

Abstract
To completely avoid ice crystal formation and thus get a higher survival rate, vitrification methods have been commonly used for cryopreservation of oocytes and embryos. However, currently used vitrification methods for oocytes and embryos are not suitable for the cryopreservation of preantral follicles (PFs). In the present study, stainless steel mesh was fabricated into mini mesh cups to vitrify isolated PFs. Moreover, isolated follicles were encapsulated and then subjected to vitreous cryopreservation to facilitate in vitro culture/maturation of follicles after warming. The results showed that the percentages of viable follicles did not differ significantly between the vitrification group and fresh group soon after warming (81.25% vs. 85.29%, P>0.05) and after a 7-day culture period (77.78% vs. 83.33%, P>0.05). No difference in mean follicular diameter was observed between cryopreserved and fresh follicles when cultured in vitro. Transmission electron microscopic analysis revealed that vitreous cryopreservation could maintain the ultrastructure of follicles in alginate beads. In conclusion, the present vitrification method could efficiently cryopreserve isolated human ovarian follicles encapsulated by calcium alginate, which could be put into immediate use (in vitro culture/ maturation) after warming. However, more follicles and some detailed biochemical analyses are required to further investigate the effects of vitrification on the long-term growth of human encapsulated PFs.
Keywords: Alginate, Cryopreservation, In vitro culture, Preantral follicle, Vitrification
Advances in the treatment of female cancers have contributed to increased patient survival rates but can cause infertility in young women. Therefore, preserving fertility before cancer treatment is necessary for many of these patients [1, 2]. Although oocyte/embryo freezing can be offered to patients [3,4,5], some patients do not get the chance to have mature oocytes/embryos cryopreserved for later use (in vitro fertilization/ embryo transfer), as radiotherapy and chemotherapy cannot always be delayed to get mature oocytes through ovarian stimulation [6]. Therefore, the freezing of a relatively large quantity of immature oocytes enclosed in preantral follicles (PFs) has emerged as a promising alternative to safeguard fertility for cancer patients in recent years [7, 8].
PFs can be frozen as isolated follicles or in situ (ovarian tissue cryopreservation) [7,8,9]. Cryopreservation of isolated ovarian follicles has been attempted in several species [10,11,12]. The most successful experiments, carried out in mice, showed that it is possible to obtain normal offspring after isolated follicle cryopreservation and in vitro culture [13]. However, only a few studies have reported the cryopreservation of isolated PFs from large mammals and humans because of the relatively large size, fragile architecture, and difficulties of in vitro culture.
In recent years, there has been great progress with respect to the in vitro culture of isolated ovarian follicles. Follicle encapsulation within alginate hydrogels, a common tissue engineering scaffold, mimics the ovary by providing the appropriate three-dimensional (3D) context while supporting somatic cell and egg interactions to optimize oocyte development [14,15,16,17,18]. If a 3D scaffold is provided to the freshly isolated human PF and the encapsulated follicles are cryopreserved, the isolated ovarian follicles could be ready for use at any time, i.e., put into direct culture or transplanted after thawing without a long and complicated process of follicle preparation.
Slow-freezing methods are most commonly used for the cryopreservation of cells and tissues [19]. However, it is extremely difficult to preserve the intactness and integrity of cells and scaffolds, as the formation of ice crystals would destroy the complicated 3D constructs [20]. Vitrification is defined as glass-like solidification and/or complete avoidance of ice crystal formation during cooling and warming [21,22,23]. Therefore, cryopreservation of 3D constructs could be more efficient if the cryopreservation solutions enclosed in constructs and cells were ice-free during the entire procedure [24,25,26].
In the present study, we investigated the prospect of vitreous cryopreservation of isolated human PFs encapsulated with calcium alginate. The viability, growth, and ultrastructure of the ovarian follicles were compared with those of fresh controls that were encapsulated but without cryopreservation.
Materials and Methods
Collection of human ovarian tissue
This study was approved by the Ethics Committee of The First Affiliated Hospital of Sun Yat-Sen University. Ovarian tissues were obtained from 27 women aged 22 to 38 years (28.52 ± 4.90), who had undergone laparoscopic surgery or laparotomy for nonovarian benign gynecological disease, such as myomas and tubal ligation. Ovarian tissue (about 5×5×3 mm to 10×10×3 mm) obtained from each woman was placed into a sterile glass cup incubated with HEPES-buffered modified Eagle's medium (HEPES-MEM, Lonza Walkersville, Walkersville, MD, USA) and transported to the laboratory on ice. All patients recruited into the study provided written informed consent.
PFs isolation
Human PFs were isolated from ovarian tissue according to the protocol of Dolmans et al. [27]. Briefly, the ovarian tissue was cut into 1×1×1-mm fragments using a tissue chopper (McIlwain Tissue Chopper, The Mickle Laboratory Engineering, Guildford, UK) adjusted to yield 1-mm serial sections. The tissue fragments were put in PBS solution containing 0.07 mg/ml Liberase enzyme (Roche, Indianapolis, IN, USA) and 20 U/ml DNA enzyme (Sigma-Aldrich, Carlsbad, CA, USA) in 14-ml test tubes and incubated in a water bath at 37 C for 1 h with gentle agitation. During incubation, the ovarian tissue was blown and aspirated by Pasteur pipettes every 15 min to facilitate the digestion of tissue. Digestion was terminated by the addition of an equal volume of HEPES-MEM at 4 C supplemented with 10% human serum albumin (HSA; SAGE In Vitro Fertilization) and centrifuged at 50 g for 10 min at 4 C. The supernatant was discarded, and the pellet was transferred to culture dishes for investigation of PFs under a stereomicroscope (Leica, Wetzlar, Germany). Morphologically normal PFs, with two or three layers of granulosa cells and centrally located spherical oocytes, were washed three times in Dulbecco's phosphate-buffered saline (D-PBS) supplemented with 10% HSA and used in the present study.
