

Abstract
ARF is a multifunctional tumor suppressor that acts as both a sensor of oncogenic stimuli and as a key regulator of ribosome biogenesis. Recently, our group established the DEAD-box RNA helicase and microRNA (miRNA) microprocessor accessory subunit, DDX5, as a critical target of basal ARF function. To identify other molecular targets of ARF, we focused on known interacting proteins of DDX5 in the microprocessor complex. Drosha, the catalytic core of the microprocessor complex, plays a critical role in the maturation of specific non-coding RNAs, including miRNAs and rRNAs. Here, we report that chronic or acute loss of Arf enhanced Drosha protein expression. This induction did not involve Drosha mRNA transcription or protein stability but rather relied on the increased translation of existing Drosha mRNAs. Enhanced Drosha expression did not alter global miRNA production, but rather modified expression of a subset of miRNAs in the absence of Arf. Elevated Drosha protein levels were required to maintain the increased rRNA synthesis and cellular proliferation observed in the absence of Arf. Arf-deficient cells transformed by oncogenic RasV12 were dependent on increased Drosha expression as Drosha knockdown was sufficient to inhibit Ras-dependent cellular transformation. Thus, we propose that ARF regulates Drosha mRNA translation to prevent aberrant cell proliferation and Ras-dependent transformation.
Keywords: ARF, Drosha, translation, tumor suppression, rRNA
Introduction
Cancer initiation and progression are hallmarked by the loss of regulatory mechanisms that control cellular growth and proliferation. The CDKN2A (Ink4a/Arf) locus encodes two distinct tumor suppressors, p16INK4a and p19ARF (p14ARF in humans) (1). ARF has classically been regarded as an activator of p53 through its ability to bind and sequester Mdm2, the E3 ubiquitin ligase for p53, in the nucleolus (2-5). Recently, numerous p53-independent functions have been attributed to ARF (6), including its regulation of rRNA synthesis and ribosome biogenesis (7). ARF accomplishes this, at least in part, through its repressive interaction with the ribosome chaperone nucleophosmin (NPM) thereby limiting steady-state ribosome biogenesis and growth (1, 8-10).
Recently, a nucleolar proteomic screen expanded on the mechanism through which ARF controls rRNA synthesis by identifying a novel relationship between ARF and the DEAD-box RNA helicase, DDX5 (11). Basal ARF proteins restrict DDX5 access to the nucleolus and antagonize the DDX5-NPM interaction. This interference is sufficient to disrupt DDX5’s role in rRNA transcription and rRNA processing. An assessment of other proteins that are involved in ribosome biogenesis, especially those that associate with NPM and/or DDX5 could reveal additional modes through which ARF acts as a potent tumor suppressor.
Drosha is an RNase III endonuclease that was originally linked with the processing of rRNAs and is now widely studied in the context of microRNA (miRNA) biogenesis (12, 13). miRNAs represent a class of short, endogenous, noncoding RNAs that control gene expression by inhibiting translation and/or inducing degradation of specific target mRNAs (14). Similar to rRNAs, most miRNAs initially exist as long primary transcripts (pri-miRNAs) that are processed through a series of enzymatic cleavage steps to generate mature miRNAs (15-17). Drosha forms two different complexes, a small microprocessor complex that contains only Drosha and DGCR8, and a larger complex that contains accessory proteins including DDX5 (18, 19). While both complexes are capable of processing pri-miRNAs, only the latter has demonstrated the ability to process pre-rRNAs (20).
Abnormalities in mature miRNA levels as well as in the expression of miRNA processing enzymes have been linked with various types of disease (21). While some studies suggest that there are correlations between decreased Drosha expression and tumor incidence and prognosis, other studies have shown that reducing Drosha expression impairs rRNA processing and cellular proliferation (22-26). Despite Drosha’s apparent link to human diseases, little is known about its regulation. Previous studies have shown that Drosha is stabilized through its protein-protein interaction with DGCR8, but outside of this, no other forms of transcriptional or post-transcriptional regulation are known (27).
Given ARF’s multifaceted involvement in mediating ribosome biogenesis and its newly characterized relationship with DDX5, a component of the Drosha processing complex, we sought to determine whether ARF impacted Drosha. In this report, we show that Drosha is post-transcriptionally regulated in an ARF-dependent manner. We identify Drosha as a unique translational target of the ARF tumor suppressor. Moreover, we show that the increased Drosha expression in the absence of Arf is required for efficient Ras-mediated cell transformation through Drosha’s ability to regulate rRNA synthesis and cell proliferation.
Results
Loss of Arf induces Drosha expression
Previous reports have demonstrated that ARF serves as a major regulator of ribosome biogenesis (7) and that this is due at least in part to its modulation of DDX5 localization and function (7, 11). Moreover, other studies have established a clear link between DDX5 and Drosha in the processing of double stranded RNAs including rRNAs (18, 20). To test whether a relationship exists between Drosha and ARF, we first compared Drosha protein levels using WT and Arf −/− MEFs. Higher levels of Drosha protein were observed in cells with genetic ablation of Arf exon 1β (Figure 1a, left). We explored this result further by acutely manipulating the expression of ARF using either a lentivirus encoding an shRNA targeting Arf exon 1β, the ARF-specific exon of the CDKN2A locus, or an ARF overexpressing retrovirus. Consistent with the previous finding, acute knockdown of basal ARF expression resulted in heightened Drosha expression (Figure 1b). Conversely, ectopic overexpression of ARF lowered Drosha expression (Figure 1c). ARF-mediated regulation of Drosha protein expression was not MEF-specific as similar trends were observed using Arf flox/flox mouse astrocytes infected with adenoviruses encoding Cre recombinase (Figure 1d). The array of genetic techniques employed to disrupt ARF activity demonstrates a novel link between these two proteins and warranted a closer examination into the mechanism through which ARF suppresses Drosha.
