the data generated by the assay? Can we start from these user requirements and custom design the assays and their validation requirements with that use in mind?
It is worth considering the intersection of our proposals on validation practice with ideas coming from the area of Evidence-Based Toxicology (EBT) (Hartung, 2009b, a). EBT aims to build off of the success of Evidence-Based Medicine (EBM). EBM relies on the use of rigorous, unbiased statistically-based meta-analyses of extensive preclinical and clinical data to determine best practices for clinicians. EBM has 3 pillars which are relevant here: (1) method assessment; (2) meta-analysis of studies; and (3) causation of health effects. In #1, EBT aims to compare different options to determine the toxicity of compounds, which is consistent with our aim to quickly develop and evaluate new tests. Under this heading, EBT could help us better understand which tests we should be developing, how they will be used, and new questions we might want to ask of the method development and validation processes. Items 2 and 3 are relevant to the current discussion because we need to know the linkage between a KE and the ultimate adverse outcome, forming the basis of our relevance and fitness for purpose tasks. In the preceding discussion, we simply assumed that knowledge of a chemical interaction with a particular molecular target was sufficient to trigger a pathway with a causal linkage to an adverse outcome. However, for each assay-endpoint pair, significant study and analysis from the literature will be required to determine these linkages. This effort is one subject of the field of systems toxicology, which is closely tied to EBT.
Finally, we discuss one counter-productive aspect of current assay development, validation and acceptance, which is that validated assays tend to become “frozen in time” because of the lengthy process and high costs involved. Many of our current guideline assays took years to a decade or more to go through this process (and in fact a number of the currently used guideline tests were never validated through a formal process), leaving us to rely on old technology. For standard in vivo tests, this is understandable and necessary due to the complexity and lengthy gestation of such tests. With HTS assays, on the other hand, the development time from conception to production in high-throughput format can be months. One can then imagine a process in which there is an ongoing competition to develop increasingly better assays or more complete batteries of assays to assess the ability of chemicals to trigger particular AOPs or impact specified toxicity pathways. This could lead to a rolling development-validation-acceptance-use process that is iteratively applied to a large, pre-plated library of chemicals. Figure 1 illustrates this iterative process. If we can implement a streamlined process for rigorous, yet practical validation of HTS assays, enabling us to employ new HTS technologies in almost real-time from when they are developed, we will have made significant progress in realizing the promise of 21st century toxicology.
Footnotes
Disclaimer: The views expressed in this paper are those of the authors and do not necessarily reflect the views or policies of the U.S. Environmental Protection Agency, the U.S. Food and Drug Administration or the U.S. National Institutes of Health.
This paper was presented by Richard Judson at the workshop on “Evidence-based Toxicology for the 21st Century: Opportunities and Challenges,” held on January 24–25, 2012 at the Environmental Protection Agency campus in Research Triangle Park, NC, USA (see www.ebtox.com).
References
- Ankley GT, Bennett RS, Erickson RJ, et al. Adverse outcome pathways: a conceptual framework to support ecotoxicology research and risk assessment. Environ Toxicol Chem. 2010;29:730–741. doi: 10.1002/etc.34. [DOI] [PubMed] [Google Scholar]
- Auld DS, Thorne N, Nguyen DT, et al. A specific mechanism for nonspecific activation in reporter-gene assays. ACS Chem Biol. 2008;3:463–470. doi: 10.1021/cb8000793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatori N, Boyer JL, Rockett JC. Exploiting genome data to understand the function, regulation, and evolutionary origins of toxicologically relevant genes. EHP Toxicogenomics. 2003;111:61–65. [PubMed] [Google Scholar]