Calcium alginate embedding
A 1.5% (w/v) solution of sodium alginate (55–65% guluronic acid, FMC BioPolymer, Philadelphia, PA, USA) in PBS was prepared and autoclaved [28]. The isolated follicles were transferred with a micropipette to droplets (2 μl) of alginate solution. To form beads, the droplets were slowly released into a small beaker containing a solution of CaCl2 (0.1 M). The droplets immediately gelled to form beads. Beads containing individual follicles were then immediately removed from the beaker using glass pipettes 2 min after adding CaCl2 and then washed three times in culture medium.
Experimental design
The encapsulated follicles from each patient were randomly distributed into two groups. In group 1, the encapsulated follicles were vitrified, stored in liquid nitrogen for 4 h and then cultured in vitro for 2 h or 7 days after warming (vitrification group). In group 2, the follicles were placed directly into culture without cryopreservation. In both groups, we assessed the diameter and survival rate of part of the follicles after 2 h of in vitro culture, while other follicles were allowed to grow in vitro for 7 days. The diameter, viability and ultrastructure of PFs and the proliferation of granulosa cells were compared between the two groups after 7 days of in vitro culture.
Vitrification and warming procedures
A small piece of stainless steel mesh (mesh size, 50 µm; Zhenxing Hardware Sifting Screen Factory, Guangzhou, China) was carefully molded into a cup shap (d = 0.6–1 mm; h = 0.2–0.4 mm). This homemade cryo-container was then sterilized and ready for use [29]. The encapsulated preantral follicles to be vitrified were placed into the cup-shaped stainless steel mesh (Fig. 1), immersed in an equilibration solution consisting of 10% ethylene glycol (EG; Sigma-Aldrich) in Dulbecco's phosphate-buffered saline (D-PBS) with 10% HSA for 3 min, transferred to 25% EG in D-PBS for 3 min (vitrification solution 1, VS1), and then immersed in a vitrification solution consisting of 40% EG (v/v), 0.6 mol/l sucrose, and 20% HSA in D-PBS (VS2) for 3 min. After the final step, the stainless steel mesh loaded with encapsulated PFs was submerged immediately into liquid nitrogen. The vitrification procedures were carried out at room temperature (23–25 C).
Fig. 1.
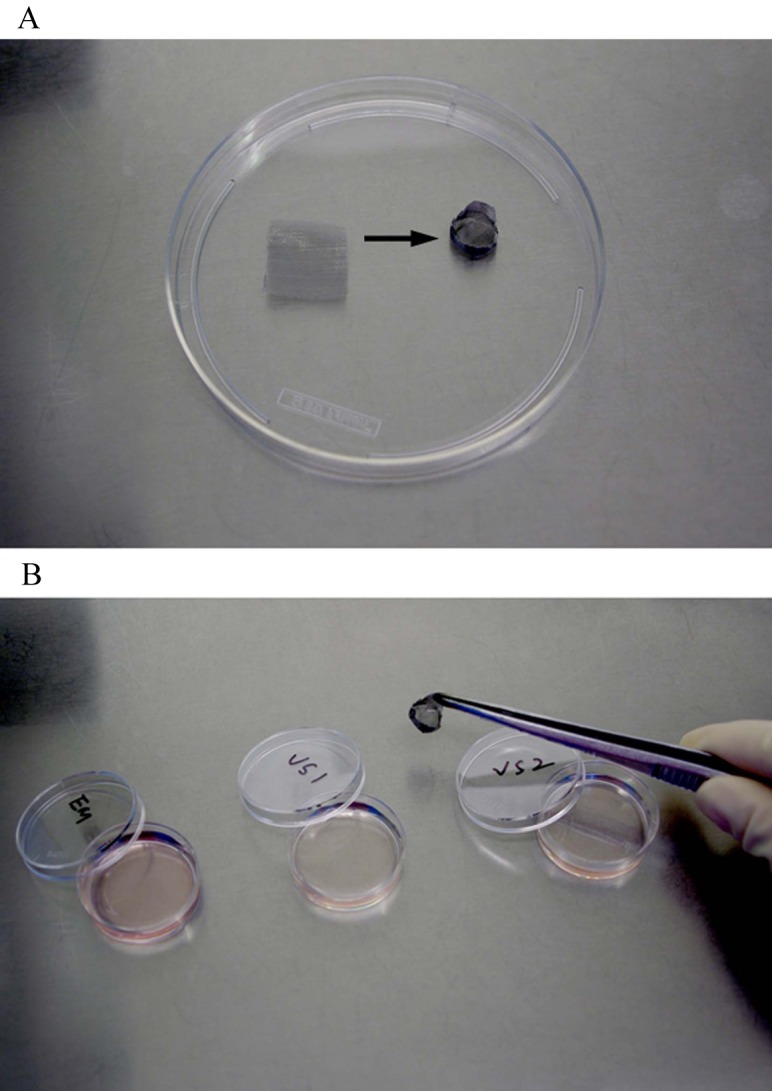
Fabrication of the vitrification carrier and the manipulation procedures for encapsulated human preantral follicles. A: A small piece of stainless-steel mesh (mesh size, 50 µm) was molded into a cup shape. B: Encapsulated human follicles loaded in the cup-shaped stainless-steel mesh could be easily transferred from one vitrification medium to another with a pair of tweezers.