Figure 1. Arf negatively regulates Drosha protein expression in a transcriptionally independent manner.

(a-d, left column) Cells of the indicated genotype were lysed, and separated proteins were immunoblotted for the indicated proteins. Arf flox/flox astrocytes were infected with adenoviruses encoding β-galactosidase (LacZ) or Cre recombinase and were harvested at 5 days post-infection for gene expression analysis. Drosha expression fold change relative to WT or control infected cells is indicated. (a-d, right column) Quantitative RT-PCR analysis was performed. Drosha mRNA levels were normalized to Gadph mRNA levels. Fold change was calculated using the ΔΔCT method. Data are the mean ± SEM (N=3).
ARF suppresses the translation of Drosha mRNA
To determine how basal ARF modulates Drosha expression, we assessed different aspects of Drosha gene expression in response to ARF manipulation. Despite the increases in Drosha protein expression, no significant changes were observed in Drosha mRNA levels following Arf loss, acute ARF knockdown, or ectopic overexpression of ARF as determined by quantitative RT-PCR (Figure 1a-d, right; Supplementary Figure 1). Next, we treated WT and Arf −/− MEFs with Actinomycin D to evaluate Drosha mRNA stability and observed similar rates of Drosha mRNA decay in the presence or absence of Arf (Figure 2a). Taken together, this data indicates that ARF may post-transcriptionally regulate Drosha expression.
Figure 2. Loss of Arf has no effect on Drosha mRNA or protein stability.

(a) WT and Arf −/− MEFs were treated with 4 μg/ml actinomycin D for the indicated times. Quantitative RT-PCR analysis was performed. Data are represented as the percent of Drosha mRNA remaining after normalization to Gadph levels at t=0. (b-c) Cells were treated with 25 μg/ml cycloheximide (CHX) and were harvested at the indicated times for immunoblot analysis. Densitometry quantification is depicted in panel B and data are represented as percent remaining of Drosha protein levels normalized to γ-tubulin. (d) WT MEFs were treated with 40 μM MG-132 or DMSO for 8 hours and changes in Drosha and p21 (positive control) protein levels were measured.
Previous reports identified a positive relationship between Drosha and its microprocessor partner, DGCR8, such that Drosha protein is stabilized when bound to DGCR8 (27). To address potential changes in Drosha protein stability in response to Arf loss, Drosha protein expression was measured in WT and Arf −/− MEFs following cycloheximide treatment. The rate of Drosha turnover remained unchanged in the presence or absence of Arf despite the fact that we observed significantly more Drosha at the original time of treatment in cells lacking Arf (Figures 2b-c). Furthermore, treatment of WT MEFs with the proteosomal inhibitor MG-132 failed to induce Drosha protein levels to those observed in Arf −/− MEFs (Figure 2d). This data suggests that the differences in Drosha protein expression were not caused by altered protein stability that led us to hypothesize that Drosha might be translationally regulated by ARF.
Basal ARF negatively regulates multiple aspects of ribosome biogenesis, including rDNA transcription, rRNA processing, and ribosome nuclear export (28, 29, 9, 7, 30). While it is possible that ARF’s antagonizing role in these processes may disrupt the global translation of all mRNAs, existing data suggests that select genes may be translationally regulated by ARF (31). Having ruled out ARF regulation of Drosha transcription, mRNA and protein stability, we sought to determine the effects of Arf loss on the translation of existing Drosha mRNAs. We compared the percentage of Drosha mRNA transcripts associated with actively translating polyribosomes (polysomes) in WT and Arf −/− MEFs. Ribosomes were detected in lysates separated in sucrose gradients by continuous measurement of RNA absorbance (A254nm). Loss of Arf enhanced the overall formation of polysomes actively engaged in mRNA translation as previously described (Figure 3b) (9). Quantitative RT-PCR was performed to evaluate the distribution of Drosha mRNA transcripts in monosome-, disome-, and polysome-containing fractions. Drosha mRNAs were abundant in the heavier polysome fractions 11-13 in the absence of Arf, shifting away from lighter polysomes in fraction 9 from WT cells (Figure 3c). Importantly, GAPDH mRNA transcript distribution remained unchanged across polysomes (Figure 3d), suggesting that Arf loss does not globally affect the translation of every cellular transcript, but rather leads to selective mRNA translation.
Figure 3. Translation of Drosha mRNAs is augmented upon loss of Arf.

(a) Endogenous Drosha protein levels are elevated in MEFs that lack Arf compared to WT MEFs. (b) Cytosolic extracts were prepared from equal number WT and Arf −/− MEFs that had been treated for 5 min with CHX (10 μg/ml). Extracts were then subjected to differential density centrifugation and analyzed via constant UV monitoring (254 nm). (c-d) Monosome-, disome-, and polysome-associated Drosha mRNA levels were measured with qRT-PCR and were calculated as a percentage of total Drosha mRNA present in all fractions. Data are the mean ± SEM (N=3).