- Berg EL, Yang J, Melrose J, et al. Chemical target and pathway toxicity mechanisms defined in primary human cell systems. J Pharmacol Toxicol Methods. 2010;61:3–15. doi: 10.1016/j.vascn.2009.10.001. [DOI] [PubMed] [Google Scholar]
- Birnbaum LS, Stokes WS. Safety testing: moving toward alternative methods. Environ Health Perspect. 2010;118:A12–13. doi: 10.1289/ehp.0901704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boobis AR, Doe JE, Heinrich-Hirsch B, et al. IPCS framework for analyzing the relevance of a noncancer mode of action for humans. Crit Rev Toxicol. 2008;38:87–96. doi: 10.1080/10408440701749421. [DOI] [PubMed] [Google Scholar]
- Bouhifd M, Bories G, Casado J, et al. Automation of an in vitro cytotoxicity assay used to estimate starting doses in acute oral systemic toxicity tests. Food Chem Toxicol. 2012;50:2084–2096. doi: 10.1016/j.fct.2012.03.046. [DOI] [PubMed] [Google Scholar]
- Bradbury SP, Feijtel TC, Van Leeuwen CJ. Meeting the scientific needs of ecological risk assessment in a regulatory context. Environ Sci Technol. 2004;38:463A–470A. doi: 10.1021/es040675s. [DOI] [PubMed] [Google Scholar]
- Brazma A, Hingamp P, Quackenbush J, et al. Minimum information about a microarray experiment (MIAME)-toward standards for microarray data. Nat Genet. 2001;29:365–371. doi: 10.1038/ng1201-365. [DOI] [PubMed] [Google Scholar]
- Collins FS, Gray GM, Bucher JR. Toxicology. Transforming environmental health protection. Science. 2008;319:906–907. doi: 10.1126/science.1154619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper-Hannan R, Harbell JW, Coecke S, et al. The Principles of Good Laboratory Practice: Application to In Vitro Toxicology Studies The Report and Recommendations of ECVAM Workshop. ATLA. 1999;27:539–577. doi: 10.1177/026119299902700410. [DOI] [PubMed] [Google Scholar]
- Ding D, Xu L, Fang H, et al. The EDKB: an established knowledge base for endocrine disrupting chemicals. BMC Bioinformatics. 2010;11(Suppl 6):S5. doi: 10.1186/1471-2105-11-S6-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dix DJ, Houck KA, Martin MT, et al. The ToxCast program for prioritizing toxicity testing of environmental chemicals. Toxicol Sci. 2007;95:5–12. doi: 10.1093/toxsci/kfl103. [DOI] [PubMed] [Google Scholar]
- Doull J, Borzelleca JF, Becker R, et al. Framework for use of toxicity screening tools in context-based decision-making. Food Chem Toxicol. 2007;45:759–796. doi: 10.1016/j.fct.2006.10.025. [DOI] [PubMed] [Google Scholar]
- Gohlke JM, Thomas R, Zhang Y, et al. Genetic and environmental pathways to complex diseases. BMC Syst Biol. 2009;3:46. doi: 10.1186/1752-0509-3-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamadeh HK, Bushel PR, Jayadev S, et al. Gene expression analysis reveals chemical-specific profiles. Toxicol Sci. 2002;67:219–231. doi: 10.1093/toxsci/67.2.219. [DOI] [PubMed] [Google Scholar]
- Hartung T, Bremer S, Casati S, et al. A modular approach to the ECVAM principles on test validity. Altern Lab Anim. 2004;32:467–472. doi: 10.1177/026119290403200503. [DOI] [PubMed] [Google Scholar]
- Hartung T. Food for thought … on validation. ALTEX. 2007;24:67–80. doi: 10.14573/altex.2007.2.67. [DOI] [PubMed] [Google Scholar]
- Hartung T. A toxicology for the 21st century–mapping the road ahead. Toxicol Sci. 2009a;109:18–23. doi: 10.1093/toxsci/kfp059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartung T. Food for thought… on evidence-based toxicology. ALTEX. 2009b;26:75–82. doi: 10.14573/altex.2009.2.75. [DOI] [PubMed] [Google Scholar]
- ICCVAM. Validation and Regulatory Acceptance of Toxicological Test Methods: A Report of the ad hoc Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) (NIH Publication No. 97-3981).NIEHS. 1997 http://iccvam.niehs.nih.gov/docs/guidelines/validate.pdf.
- ICCVAM. ICCVAM Guidelines for the Nomination and Submission of New, Revised, and Alternative Test Methods. (NIH Publication No. 03-4508).NIEHS. 2003 http://iccvam.niehs.nih.gov/docs/guidelines/subguide.htm.