After 4 h of cryopreservation in liquid nitrogen, the cryopreserved follicles were taken out of the liquid nitrogen box and warmed immediately. The five-step cryoprotectant dilution method was performed to warm the encapsulated PFs. Briefly, the mesh cup loaded with encapsulated preantral follicles was placed in a dish containing 10% HSA and 1 mol/l sucrose in D-PBS for 1 min at 37 C. After incubation, the preantral follicles were transferred sequentially to 0.75 mol/l, 0.5 mol/l, 0.2 mol/l, and 0.1 mol/l sucrose at 23–25 C and then washed twice in D-PBS. Finally, the follicles were picked out of the mesh cup and equilibrated for 15 min in the culture medium at 37 C in a 5% CO2 environment before culture. From this point on, these encapsulated follicles were handled the same way as the control nonfrozen encapsulated follicles.
In vitro culture of encapsulated follicles
The follicles were grown individually in 96-well plates (1 follicle per well) in 100 μl culture medium at 37 C in a humidified atmosphere of 5% CO2 [30]. The culture medium consisted of alpha MEM (Sigma) supplemented with 100 μg/ml penicillin, 100 μg/ml streptomycin, 0.25 μg/ml amphotericin B, 10% HSA, ITS (insulin 5 μg /ml, transferrin 5.5 μg/ml, selenium 5 ng/ml), 0.23 mM pyruvate, 2 mM glutamine and 2 mM hypoxanthine). The medium was exchanged every 2 days.
Follicles were cultured at 37 C in 5% CO2 for 7 days. Every 2 days, half of the media volume was exchanged, and the follicles were examined for survival and size measurements after 2 h and 7 days of in vitro culture. Two diameters were measured for each follicle, and selected images were captured. The integrated measuring tools in the ImageJ software were used in the present study (Java-based image processing program developed at the National Institutes of Health) [31].
Assessment of follicle viability
The viability of morphologically normal encapsulated follicles was analyzed using live (calcein-AM) and dead (ethidium homodimer-1) markers. The vitrified/warmed and fresh follicles were transferred to PBS containing 2 μmol/l of calcein-AM and 5 μmol/l of ethidium homodimer-1 (Molecular Probes, Leiden, The Netherlands). They were incubated with the fluorescent dyes for 20 min at 37 C in the dark. Then the follicles were washed in PBS and observed under an inverted fluorescence microscope (Leica). The encapsulated follicles were classified into three categories depending on the percentage of dead granulosa cells and oocytes, as follows: live follicles (follicles with oocytes and all the granulosa cells viable), partially damaged follicles (follicles with viable oocytes and some dead granulosa cells) and dead follicles (follicles with dead oocytes or most of the granulosa cells dead; Fig. 2).
Fig. 2.
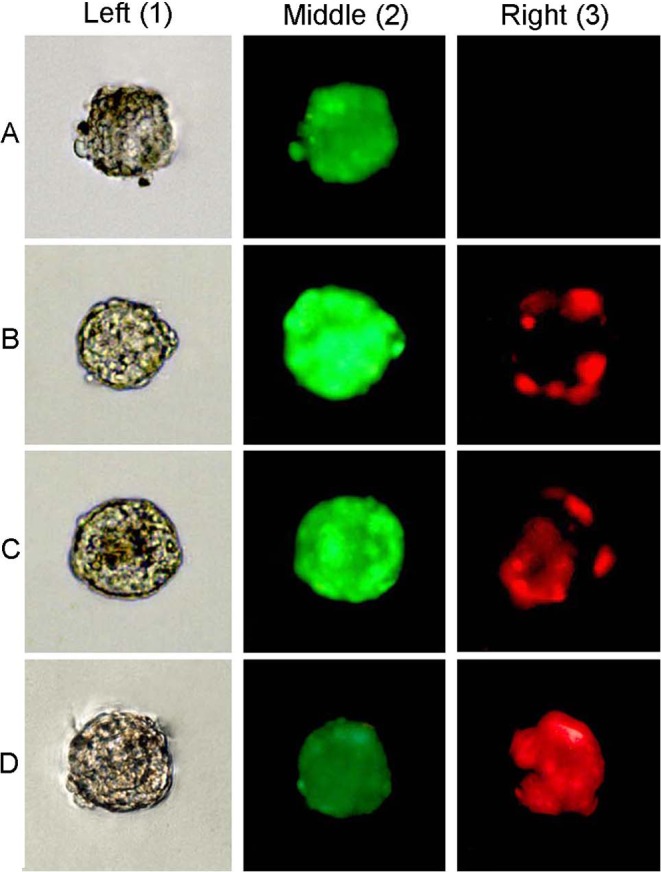
The viability classification of the isolated follicles. Encapsulated follicles were stained with calcein-AM, which indicated the viability of the cell cytoplasm, and with ethidium homodimer-1 to show nuclei of dead cells. The left column (1) shows a follicle in a bright field; the middle column (2) shows images obtained for the same follicle with a filter to visualize calcein-AM staining; and the right column (3) shows ethidium homodimer-1 staining. Follicles were classified into three categories depending on the viability of granulosa cells (GCs) and oocytes. Live follicles (A): follicles with the oocyte and all the GCs viable. Partially damaged follicles (B, C): follicles with the oocyte viable and <50% of dead GCs. Dead follicles (D): follicles with both the oocyte or >50% GCs dead, as photo D3 shows using the filter to visualize red fluorescence. 200×.