ARF-knockdown alters the expression of only a subset of mature miRNAs despite higher levels of Drosha
To begin to understand the downstream effects of heightened Drosha translation, we sought to compare global miRNA expression patterns upon ARF knockdown. It was previously reported that alterations in known miRNA processing factors failed to alter specific mature miRNA expression, suggesting that the microprocessor itself does not act at a rate-limiting stage of this process in some instances (32). Nevertheless, in order measure the impact of ARF knockdown on the miRNA signature of these cells, a Taqman array platform was used to quantify the changes in expression of over 300 mouse-specific miRNAs. The goal was to identify miRNA expression that was significantly altered (>1.4 fold change). Although approximately 50% of all miRNAs examined on the array were either undetectable or present at very low levels (CT > 31) in MEFs, there were 34 miRNAs that underwent significant changes in expression (11 upregulated and 23 downregulated) upon ARF knockdown and subsequent Drosha elevation (Figures 4a-b; Supplementary Figure 2). These findings imply that gains in Drosha expression, at least via loss of ARF, can significantly modify the miRNA landscape within the cell.
Figure 4. The expression of only a subset of miRNAs is altered upon ARF knockdown.

(a) Global miRNA expression profiles of WT MEFs infected with shLuc or shArf-encoded lentivirus were determined by TaqMan MicroRNA qRT-PCR in three separate experiments. Only miRNAs (N = 147), that were present at appreciable quantities in at least one condition (CT value is less than 31) were used for analysis. miRNA expression fold changes were calculated for each replicate and then averaged. The heat map shows the fold-changes in expression for a subset of miRNAs in WT shArf MEFs relative to WT shLuc MEFs. Each colored block represents the expression of 1 miRNA (labeled on the left). Expression signals are converted into color (red, high signal; green, low signal). Color intensities are proportional to the variation of expression as indicated in the scale bar. (b) Table depicting the 10 most up- and down-regulated miRNAs in WT shArf cells relative to WT shLuc cells.
Reduced Drosha expression impairs rRNA processing and cellular proliferation in the absence of Arf
Similar to its accessory protein partner, DDX5, Drosha has been implicated in rRNA processing (8, 33). Since it has been shown that cells lacking Arf process rRNA precursors more efficiently than their WT counterparts (7, 9), we hypothesized that reducing Drosha expression in Arf −/− MEFs would impair rRNA processing. Two independent lentiviral shRNA constructs encoding different Drosha-specific shRNAs were used to obtain sufficient knockdown relative to the luciferase control hairpin (Figure 5a). We monitored the processing of the initial 47S pre-rRNA transcript via [methyl-3H]-methionine pulse-chase analysis (7) revealing a delayed accumulation of mature 28S and 18S rRNAs in cells following Drosha knockdown (Figure 5b). In a separate experiment, WT MEFs were infected with a retrovirus encoding Drosha to determine whether elevated Drosha levels could accelerate ribosome biogenesis. Here, we discovered a more rapid accumulation of mature 28S and 18S rRNAs in cells expressing Drosha versus vector transduced cells (Supplementary Figure 3). Together, this data imply that Drosha is not only required to enhance the processing of nascent rRNA transcripts in the absence of Arf but also that the upregulation of Drosha associated with Arf loss is sufficient for increased rRNA maturation.
Figure 5. Drosha knockdown reduces proliferation and impairs ribosomal RNA maturation.

(a) Infected Arf −/− MEFs were lysed, and separated proteins were immunoblotted to confirm Drosha knockdown. (b) shLuc and shDrosha Arf −/− MEFs were labeled with [methyl-3H]-methionine and chased for the indicated times. Radiolabeled RNA was separated on an agarose gel, transferred to a membrane and visualized by autoradiography (left panel). Relative band intensities were determined for rRNA in the processing assay and plotted over time (right panels). The band intensities for all conditions were first individually normalized to their respective 47S levels at T=0 and then fold change was calculated. (c-d) Following Drosha knockdown, cells were plated in triplicate at a density of 5×104 per well in a 6-well plate for a proliferation assay and counted over a 4-day time period (c). These cells were also seeded in triplicate at 5×103 cells per dish in parallel and grown for 12 days. Foci were fixed in methanol, stained with Giemsa and counted (d).
Drosha’s role in facilitating rRNA synthesis suggested that it might also be critical for cell proliferation. In cells depleted of Drosha by shRNA knockdown, we observed a dramatic decrease in proliferation rates relative to control-infected cells (Figure 5c). Furthermore, Arf −/− MEFs were dependent on elevated Drosha expression for long-term proliferation; Arf −/− MEFs plated at low density formed fewer colonies following Drosha knockdown after twelve days in culture (Figure 5d). Taken together, our results show that cells lacking Arf rely on augmented Drosha expression to maintain aberrant and rapid cellular proliferation rates.
Drosha knockdown promotes apoptosis
To investigate whether the decrease in proliferation was linked to a change in cell viability, we analyzed the cell cycle distribution of both knockdown and control cells using flow cytometry (Supplementary Figure 4a). In accordance with the aforementioned proliferation data, the G1 and G2/M distribution of Drosha-depleted cells was significantly reduced compared to control knockdown cells. In addition, the population of sub-G1 cells was significantly larger upon Drosha knockdown (5.19% shLuc versus 49.53% shDrosha 1 or 37.78% shDrosha 2), which represented cells with a hypodiploid genome because of DNA degradation, a commonly associated feature of cells undergoing apoptosis (Figure 6a).