- ICCVAM. The LUMI-CELL® ER (BG1Luc ER TA) Test Method: An In Vitro Assay for Identifying Human Estrogen Receptor Agonist and Antagonist Activity of Chemicals. (NIH Publication No. 11-7814).NIEHS. 2011 [Google Scholar]
- Inglese J, Auld DS, Jadhav A, et al. Quantitative high-throughput screening: a titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc Natl Acad Sci U S A. 2006;103:11473–11478. doi: 10.1073/pnas.0604348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworska J, Nikolova-Jeliazkova N, Aldenberg T. QSAR applicabilty domain estimation by projection of the training set descriptor space: a review. Altern Lab Anim. 2005;33:445–459. doi: 10.1177/026119290503300508. [DOI] [PubMed] [Google Scholar]
- Judson R, Richard AM, Dix DJ, et al. The Toxicity Data Landscape for Environmental Chemicals. Environ Health Perspect. 2009;117:685–695. doi: 10.1289/ehp.0800168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judson RS, Houck KA, Kavlock RJ, et al. Predictive In Vitro Screening of Environmental Chemicals – The ToxCast Project. Environ Health Perspect. 2010;118:485–492. doi: 10.1289/ehp.0901392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavlock RJ, Austin CP, Tice RR. Toxicity testing in the 21st century: implications for human health risk assessment. Risk Anal. 2009;29:485–487. doi: 10.1111/j.1539-6924.2008.01168.x. discussion 492–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leist M, Efremova L, Karreman C. Food for thought … considerations and guidelines for basic test method descriptions in toxicology. ALTEX. 2010;27:309–317. doi: 10.14573/altex.2010.4.309. [DOI] [PubMed] [Google Scholar]
- Linge JP, Hartung T. ECVAM’s approach to intellectual property rights in the validation of alternative methods. Altern Lab Anim. 2007;35:441–446. doi: 10.1177/026119290703500411. [DOI] [PubMed] [Google Scholar]
- Meek ME, Bucher JR, Cohen SM, et al. A framework for human relevance analysis of information on carcinogenic modes of action. Crit Rev Toxicol. 2003;33:591–653. doi: 10.1080/713608373. [DOI] [PubMed] [Google Scholar]
- Netzeva TI, Worth A, Aldenberg T, et al. Current status of methods for defining the applicability domain of (quantitative) structure-activity relationships. The report and recommendations of ECVAM Workshop 52. Altern Lab Anim. 2005;33:155–173. doi: 10.1177/026119290503300209. [DOI] [PubMed] [Google Scholar]
- NRC. Toxicity Testing in the 21st Century: A Vision and a Strategy. Washington DC: National Academies Press; 2007. Vol. [Google Scholar]
- Nuwaysir EF, Bittner M, Trent J, et al. Microarrays and toxicology: the advent of toxicogenomics. Mol Carcinog. 1999;24:153–159. doi: 10.1002/(sici)1098-2744(199903)24:3<153::aid-mc1>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Guidance document on the validation and international acceptance of new or updated test methods for hazard assessment: OECD Series on testing an assessment. Number 34. E Directorate 2005 [Google Scholar]
- GUIDANCE DOCUMENT ON USING CYTOTOXICITY TESTS TO ESTIMATE STARTING DOSES FOR ACUTE ORAL SYSTEMIC TOXICITY TESTS. 2010 iccvam.niehs.nih.gov/SuppDocs/FedDocs/OECD/OECD-GD129.pdf.