Ultrastructure
The ultrastructure of cryopreserved and fresh encapsulated follicles was assessed as described by Matos et al. [32, 33]. Briefly, 61 ovarian follicles embedded in calcium alginate were fixed in 1.5% glutaraldehyde in PBS solution containing 5% BSA (29 cryopreserved PFs and 32 fresh PFs). After fixation for 2 to 5 days at 4 C, the samples were rinsed in PBS, postfixed with 1% osmium tetroxide (Agar Scientific, Elektron Technology UK, Stansted, Essex, England) in PBS and rinsed again in PBS. The samples were dehydrated through increasing concentrations of ethanol, immersed in propylene oxide (solvent substitution), embedded in Epon 812 and sectioned using a Reichert-Jung Ultracut E ultramicrotome (Leica Mikrosysteme GmbH, Vienna, Austria). Ultrathin sections (60–80 nm) were cut with a diamond knife, mounted on copper grids and contrasted with saturated uranyl acetate followed by lead citrate. They were examined and photographed using Zeiss EM109 and Zeiss EM 10 electron microscopes at 80 kV.
The following ultrastructural alterations of the selected cell organelles and structures of encapsulated follicles were recorded: (1) reduced abundance of microvilli, (2) reduction of the rough endoplasmic reticulum, (3) existence of swollen mitochondria, with few or no crista, (4) changes of granulosa cells, (5) rupture of the oolemma, (6) fracture or delamination of the zona pellucida and (7) enlarged perivitelline space. We recorded the ultrastructural modifications one by one, calculated the proportions and compared them between the cryopreserved and fresh follicles groups respectively.
3H-thymidine incorporation capability of granulosa cells
After 7 days of in vitro culture, culture media were exchanged and replaced with media supplemented with 0.4-Ci methyl-3H-thymidine (PerkinElmer, Boston, MA, USA; 1 μci/100 μl) per well. After 16 h, 4 follicles in beads were collected for each replicate (3 replicates), washed twice with D-PBS, and then dissolved in 10 mM EDTA. Next, 3H-thymidine incorporation was assayed as described previously [34,35,36,37]. Nonspecific incorporation was determined using empty alginate gels.
Statistical analysis
The follicular viability was compared using the chi-square test. The diameters of the follicles were expressed as means ± SD and compared using the paired sample t-test. The counts per minute of 3H-thymidine incorporation in granulosa cells were converted by log, expressed as means ± SD and compared using the paired sample t-test. Differences were considered significant when P<0.05.
Results
Viability of encapsulated preantral follicles after vitrification/warming and in vitro culture
The majority of the human preantral follicles were intact after isolation and encapsulation, with a central oocyte and surrounding layers of granulosa cells. The damaged follicles or follicles with partially injured granulosa cells detached from the follicle, likely a result of the mechanical isolation procedure, were excluded from the study. The number of follicles isolated from each patient sample ranged from 0 to 16 (Table 1).
Table 1. Secondary follicle isolation from each patient.
| Patient | Age | Diagnosis | Methods of collection | VG (n) | FG (n) | Total (n) |
| Pat1 | 23 | Myomas | Laparotomy | 5 | 7 | 12 |
| Pat2 | 26 | Tubal ligation | Laparoscopic | 5 | 6 | 11 |
| Pat3 | 31 | Myomas | Laparotomy | 4 | 5 | 9 |
| Pat4 | 34 | Tubal ligation | Laparoscopic | 4 | 6 | 10 |
| Pat5 | 27 | Myomas | Laparotomy | 0 | 0 | 0 |
| Pat6 | 23 | Myomas | Laparotomy | 4 | 6 | 10 |
| Pat7 | 38 | Tubal ligation | Laparoscopic | 3 | 5 | 8 |
| Pat8 | 35 | Tubal ligation | Laparoscopic | 0 | 0 | 0 |
| Pat9 | 24 | Myomas | Laparotomy | 5 | 7 | 12 |
| Pat10 | 26 | Myomas | Laparotomy | 3 | 6 | 9 |
| Pat11 | 28 | Tubal ligation | Laparotomy | 0 | 0 | 0 |
| Pat12 | 34 | Tubal ligation | Laparoscopic | 6 | 5 | 11 |
| Pat13 | 22 | Myomas | Laparotomy | 7 | 6 | 13 |
| Pat14 | 23 | Myomas | Laparotomy | 0 | 0 | 0 |
| Pat15 | 25 | Tubal ligation | Laparoscopic | 7 | 6 | 13 |
| Pat16 | 33 | Myomas | Laparotomy | 3 | 3 | 6 |
| Pat17 | 29 | Tubal ligation | Laparoscopic | 6 | 5 | 11 |
| Pat18 | 26 | Myomas | Laparotomy | 5 | 5 | 10 |
| Pat19 | 24 | Myomas | Laparotomy | 9 | 5 | 14 |
| Pat20 | 28 | Tubal ligation | Laparoscopic | 0 | 0 | 0 |
| Pat21 | 27 | Myomas | Laparotomy | 7 | 6 | 13 |
| Pat22 | 28 | Tubal ligation | Laparoscopic | 0 | 0 | 0 |
| Pat23 | 23 | Myomas | Laparotomy | 5 | 5 | 10 |
| Pat24 | 34 | Tubal ligation | Laparoscopic | 6 | 7 | 13 |
| Pat25 | 38 | Myomas | Laparotomy | 0 | 0 | 0 |
| Pat26 | 27 | Myomas | Laparotomy | 8 | 8 | 16 |
| Pat27 | 34 | Tubal ligation | Laparoscopic | 4 | 6 | 10 |
VG, human preantral follicles subjected to vitreous cryopreservation after encapsulation by calcium alginate; FG, encapsulated follicles directly subjected to in vitro culture without cryopreservation.