Figure 6. shRNA-mediated knockdown of Drosha in MEFs promotes cell death via apoptosis.
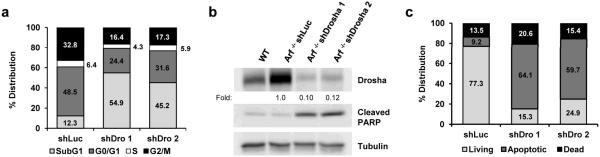
(a) Quantification of the cell cycle distribution of shLuc and shDrosha Arf −/− MEFs as determined by flow cytometry. (b) Immunoblot analysis examining PARP cleavage in response to Drosha knockdown. (c) Percentage of living, apoptotic (annexin V-positive), and dead (PI-positive and double-positive) shLuc and shDrosha cells determined by flow cytometry. Data are expressed as the mean ± SD of 10 000 events performed in triplicate.
In order to determine if apoptosis accounted for the differences in Arf −/− cell proliferation upon Drosha knockdown, we sought to quantify the population of cells undergoing apoptosis by flow cytometric analyses with FITC Annexin-V and propidium idiodine (PI) double staining. Arf −/− cells maintain an active p53 response to DNA damaging agents, and transient etoposide treatment properly induced apoptosis in these cells (Supplementary Figure 4b). Approximately 60-70% of Drosha-depleted Arf −/− MEFs stained positive for Annexin V compared to only 10% of control cells (Figure 6c), indicating that Drosha knockdown decreases cell proliferation at least in part by greatly increasing apoptosis. Caspase-mediated cleavage of the PARP protein is an indicator of cells undergoing apoptosis. In agreement with our observed Annexin-V staining, enhanced cleavage of PARP was observed in Drosha-knockdown cells relative to control knockdown (Figure 6b).
RasV12-induced transformation of Arf −/− MEFs requires Drosha
ARF protects normal cells from oncogenic RasV12 transformation by activating a p53-dependent growth arrest or apoptotic response (34). However, in the absence of Arf, MEFs transduced with RasV12 undergo cellular transformation, an event that can be both observed and quantified by anchorage independent growth in soft agar (35). To determine whether elevated Drosha levels phenocopied Arf loss, WT MEFs ectopically expressing Drosha and oncogenic RasV12 were plated in soft agar. Unlike RasV12-transduced Arf −/− MEFs, Drosha did not cooperate with RasV12 to transform WT cells. Furthermore, unlike RasV12, Drosha alone was unable to transform Arf −/− MEFs suggesting that Drosha does not act as a bona fide oncogene to drive cellular transformation (Supplementary Figure 5).
While overexpression of Drosha alone was not sufficient to transform immortal Arf −/− cells, we hypothesized that Drosha might be necessary for RasV12 transformation in the absence of Arf. To test this hypothesis, Arf −/− MEFs were first infected with retroviruses encoding oncogenic RasV12 followed by transduction of Drosha-specific shRNAs (Figure 7a). Reduction of Drosha protein expression was sufficient to impair RasV12-driven colony formation and anchorage-independent growth as indicated by a reduction in both the number of colonies and their overall size (Figures 7b-c), implying that Arf-deficient cells transformed by oncogenic RasV12 require elevated Drosha expression to maintain the transformed phenotype.
Figure 7. Drosha knockdown significantly inhibits Ras-induced colony formation of Arf −/− MEFs.
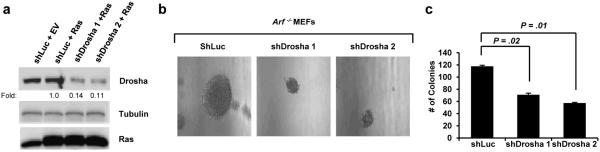
(a) Immunoblot analysis to confirm Ras overexpression and Drosha knockdown in Arf −/− MEFs. (b-c) A total of 5×104 infected cells per condition were seeded in triplicate onto soft agar plates and were grown for 3 weeks. Colonies were examined under a microscope and counted. Colony number is expressed as the mean ± SEM.
Discussion
The tumor suppressor nature of ARF was originally ascribed to its ability to stabilize and activate p53 in the presence of oncogenic stress. Over the last decade, numerous groups have established ARF as a potent multifaceted tumor suppressor that is not only crucial for the cellular response to oncogene activation, but that is also capable of monitoring steady-state ribosome synthesis and growth in a p53-independent manner (2-5, 8, 9, 28, 29). Aside from the p53-MDM2 network, NPM was one of the first proteins to be associated with ARF (8, 30); this novel interaction suppresses ribosome nuclear export, a rate limiting step of ribosome biogenesis (10, 36). More recently, a dynamic relationship between ARF and the DDX5 RNA helicase was revealed, further illustrating how ARF is able to control ribosome output through the coordinated regulation of rRNA transcription and rRNA processing (7, 11). Given that loss of Arf, a common event in cancer, enhances several important steps of ribosome maturation, one might predict that a global increase in protein translation would ensue under these conditions. Although future work pertaining to this hypothesis is required, a previous study as well as the data presented here, present a scenario that disruption of Arf expression likely initiates a selective translational program that accounts for an overall pro-growth phenotype (31). The initiation of this selective translational program could provide a more permissive cellular environment for secondary oncogenic driver mutations, resulting in a more robust transformative phenotype.