- Reynolds VL. Applications of emerging technologies in toxicology and safety assessment. Int J Toxicol. 2005;24:135–137. doi: 10.1080/10915810590948280. [DOI] [PubMed] [Google Scholar]
- Rogers JM, Denison MS. Recombinant cell bioassays for endocrine disruptors: development of a stably transfected human ovarian cell line for the detection of estrogenic and anti-estrogenic chemicals. In Vitr Mol Toxicol. 2000;13:67–82. [PubMed] [Google Scholar]
- Sailstad DM, Hattan D, Hill RN, et al. ICCVAM evaluation of the murine local lymph node assay. The ICCVAM review process. Regul Toxicol Pharmacol. 2001;34:249–257. doi: 10.1006/rtph.2001.1496. [DOI] [PubMed] [Google Scholar]
- Schechtman LM, Wind M, Stokes WS. Streamlining the validation process: the ICCVAM nomination and submission process and guidelines for new, revised, and alternative test methods. ALTEX. 2006;23:336–341. [Google Scholar]
- Schurer SC, Vempati U, Smith R, et al. BioAssay ontology annotations facilitate cross-analysis of diverse high-throughput screening data sets. J Biomol Screen. 2011;16:415–426. doi: 10.1177/1087057111400191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seed J, Carney EW, Corley RA, et al. Overview: Using mode of action and life stage information to evaluate the human relevance of animal toxicity data. Crit Rev Toxicol. 2005;35:664–672. doi: 10.1080/10408440591007133. [DOI] [PubMed] [Google Scholar]
- Shukla SJ, Huang R, Austin CP, et al. The future of toxicity testing: a focus on in vitro methods using a quantitative high-throughput screening platform. Drug Discov Today. 2010;15:997–1007. doi: 10.1016/j.drudis.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simeonov A, Jadhav A, Thomas CJ, et al. Fluorescence spectroscopic profiling of compound libraries. J Med Chem. 2008;51:2363–2371. doi: 10.1021/jm701301m. [DOI] [PubMed] [Google Scholar]
- Singh AV, Yang C, Kavlock RJ, et al. Developmental Toxicology Research Strategies: Computational Toxicology. In: Daston GP, Knudsen TB, editors. Developmental Toxicology. New York: Elsevier; 2010. [Google Scholar]
- Sonich-Mullin C, Fielder R, Wiltse J, et al. IPCS conceptual framework for evaluating a mode of action for chemical carcinogenesis. Regul Toxicol Pharmacol. 2001;34:146–152. doi: 10.1006/rtph.2001.1493. [DOI] [PubMed] [Google Scholar]
- Stokes WS, Schechtman LM, Rispin A, et al. The Use of Test Method Performance Standards to Streamline the Validation Process. ALTEX. 2006;23:342–345. [Google Scholar]
- Stokes WS, Wind M. Recent progress and future directions at NICEATM-ICCVAM: Validation and regulatory acceptance of alternative test methods that reduce, refine, and replace animal use. ALTEX. 2010a;27:221–232. [Google Scholar]
- Stokes WS, Wind M. Validation of innovative technologies and strategies for regulatory safety assessment methods: challenges and opportunities. ALTEX. 2010b;27:87–95. [PubMed] [Google Scholar]
- Stokes WS, Wind M. NICEATM and ICCVAM participation in the International Cooperation on Alternative Test Methods. ALTEX. 2010c;27:211–219. [Google Scholar]
- Stokes WS, S LM. Validation and regulatory acceptance of new, revised, and alternative toxicological methods. In: Hayes AW, editor. Principles and Methods of Toxicology. Philadelphia, Pennsylvania: Taylor and Francis; 2007. [Google Scholar]
- Takeuchi S, Matsuda T, Kobayashi S, et al. In vitro screening of 200 pesticides for agonistic activity via mouse peroxisome proliferator-activated receptor (PPAR)alpha and PPARgamma and quantitative analysis of in vivo induction pathway. Toxicol Appl Pharmacol. 2006;217:235–244. doi: 10.1016/j.taap.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Toxic Substances Control Act of 1976. 1976. 15 U.S.C. §2601 et seq. [Google Scholar]
- Visser U, Abeyruwan S, Vempati U, et al. BioAssay Ontology (BAO): a semantic description of bioassays and high-throughput screening results. BMC Bioinformatics. 2011;12:257. doi: 10.1186/1471-2105-12-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wind M, Stokes WS. Developing performance standards to expedite validation of innovative and improved test methods. ALTEX. 2010;27:97–102. [PubMed] [Google Scholar]
- Zhou T, Chou J, Watkins PB, et al. Toxicogenomics: transcription profiling for toxicology assessment. EXS. 2009;99:325–366. doi: 10.1007/978-3-7643-8336-7_12. [DOI] [PubMed] [Google Scholar]