A total of 144 encapsulated human preantral follicles were examined for their viability based on the live (calcein-AM) and dead (ethidium homodimer-1) markers (Fig. 2). The influence of the cryopreservation procedure on the viability of encapsulated follicles is demonstrated in Table 2. The vitrification group had a viability of 81.2% immediately after warming and 2 h of in vitro culture, which was not significantly different from that of the fresh group (85.3%, P>0.05). The percentage of viable follicles did not differ significantly between the vitrification and fresh groups after a 7-day culture period (77. 8 vs. 83.3%, P>0.05).
Table 2. Viability of encapsulated preantral follicles of the vitrification group after freezing/warming and in vitro culture compared with the fresh group without cryopreservation.
| 2 h of in vitro culture | 7 days of in vitro culture | |||
| VG | FG | VG | FG | |
| No. of encapsulated follicles | 32 | 34 | 36 | 42 |
| Live follicles | 26 | 29* | 28 | 35* |
| Partially damaged follicles | 5 | 4 | 6 | 4 |
| Dead follicles | 1 | 1 | 2 | 3 |
VG, vitrification group after 2 h or 7 days of in vitro culture; FG, fresh group after 2 h or 7 days of in vitro culture. * No significant differences between the vitrification and fresh groups after 2 h and 7 days of in vitro culture.
Follicle diameter and granulosa cell proliferation
The diameters and functions of vitrified and noncryopreserved encapsulated follicles were recorded over 7 days of continuous culture. Table 3 demonstrates that the diameter of encapsulated follicles from both groups increased after the continuous in vitro culture period. The mean follicle diameter of vitrified/warmed human preantral follicles in the study was 96.5 ± 8.3 µm at the onset of the culture period and 123.1 ± 10.5 µm after the 7-day culture period. Oocyte diameter also increased from 66.3 ± 1.5 µm to 94.7 ± 1.6 µm in the vitrification group. There was a significant increase in follicular and oocyte diameter after 7 days of in vitro culture in both groups. There was no significant difference in mean follicular diameter between the cryopreserved follicles and those cultured without cryopreservation at the beginning and end of the 7-day culture period.
Table 3. Comparison of diameter (µm) of encapsulated preantral follicles between the vitrification group and fresh group before and after in vitro culture.
| Group | No. of follicles | Before culture | After 7 days of in vitro culture | ||
| Diameter of follicle | Diameter of oocyte | Diameter of follicle | Diameter of oocyte | ||
| VG | 36 | 96.5 ± 8.3a | 66.3 ± 1.5b | 123.1 ± 10.5*,c | 94.7 ± 1.6*,d |
| FG | 42 | 93.2 ± 9.5e | 69.7 ± 1.8f | 131.5 ± 12.3*,g | 96.2 ± 12.3*,h |
VG, vitrification group after 7 days in vitro culture; FG, fresh group after 7 days in vitro culture. * Significantly higher than their initial size (P<0.05) respectively in VG and FG after 7 days of in vitro culture. There were no significant differences between the VGa,b and FGe,f groups before culture (P>0.05). There were also no significant differences between the VGc,d and FGg, h groups after in vitro culture for 7 days (P>0.05).
Moreover, vitrification/warming did not significantly affect the proliferation of granulosa cells. After the 7-day culture period, the mean counts per minute (converted by log) of 3H-thymidine incorporation in granulosa cells in the fresh group and vitrification group follicles were 3.4 ± 0.3 and 3.3 ± 0.5 cpm, respectively, with no significant difference found between the two groups (P>0.05).
Ultrastructure of encapsulated human preantral follicles after vitrification and warming
By transmission electron microscopy, most of the cryopreserved follicles appeared healthy looking, presenting normal ultrastructural features of human follicles immediately after warming (Fig. 3): intact nuclear and cellular membranes, normally arranged chromatin, abundant amount of rough endoplasmic reticulum and normal cristae and an electron-dense matrix in most mitochondria. Some minor ultrastructural alterations were observed, and these alterations were different from follicles after 7 days of in vitro culture. For example, Fig. 3A and 3C show an enlarged space between follicular cells and oocytes, which was observed mainly in the follicles immediately after warming. On the other hand, a vacuolated cytoplasmic organelle in the oocyte cytoplasm was only observed after 7 days of in vitro culture (Fig. 3F) in both cryopreserved and fresh follicles, which might be related to the nonoptimal in vitro culture conditions.
Fig. 3.

Transmission electron microscopic assessment of vitrified human encapsulated follicles after 2 h (A–D) and 7 days (E–J) of in vitro culture. A continuous layer of flattened/irregular cuboid follicular cells surrounds the oocyte. The membrane of the nucleus is intact and surrounded by a perinuclear cluster of cytoplasmic organelles including mitochondria, electron-dense lipid bodies and endoplasmic reticulum (ER). GC, granulosa cells; M, mitochondria; NM, nuclear membrane; BM, base membrane; MV, microvilli.