The RNase III endonuclease, Drosha, participates in several essential cellular processes, most notably, the processing of pre-rRNA and pri-miRNA intermediate species. Given the role of these small non-coding RNAs in development and disease, it is conceivable that the machinery responsible for their maturation must be tightly monitored. To date, very little is known about the mechanisms through which Drosha is regulated. Here, we presented evidence that Drosha expression is controlled at the level of translation in an ARF-dependent manner. Although we have demonstrated that existing Drosha mRNAs are excluded from polysomes in the presence of ARF, further studies will be needed to provide insight into the precise mechanism through which ARF antagonizes Drosha transcript association with polyribosomes. Given Drosha’s ability to promote rRNA processing and increase cytosolic ribosome availability, this could represent a feed-forward loop. Heighted Drosha levels would stimulate ribosome production that in turn would enhance Drosha mRNA translation. However, this over-simplified loop does not take into account any selective translation. Rather, translational selectivity could occur through miRNA-directed translation. Here, we show that loss of Arf and the concomitant increase in Drosha levels impacts the miRNA profile of these cells, albeit not globally. This is in agreement with previous findings that the Drosha-containing microprocessor does not serve as a rate-limiting factor in miRNA processing (32). It is possible that one or more of the 23 miRNAs that were repressed upon ARF knockdown might target the Drosha transcript. This could account for the lack of Drosha translational repression under these conditions.
Preceding studies have yielded conflicting results regarding Drosha’s role in cell growth, proliferation, and transformation (22-26). Alterations in RNASEN (gene encoding mouse and human Drosha) copy number have been correlated with specific types of cancer, but there is no clear trend that exclusively establishes this RNA processing enzyme as a tumor suppressor or oncogene. Our findings indicate that in Arf-deficient primary mouse fibroblasts, Drosha plays an important role in mediating enhanced cell growth and proliferation. Drosha knockdown impaired rRNA processing, ribosome biogenesis and reduced the proliferation rate of cells while activating an apoptotic cell death response. Furthermore, we uncovered a critical role for elevated Drosha expression in maintaining RasV12-induced cellular transformation. Given the well-established association between increased translation rates, proliferation, and neoplastic transformation, perhaps Drosha makes a required cellular process, such as ribosome biogenesis, more efficient to accommodate the overwhelming protein synthesis demands following exposure to oncogenic stimuli. In this setting, oncogenic Ras requires the elevated ribosome biogenesis that heightened Drosha provides. In the absence of Arf and presence of activated RasV12, loss of Drosha expression acts as a synthetic lethal event triggering apoptosis. Thus, we have established a novel regulatory link between Drosha and ARF that not only defines the growth properties of these two proteins but also highlights new mechanisms through which they function to establish a pro- or anti-tumor regimen.
Materials/Subjects and Methods
Cell culture and reagents
Low passage (P3-P5) primary B6/129 WT and Arf −/− MEFs were isolated and cultured as previously described (10). For western blot analysis, membranes were probed with the following antibodies: rabbit anti-Drosha (ab12286) (Abcam, Cambridge, MA); rat anti-p19ARF (sc-32748), rabbit anti-p16INK4a (sc1207), mouse anti-p21 (sc6246), rabbit anti-Ras (sc520), and mouse anti-γ-tubulin (sc17787) (all from Santa Cruz Biotechnology, Santa Cruz, CA); rabbit anti-cleaved PARP (#9544) (Cell Signaling, Danvers, MA). For apoptosis assay, etoposide (Sigma, St. Louis, MO) was used at a final concentration of 50 μM.
Plasmids and viral production
For Drosha overexpression, the Drosha ORF was first PCR amplified using cDNA derived from MEF total RNA. The following primers were used: forward 5’-GACGATATCGGACGC ATCGAGATGCAAGG-3’, reverse 5’-GACGATATCCCACTCCTGCCCTCGTTTAC-3’. The Drosha ORF was then cloned into the pBabe-puro retroviral backbone. The EcoRV sites flanking the ORF allowed for blunt end ligation into the SnaBI site of pBabe-puro. pBabe-puro-H-RasV12 was a generous gift from Martine Roussel (St. Jude Children's Research Hospital, Memphis, TN) and pBabe-HA-ARF has been previously described (10). Retroviral production was performed as previously described (37) and collected retrovirus was used to infect MEFs in the presence of 10 μg/ml polybrene.
pLKO.1-puro constructs obtained from the Genome Institute at Washington University were used for RNA interference (RNAi) against Drosha and Arf. Sequences for the short hairpin RNAs are 5′-CCTGGACAAGTTGATAGGATA-3′ for Drosha (here named shDrosha 1), 5′-CTTCGAGAAGTCTGGCTCAAT-3′ also for Drosha (here named shDrosha 2), and 5′-TCTACTGGTCTGCCTAAAGGT-3′ for the luciferase control. pLKO-puro-shARF has been previously described (38). For lentiviral production, 5 × 106 293T cells were cotransfected with pCMV-VSV-G, pCMVΔR8.2, and pLKO.1-puro constructs using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Forty-eight hours post-transfection, viral supernatants were collected and pooled.