After 7 days of in vitro culture, a regularly structured zona pellucida was observed at the oocyte-follicular cell interface in both groups, and some interdigitations between oocyte microvilli and granulosa cell prolongations were also observed (Fig. 3E, H). However, more ultrastructural alterations were detected after 7 days of in vitro culture than in follicles immediately after warming. A relatively higher trend of ultrastructure alterations was observed than in the corresponding fresh follicles after 7 days of in vitro culture, although no statistical difference was observed (Fig. 4).
Fig. 4.

Proportions of ultrastructural alterations of follicles in the vitrification Group (VG) and fresh Group (FG) after 7 days of in vitro culture. The proportions of alterations (change of microvilli, change of mitochondria cristae in the oocyte, reduced numbers of ER, rupture of the oolemma, change of the homogenous zona pellucida (ZP), change of the perivitelline space (PVS), change of the GC) in follicles were analyzed in VG and FG after 7 days of in vitro culture. No significant difference was found between the two groups.
Discussion
Compared with isolated PFs, ovarian tissue is a complex structure that needs a longer period of exposure to the cryoprotectants for vitrification. However, to decrease the toxic effects of a high concentration of cryoprotectant on cells and tissues, equilibration with the vitrification medium must be strictly controlled (for example, 1 min in vitrification medium and less than 30 sec for being loaded on cryocontainers for oocytes and embryos). Inadequate permeation of vitrification solutions might affect the number of normal follicles that survive [38,39,40]. Therefore, it is logical to speculate that vitreous cryopreservation of isolated, individual PFs might be more effective than vitrification of small sections of ovarian tissues.
Quickly and efficiently transferring embryos/cell constructs among different vitrification solutions to get them adequately permeated and minimizing the time for loading them on/into cryocontainers are critical for successful vitrification, especially for the cryopreservation of tens of encapsulated PFs, and this is closely related to the type of cryocontainer used and the efficiency of manipulation. In the present study, small pieces of stainless steel mesh were carefully molded into a cup-shaped container, and PFs were loaded into the cryocontainer in the first procedure and kept in it during the whole process of vitrification/warming. It was thus not necessary to use Pasteur pipettes to manipulate PFs among vitrification/warming solutions, and the loading procedure was also omitted. Therefore, the mesh cup behaved here not just as a cryocontainer itself but also as a transfer vehicle during the vitrification/warming process, which makes this vitrification method easier to carry out than currently used vitrification methods.
Encapsulation would reduce the cooling and warming rates a little because of the slightly increased volume of encapsulated follicles compared with “naked” follicles without encapsulation. However, encapsulation of follicles could theoretically protect fragile human follicles from some mechanical damage when they are manipulated during vitrification/warming and in vitro culture. Moreover, storage of encapsulated (pre-cultured) PFs could solve the problems related to labor, required techniques and time constraints when cryopreserved follicles are put into clinical use. In the present study, the percentage of viable follicles did not differ significantly between the vitrification and fresh groups soon after warming and after a 7-day culture period. No difference in mean follicle diameter was observed when they were cultured in vitro. These results demonstrated the effectiveness of the present vitrification method. However, transmission electron microscopic analysis revealed a slightly higher trend of ultrastructure changes in the vitrification group than in the fresh group. More follicles are required to further investigate any possible damage resulting from vitrification in encapsulated follicles. On the other hand, scaffold integrity is a primary concern for the preservation of encapsulated follicles at low temperatures [24]. Cracks in the scaffold can affect the subsequent culture and growth of follicles [41]. Although the microstructure of the scaffold was not investigated here, the increased diameters of oocytes/follicles and proliferation of cumulus cells indicated the effectiveness of the scaffold after vitrification and warming.
Alginate beads are reported to have a high porosity range and to limit the diffusion of only large proteins. Substrates with a molecular weight below 2 ×104 can diffuse freely into and from calcium alginate beads at approximately the same rate as in water. Thus, encapsulation of the follicles did not theoretically affect the diffusion of cryoprotectant throughout the follicles and thus the viability of cryopreserved follicles [42, 43]. However, high-molecular-weight proteins cannot diffuse freely into calcium alginate beads [44], possibly affecting follicular growth when subjected to in vitro culture after warming. Inclusion of hormones or other proteins in the scaffold might solve this problem.
In conclusion, the present vitreous cryopreservation method could effectively maintain the viability of encapsulated human ovarian follicles, which could be put into immediate use (in vitro culture or transplantation [9]) after warming. More follicles are required to further evaluate the effects of vitrification on the in vitro growth of cryopreserved encapsulated human preantral follicles.
Acknowledgment
This study was supported by grants from the Key Laboratory of Guangdong Province and the National Natural Science Foundation of China (81100472).
References
- 1.Agarwal SK, Chang RJ. Fertility management for women with cancer. Cancer Treat Res 2007; 138: 15–27 [DOI] [PubMed] [Google Scholar]
- 2.Lee SJ, Schover LR, Partridge AH, Patrizio P, Wallace WH, Hagerty K, Beck LN, Brennan LV, Oktay K. American Society of Clinical Oncology recommendations on fertility preservation in cancer patients. J Clin Oncol 2006; 24: 2917–2931 [DOI] [PubMed] [Google Scholar]
- 3.Gallot D, Pouly JL, Janny L, Mage G, Canis M, Wattiez A, Bruhat MA. Successful transfer of frozen-thawed embryos obtained immediately before radical surgery for stage IIIa serous borderline ovarian tumour: case report. Hum Reprod 2000; 15: 2347–2350 [DOI] [PubMed] [Google Scholar]
- 4.Levi Setti PE, Albani E, Novara PV, Cesana A, Morreale G. Cryopreservation of supernumerary oocytes in IVF/ICSI cycles. Hum Reprod 2006; 21: 370–375 [DOI] [PubMed] [Google Scholar]
- 5.D'Angelo A, Amso NN. Embryo freezing for preventing ovarian hyperstimulation syndrome: a Cochrane review. Hum Reprod 2002; 17: 2787–2794 [DOI] [PubMed] [Google Scholar]
- 6.Abir R, Ben-Haroush A, Felz C, Okon E, Raanani H, Orvieto R, Nitke S, Fisch B. Selection of patients before and after anticancer treatment for ovarian cryopreservation. Hum Reprod 2008; 23: 869–877 [DOI] [PubMed] [Google Scholar]
- 7.Donnez J, Dolmans MM, Demylle D, Jadoul P, Pirard C, Squifflet J, Martinez-Madrid B, van Langendonckt A. Livebirth after orthotopic transplantation of cryopreserved ovarian tissue. Lancet 2004; 364: 1405–1410 [DOI] [PubMed] [Google Scholar]
- 8.Meirow D, Levron J, Eldar-Geva T, Hardan I, Fridman E, Zalel Y, Schiff E, Dor J. Pregnancy after transplantation of cryopreserved ovarian tissue in a patient with ovarian failure after chemotherapy. N Engl J Med 2005; 353: 318–321 [DOI] [PubMed] [Google Scholar]
- 9.Carroll J, Gosden RG. Transplantation of frozen-thawed mouse primordial follicles. Hum Reprod 1993; 8: 1163–1167 [DOI] [PubMed] [Google Scholar]
- 10.Amorim CA, Rodrigues AP, Rondina D, Gonçalves PB, de Figueiredo JR, Giorgetti A. Cryopreservation of ovine primordial follicles using dimethyl 318 sulfoxide. Fertil Steril 2003; 79: 682–686 [DOI] [PubMed] [Google Scholar]
- 11.Amorim CA, Rondina D, Rodrigues AP, Costa SH, Gonçalves PB, de Figueiredo JR, Giorgetti A. Isolated ovine primordial follicles cryopreserved in different concentrations of ethylene glycol. Theriogenology 2003; 60: 735–742 [DOI] [PubMed] [Google Scholar]
- 12.Santos RR, van den Hurk R, Rodrigues AP, Costa SH, Martins FS, Matos MH, Celestino JJ, Figueiredo JR. Effect of cryopreservation on viability, activation and growth of in situ and isolated ovine early-stage follicles. Anim Reprod Sci 2007; 99: 53–64 [DOI] [PubMed] [Google Scholar]
- 13.dela Peña EC, Takahashi Y, Katagiri S, Atabay EC, Nagano M. Birth of pups after transfer of mouse embryos derived from vitrified preantral follicles. Reproduction 2002; 123: 593–600 [DOI] [PubMed] [Google Scholar]
- 14.Kreeger PK, Deck JW, Woodruff TK, Shea LD. The in vitro regulation of ovarian follicle development using alginate-extracellular matrix gels. Biomaterials 2006; 27: 714–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharma GT, Dubey PK, Meur SK. Survival and developmental competence of buffalo preantral follicles using three-dimensional collagen gel culture system. Anim Reprod Sci 2009; 114: 115–124 [DOI] [PubMed] [Google Scholar]
- 16.Vigo D, Villani S, Faustini M, Accorsi PA, Galeati G, Spinaci M, Munari E, Russo V, Asti A, Conte U, Torre ML. Follicle-like model by granulosa cell encapsulation in a barium alginate-protamine membrane. Tissue Eng 2005; 11: 709–714 [DOI] [PubMed] [Google Scholar]
- 17.Pangas SA, Saudye H, Shea LD, Woodruff TK. Novel approach for the three-dimensional culture of granulosa cell-oocyte complexes. Tissue Eng 2003; 9: 1013–1021 [DOI] [PubMed] [Google Scholar]
- 18.Kreeger PK, Fernandes NN, Woodruff TK, Shea LD. Regulation of mouse follicle development by follicle-stimulating hormone in a three-dimensional in vitro culture system is dependent on follicle stage and dose. Biol Reprod 2005; 73: 942–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mazur P. Kinetics of water loss from cells from subzero temperatures 343 and the likehood of intracellular freezing. J Gen Physiol 1963; 47: 347–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arav A, Zeron Y, Leslie SB, Behboodi E, Anderson GB, Crowe JH. Phase transition temperature and chilling sensitivity of bovine oocytes. Cryobiology 1996; 33: 589–599 [DOI] [PubMed] [Google Scholar]
- 21.Mukaida T, Wada S, Takahashi K, Pedro PB, An TZ, Kasai M. Vitrification of human embryos based on the assessment of suitable conditions for 8-cell mouse embryos. Hum Reprod 1998; 13: 2874–2879 [DOI] [PubMed] [Google Scholar]