Quantitative RT-PCR
Total RNA and RNA from monosome, disome, and polysome fractions were extracted using RNA-Solv (Omega Bio-tek, Norcross, GA). For polysome profiling experiments, first-strand cDNA synthesis and real-time PCR were done as previously described (39). To amplify Drosha and GAPDH mRNAs, the following primers were used: Drosha forward, 5’-CGATGGCCAATTGTTTTGAAGCC-3’; Drosha reverse, 5’-CGGACGTGAGTGAAGATCACTC-3’; GAPDH forward, 5’-GCTGGGGCTCACCTGAAGG-3’; and GAPDH reverse, 5’-GGATGACCTTGCCCACAGC-3’ Real-time PCR was performed on an iCycler apparatus (Bio-Rad, Hercules, CA) using iQ Sybr Green Supermix (Bio-Rad, Hercules, CA). Fold change was calculated using the ΔΔCT method (40). Drosha and Gapdh transcripts per cell were calculated by extrapolation from a standard curve generated from serial dilutions of a known quantity of subcloned cDNA.
RNA and protein stability
To assess mRNA stability, MEFs were treated with 4 μg/ml actinomycin D (Sigma, St. Louis, MO), harvested at 0, 2, 4, and 8 hours post-treatment, and subjected to RNA isolation, cDNA synthesis reaction, and quantitative reverse transcription-PCR (qRT-PCR) analysis using the real-time primers for Drosha and GAPDH listed above. A second pair of Drosha primers was also used to ensure specificity; Drosha forward 5’-GATTGCCAACATGCTCCAGTGG-3’; Drosha reverse, 5’-GCTAGGAGGTGGCGAAGTTTCAC-3’. To examine protein stability, cells were treated with 25 μg/ml cycloheximide (Sigma, St. Louis, MO), harvested at 0, 2, 4, 6, and 8 hours post-treatment and subjected to Western blot analysis. For proteosomal inhibition experiment, MEFs were treated with DMSO (mock) or 40 μM MG-132 (Sigma, St. Louis, MO) for eight hours and then subjected to Western blot analysis.
Ribosome fractionation
WT and Arf −/− MEFs were treated with cycloheximide (10 μg/ml) for 5 minutes prior to harvesting to stall ribosomes on mRNAs. Cells were counted and cytosolic extracts prepared from 3 × 106 cells were subjected to ribosome fractionation as previously described (41,38) using a density gradient system (Teledyne ISCO, Lincoln, NE). Drosha and Gapdh mRNA distribution per fraction was calculated as a percentage of the total number of transcripts in all collected fractions.
Ribosomal RNA Processing
Equal numbers of infected WT or Arf −/− MEFs were grown in methionine-free starvation media containing 10% dialyzed FBS for 15 minutes. Cells were treated with 50 μCi/mL [methyl-3H]-methionine for 30 minutes and chased in complete media spiked with cold methionine (10 μmol/L) for the indicated times. Extracted RNA was separated on agarose-formaldehyde gels and transferred to a Hybond XL membrane (GE Healthcare, Piscataway, NJ). The membrane was cross-linked and sprayed with En3Hance (Perkin-Elmer, Waltham, MA) prior to autoradiography. Band intensities were quantitated using ImageQuant TL (GE Healthcare, Piscataway, NJ).
Screening of miRNA Expression
WT MEFs were infected with a control- or ARF-specific shRNA for 72 hours prior to extraction of total RNA using the miRNeasy kit (Qiagen, Courtaboeuf, France). TaqMan Megaplex RT was performed using 750ng of input RNA according to the manufacturer's protocol, and real-time PCR was run on the 384-well micro-fluidic TaqMan miRNA Array Card A using the Applied Biosystems 7900HT Real-Time PCR System (Applied Biosystems now Life Technologies; Carlsbad, CA). Data were processed and exported with Applied Biosystems SDSv2.2.1 software. Once again, relative quantification was performed using the ΔΔCt method, using U6 as a reference.
Foci formation and proliferation assays
For cell proliferation assays, infected Arf −/− MEFs were plated in triplicate at 5 × 104 cells per well. Every 24 h thereafter, cells were harvested and counted using a hemacytometer. Cells were grown for 14 days in complete medium and then were fixed with 100% methanol and stained for 30 min with 50% Giemsa. Colonies were quantified using ImageQuant TL (GE Healthcare, Piscataway, NJ)
Cell cycle distribution analysis
Infected Arf −/− MEFs (1×106) were washed once in PBS (1% FBS) and then fixed in ice cold 100% ethanol. DNA was stained with propidium iodide (20 μg/mL; Sigma, St. Louis, MO) in the presence of 1 mg/mL RNase A (Sigma, St. Louis, MO). Cells were analyzed for DNA content by flow cytometry using a FACSCalibur instrument (Becton Dickinson Instruments). The data were analyzed using CELLQUEST analysis software (Becton Dickinson).
Apoptosis analysis
Equal numbers of infected Arf −/− MEFs were stained with FITC-annexin V and propidium iodide using the Vybrant Apoptosis Assay Kit #3 (V13242; Molecular Probes/Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. For a positive control, cells were treated with etoposide (50μM) for 16 hours. Cells were analyzed by flow cytometry as described above.
Soft agar
Arf −/− MEFs were first infected with RasV12 or pBabe empty vector and then selected in puromycin (2μg/ml). Following drug selection, the cells were infected with pLKO1.1 luciferase or pLKO1.1 shDrosha. For soft-agar colony formation, 1 × 104 infected cells were seeded in triplicate on 60-mm dishes and cells were relayered with soft agar on a weekly basis. After 3 weeks, plates were examined under a microscope and colonies were counted.
Supplementary Material
Acknowledgements
The authors thank the members of the Weber lab for their technical input and suggestions. The Children’s Discovery Institute and the Genome Institute at Washington University provided lentiviral knockdown constructs. Grants from the National Institutes of Health (R01 CA120436) and Department of Defense Era of Hope Scholar Award (BC075004) to J.D.W supported this work.
Footnotes
Conflict of Interest
The authors declare no conflict of interest
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
References
- 1.Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995;83:993–1000. doi: 10.1016/0092-8674(95)90214-7. [DOI] [PubMed] [Google Scholar]
- 2.Kamijo T, Weber JD, Zambetti G, Zindy F, Roussel MF, Sherr CJ. Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. Proc Natl Acad Sci U S A. 1998;95:8292–8297. doi: 10.1073/pnas.95.14.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weber JD, Taylor LJ, Roussel MF, Sherr CJ, Bar-Sagi D. Nucleolar Arf sequesters Mdm2 and activates p53. Nat Cell Biol. 1999;1:20–26. doi: 10.1038/8991. [DOI] [PubMed] [Google Scholar]
- 4.Pomerantz J, Schreiber-Agus N, Liegeois NJ, Silverman A, Alland L, Chin L, et al. The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2's inhibition of p53. Cell. 1998;92:713–723. doi: 10.1016/s0092-8674(00)81400-2. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92:725–734. doi: 10.1016/s0092-8674(00)81401-4. [DOI] [PubMed] [Google Scholar]
- 6.Sherr CJ. Divorcing ARF and p53: an unsettled case. Nat Rev Cancer. 2006;6:663–673. doi: 10.1038/nrc1954. [DOI] [PubMed] [Google Scholar]
- 7.Sugimoto M, Kuo ML, Roussel MF, Sherr CJ. Nucleolar Arf tumor suppressor inhibits ribosomal RNA processing. Mol Cell. 2003;11:415–424. doi: 10.1016/s1097-2765(03)00057-1. [DOI] [PubMed] [Google Scholar]
- 8.Bertwistle D, Sugimoto M, Sherr CJ. Physical and functional interactions of the Arf tumor suppressor protein with nucleophosmin/B23. Mol Cell Biol. 2004;24:985–996. doi: 10.1128/MCB.24.3.985-996.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Apicelli AJ, Maggi LB, Jr., Hirbe AC, Miceli AP, Olanich ME, Schulte-Winkeler CL, et al. A non-tumor suppressor role for basal p19ARF in maintaining nucleolar structure and function. Mol Cell Biol. 2008;28:1068–1080. doi: 10.1128/MCB.00484-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brady SN, Yu Y, Maggi LB, Jr., Weber JD. ARF impedes NPM/B23 shuttling in an Mdm2-sensitive tumor suppressor pathway. Mol Cell Biol. 2004;24:9327–9338. doi: 10.1128/MCB.24.21.9327-9338.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saporita AJ, Chang HC, Winkeler CL, Apicelli AJ, Kladney RD, Wang J, et al. RNA helicase DDX5 is a p53-independent target of ARF that participates in ribosome biogenesis. Cancer Res. 2011;71:6708–6717. doi: 10.1158/0008-5472.CAN-11-1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu H, Xu H, Miraglia LJ, Crooke ST. Human RNase III is a 160-kDa protein involved in preribosomal RNA processing. J Biol Chem. 2000;275:36957–36965. doi: 10.1074/jbc.M005494200. [DOI] [PubMed] [Google Scholar]
- 13.Denli AM, Tops BB, Plasterk RH, Ketting RF, Hannon GJ. Processing of primary microRNAs by the Microprocessor complex. Nature. 2004;432:231–235. doi: 10.1038/nature03049. [DOI] [PubMed] [Google Scholar]
- 14.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 15.Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, et al. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- 16.Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, et al. MicroRNA genes are transcribed by RNA polymerase II. Embo J. 2004;23:4051–4060. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, et al. The Microprocessor complex mediates the genesis of microRNAs. Nature. 2004;432:235–240. doi: 10.1038/nature03120. [DOI] [PubMed] [Google Scholar]
- 18.Shiohama A, Sasaki T, Noda S, Minoshima S, Shimizu N. Nucleolar localization of DGCR8 and identification of eleven DGCR8-associated proteins. Exp Cell Res. 2007;313:4196–4207. doi: 10.1016/j.yexcr.2007.07.020. [DOI] [PubMed] [Google Scholar]
- 19.Han J, Lee Y, Yeom KH, Kim YK, Jin H, Kim VN. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004;18:3016–3027. doi: 10.1101/gad.1262504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fukuda T, Yamagata K, Fujiyama S, Matsumoto T, Koshida I, Yoshimura K, et al. DEAD-box RNA helicase subunits of the Drosha complex are required for processing of rRNA and a subset of microRNAs. Nat Cell Biol. 2007;9:604–611. doi: 10.1038/ncb1577. [DOI] [PubMed] [Google Scholar]