- 22.Song YC, Khirabadi BS, Lightfoot F, Brockbank KG, Taylor MJ. Vitreous cryopreservation maintains the function of vascular grafts. Nat Biotechnol 2000; 18: 296–299 [DOI] [PubMed] [Google Scholar]
- 23.Fahy GM, Wowk B, Wu J, Phan J, Rasch C, Chang A, Zendejas E. Cryopreservation of organs by vitrification: perspectives and recent advances. Cryobiology 2004; 48: 157–178 [DOI] [PubMed] [Google Scholar]
- 24.Kuleshova LL, Gouk SS, Hutmacher DW. Vitrification as a prospect for cryopreservation of tissue-engineered constructs. Biomaterials 2007; 28: 1585–1596 [DOI] [PubMed] [Google Scholar]
- 25.Dahl SL, Chen Z, Solan AK, Brockbank KG, Niklason LE, Song YC. Feasibility of vitrification as a storage method for tissue-engineered blood vessels. Tissue Eng 2006; 12: 291–300 [DOI] [PubMed] [Google Scholar]
- 26.Wu Y, Yu H, Chang S, Magalhães R, Kuleshova LL. Vitreou cryopreservation of cell-biomaterial constructs involving encapsulated hepatocytes. Tissue Eng 2007; 13: 649–658 [DOI] [PubMed] [Google Scholar]
- 27.Dolmans MM, Michaux N, Camboni A, Martinez-Madrid B, Van Langendonckt A, Nottola SA, Donnez J. Evaluation of Liberase, a purified enzyme blend, for the isolation of human primordial and primary ovarian follicles. Hum Reprod 2006; 21: 413–420 [DOI] [PubMed] [Google Scholar]
- 28.Amorim CA, Van Langendonckt A, David A, Dolmans MM, Donnez J. Survival of human pre-antral follicles after cryopreservation of ovarian tissue, follicular isolation and in vitro culture in a calcium alginate matrix. Hum Reprod 2009; 24: 92–99 [DOI] [PubMed] [Google Scholar]
- 29.Li T, Mai Q, Gao J, Zhou C. Cryopreservation of human embryonic stem cells with a new bulk vitrification method. Biol Reprod 2010; 82: 848–853 [DOI] [PubMed] [Google Scholar]
- 30.Xing W, Zhou C, Bian J, Montag M, Xu Y, Li Y, Li T. Solid-surface vitrification is an appropriate and convenient method for cryopreservation of isolated rat follicles. Reprod Biol Endocrinol. 2010; 8: 42–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Collins TJ. ImageJ for microscopy. Biotechniques 2007; 43(Suppl): 25–30 [DOI] [PubMed] [Google Scholar]
- 32.Matos MH, Andrade ER, Lucci CM, Báo SN, Silva JR, Santos RR, Ferreira MA, Costa SH, Celestino JJ, Figueiredo JR. Morphological and ultrastructural analysis of sheep primordial follicles preserved in 0.9% saline solution and TCM 199. Theriogenology 2004; 62: 65–80 [DOI] [PubMed] [Google Scholar]
- 33.Martinez-Madrid B, Camboni A, Dolmans MM, Nottola S, Van Langendonckt A, Donnez J. Apoptosis and ultrastructural assessment after cryopreservation of whole human ovaries with their vascular pedicle. Fertil Steril 2007; 87: 1153–1165 [DOI] [PubMed] [Google Scholar]
- 34.Hirota Y, Osuga Y, Nose E, Koga K, Yoshino O, Hirata T, Yano T, Tsutsumi O, Sakuma S, Muramatsu T, Taketani Y. The presence of midkine and its possible implication in human ovarian follicles. Am J Reprod Immunol 2007; 58: 367–373 [DOI] [PubMed] [Google Scholar]
- 35.Gilchrist RB, Ritter LJ, Myllymaa S, Kaivo-Oja N, Dragovic RA, Hickey TE, Ritvos O, Mottershead DG. Molecular basis of oocyte-paracrine signalling that promotes granulosa cell proliferation. J Cell Sci 2006; 119: 3811–3821 [DOI] [PubMed] [Google Scholar]
- 36.Vacková I, Engelová M, Marinov I, Tománek M. Cell cycle synchronization of porcine granulosa cells in G1 stage with mimosine. Anim Reprod Sci 2003; 77: 235–245 [DOI] [PubMed] [Google Scholar]
- 37.Edwards SJ, Reader KL, Lun S, Western A, Lawrence S, McNatty KP, Juengel JL. The cooperative effect of growth and differentiation factor-9 and bone morphogenetic protein (BMP)-15 on granulosa cell function is modulated primarily through BMP receptor II. Endocrinology 2008; 149: 1026–1030 [DOI] [PubMed] [Google Scholar]
- 38.Rall WF. Factors affecting the survival of mouse embryos cryopreserved by vitrification. Cryobiology 1987; 24: 387–402 [DOI] [PubMed] [Google Scholar]
- 39.Candy CJ, Wood MJ, Whittingham DG. Effect of cryoprotectants on the survival of follicles in frozen mouse ovaries. J Reprod Fertil 1997; 110: 11–19 [DOI] [PubMed] [Google Scholar]
- 40.Newton H, Picton H, Gosden RG. In vitro growth of oocyte granulosa cell complexes isolated from cryopreserved ovine tissue. J Reprod Fertil 1999; 115: 141–150 [DOI] [PubMed] [Google Scholar]
- 41.Anseth KS, Bowman CN, Brannon-Peppas L. Mechanical properties of hydrogels and their experimental determination. Biomaterials 1996; 17: 1647–1657 [DOI] [PubMed] [Google Scholar]
- 42.Tanaka H, Matsumura M, Veliky IA. Biotechnol Bioeng 1984; 26: 53–58 [DOI] [PubMed] [Google Scholar]
- 43.Amsden, Turner N. Biotechnol Bioeng 1999; 65: 605–610 [DOI] [PubMed] [Google Scholar]
- 44.Heise M, Koepsel R, Russell AJ, McGee EA. Reprod Biol Endocrinol 2005; 3: 47 [DOI] [PMC free article] [PubMed] [Google Scholar]