- 21.Kloosterman WP, Plasterk RH. The diverse functions of microRNAs in animal development and disease. Dev Cell. 2006;11:441–450. doi: 10.1016/j.devcel.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 22.Merritt WM, Lin YG, Han LY, Kamat AA, Spannuth WA, Schmandt R, et al. Dicer, Drosha, and outcomes in patients with ovarian cancer. N Engl J Med. 2008;359:2641–2650. doi: 10.1056/NEJMoa0803785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin RJ, Lin YC, Chen J, Kuo HH, Chen YY, Diccianni MB, et al. microRNA signature and expression of Dicer and Drosha can predict prognosis and delineate risk groups in neuroblastoma. Cancer Res. 2010;70:7841–7850. doi: 10.1158/0008-5472.CAN-10-0970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sand M, Gambichler T, Skrygan M, Sand D, Scola N, Altmeyer P, et al. Expression levels of the microRNA processing enzymes Drosha and dicer in epithelial skin cancer. Cancer Invest. 2010;28:649–653. doi: 10.3109/07357901003630918. [DOI] [PubMed] [Google Scholar]
- 25.Sugito N, Ishiguro H, Kuwabara Y, Kimura M, Mitsui A, Kurehara H, et al. RNASEN regulates cell proliferation and affects survival in esophageal cancer patients. Clin Cancer Res. 2006;12:7322–7328. doi: 10.1158/1078-0432.CCR-06-0515. [DOI] [PubMed] [Google Scholar]
- 26.Muralidhar B, Winder D, Murray M, Palmer R, Barbosa-Morais N, Saini H, et al. Functional evidence that Drosha overexpression in cervical squamous cell carcinoma affects cell phenotype and microRNA profiles. J Pathol. 2011;224:496–507. doi: 10.1002/path.2898. [DOI] [PubMed] [Google Scholar]
- 27.Han J, Pedersen JS, Kwon SC, Belair CD, Kim YK, Yeom KH, et al. Posttranscriptional crossregulation between Drosha and DGCR8. Cell. 2009;136:75–84. doi: 10.1016/j.cell.2008.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lessard F, Morin F, Ivanchuk S, Langlois F, Stefanovsky V, Rutka J, et al. The ARF tumor suppressor controls ribosome biogenesis by regulating the RNA polymerase I transcription factor TTF-I. Mol Cell. 2010;38:539–550. doi: 10.1016/j.molcel.2010.03.015. [DOI] [PubMed] [Google Scholar]
- 29.Ayrault O, Andrique L, Larsen CJ, Seite P. Human Arf tumor suppressor specifically interacts with chromatin containing the promoter of rRNA genes. Oncogene. 2004;23:8097–8104. doi: 10.1038/sj.onc.1207968. [DOI] [PubMed] [Google Scholar]
- 30.Itahana K, Bhat KP, Jin A, Itahana Y, Hawke D, Kobayashi R, et al. Tumor suppressor ARF degrades B23, a nucleolar protein involved in ribosome biogenesis and cell proliferation. Mol Cell. 2003;12:1151–1164. doi: 10.1016/s1097-2765(03)00431-3. [DOI] [PubMed] [Google Scholar]
- 31.Kawagishi H, Nakamura H, Maruyama M, Mizutani S, Sugimoto K, Takagi M, et al. ARF suppresses tumor angiogenesis through translational control of VEGFA mRNA. Cancer Res. 2010;70:4749–4758. doi: 10.1158/0008-5472.CAN-10-0368. [DOI] [PubMed] [Google Scholar]
- 32.Diederichs S, Haber DA. Dual role for argonautes in microRNA processing and posttranscriptional regulation of microRNA expression. Cell. 2007;131:1097–1108. doi: 10.1016/j.cell.2007.10.032. [DOI] [PubMed] [Google Scholar]
- 33.Oskowitz AZ, Penfornis P, Tucker A, Prockop DJ, Pochampally R. Drosha regulates hMSCs cell cycle progression through a miRNA independent mechanism. Int J Biochem Cell Biol. 2011;43:1563–1572. doi: 10.1016/j.biocel.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Palmero I, Pantoja C, Serrano M. p19ARF links the tumour suppressor p53 to Ras. Nature. 1998;395:125–126. doi: 10.1038/25870. [DOI] [PubMed] [Google Scholar]
- 35.Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, et al. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997;91:649–659. doi: 10.1016/s0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- 36.Maggi LB, Jr., Kuchenruether M, Dadey DY, Schwope RM, Grisendi S, Townsend RR, et al. Nucleophosmin serves as a rate-limiting nuclear export chaperone for the Mammalian ribosome. Mol Cell Biol. 2008;28:7050–7065. doi: 10.1128/MCB.01548-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roussel MF, Theodoras AM, Pagano M, Sherr CJ. Rescue of defective mitogenic signaling by D-type cyclins. Proc Natl Acad Sci U S A. 1995;92:6837–6841. doi: 10.1073/pnas.92.15.6837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miceli AP, Saporita AJ, Weber JD. Hypergrowth mTORC1 signals translationally activate the ARF tumor suppressor checkpoint. Mol Cell Biol. 2012;32:348–364. doi: 10.1128/MCB.06030-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Olanich ME, Moss BL, Piwnica-Worms D, Townsend RR, Weber JD. Identification of FUSE-binding protein 1 as a regulatory mRNA-binding protein that represses nucleophosmin translation. Oncogene. 2010;30:77–86. doi: 10.1038/onc.2010.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 41.Strezoska Z, Pestov DG, Lau LF. Bop1 is a mouse WD40 repeat nucleolar protein involved in 28S and 5. 8S RRNA processing and 60S ribosome biogenesis. Mol Cell Biol. 2000;20:5516–5528. doi: 10.1128/mcb.20.15.5516-5528.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.