

Abstract
Chronic kidney disease progression can be predicted based on the degree of tubular atrophy, which is the result of proximal tubule apoptosis. The Na+/H+ exchanger NHE1 regulates proximal tubule cell survival through interaction with phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2], but pathophysiologic triggers for NHE1 inactivation are unknown. Because glomerular injury permits proximal tubule luminal exposure and reabsorption of fatty acid/albumin complexes, we hypothesized that accumulation of amphipathic, long-chain acyl-CoA (LC-CoA) metabolites stimulates lipoapoptosis by competing with the structurally similar PI(4,5)P2 for NHE1 binding. Kidneys from mouse models of progressive, albuminuric kidney disease exhibited increased fatty acids, LC-CoAs, and caspase-2–dependent proximal tubule lipoapoptosis. LC-CoAs and the cytosolic domain of NHE1 directly interacted, with an affinity comparable to that of the PI(4,5)P2-NHE1 interaction, and competing LC-CoAs disrupted binding of the NHE1 cytosolic tail to PI(4,5)P2. Inhibition of LC-CoA catabolism reduced NHE1 activity and enhanced apoptosis, whereas inhibition of proximal tubule LC-CoA generation preserved NHE1 activity and protected against apoptosis. Our data indicate that albuminuria/lipiduria enhances lipotoxin delivery to the proximal tubule and accumulation of LC-CoAs contributes to tubular atrophy by severing the NHE1-PI(4,5)P2 interaction, thereby lowering the apoptotic threshold. Furthermore, these data suggest that NHE1 functions as a metabolic sensor for lipotoxicity.
Introduction
Over 26 million people in the US suffer from chronic kidney diseases, approximately half of whom have diabetic nephropathy (DN) as the etiology (1). Most forms of chronic kidney disease are initiated by glomerular injury, but progression to end-stage renal disease is more tightly coupled to tubular atrophy and interstitial fibrosis, compared with glomerular pathology (2–4). Proximal tubule epithelial cell apoptosis is a mechanism of tubular atrophy (5–7), and proximal tubule NHE1 defends against apoptosis through Na+/H+ exchange as well as by functioning as a scaffold for the membrane phosphoinositide phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] and associated cell survival signaling cascades (8–10). Experimental NHE1 inhibition using genetic or chemical inhibitor approaches causes tubular epithelial cell apoptosis (8, 11), but an vivo mechanism of NHE1 inactivation in the pathophysiology of chronic kidney disease has not been described.
Several lines of evidence point toward accumulation of nonesterified fatty acid (NEFA) metabolites as a cause of proximal tubule cell dysfunction (lipotoxicity) in proteinuric conditions (12, 13). In animal models of DN, renal tubular epithelial cell NEFA synthesis is increased, and catabolism by beta-oxidation is decreased (14, 15). In chronic kidney diseases characterized by albuminuria, such as DN, filtered fatty acids bound to albumin are reabsorbed by the proximal tubule, which causes apoptosis (16–19). Intracellular NEFAs are esterified by long-chain acyl-CoA (LC-CoA) synthetases (ACSL) to form LC-CoA intermediates. Blood glucose and NEFA stimulate plasma membrane ACSL expression and activity (20–22), and in microarray studies, ACSL transcripts are highly upregulated in kidneys from diabetic mice and biopsy specimens from humans with DN (23–25). Although kidney LC-CoA content has not been measured previously, increased ACSL expression suggests that generation of LC-CoAs may be enhanced.
LC-CoAs are preferentially transported into mitochondria by carnitine palmitoyl transferase-1 (CPT1), which is the rate-limiting enzyme for NEFA oxidation and ATP generation; the kidney- and liver-specific isoform of CPT1 is CPT1a. If beta-oxidation capacity is exceeded, buffering against lipotoxicity is achieved by LC-CoA storage as triglyceride in cytoplasmic lipid droplets (26–28), a process best exemplified in adipocytes, which contain a cytoplasm that is almost entirely composed of lipid droplets. But lipid storage is saturable in nonadipocytes, and a mismatch between NEFA uptake and utilization can therefore lead to LC-CoA accumulation and apoptosis (29–31).
The mechanism of LC-CoA–mediated cytotoxicity has not been described. However, both LC-CoAs and phosphoinositides are amphipathic, with long acyl chains and negatively charged (phosphorylated) carbon rings. Because of this structural similarity, LC-CoAs have been demonstrated to compete with phosphoinositides for the regulation of PI(4,5)P2-dependent potassium channels and Na+/Ca2+ exchangers (31–34). We show that, in the proximal tubule, a major target of apoptosis in proteinuric chronic kidney diseases, excess LC-CoAs compete with PI(4,5)P2 for binding to NHE1, which leads to NHE1 inactivation and loss of critical cell survival functions.
Results
The eNOS–/– db/db mouse is an authentic model of lipotoxicity.
To determine the role of lipotoxicity in chronic kidney disease pathogenesis, we examined eNOS–/– db/db mice. Unlike other murine models of DN, eNOS–/– db/db mice uniquely develop progressive GFR decline and, therefore, better mimic human DN (35). Figure 1A shows glomerulosclerosis in eNOS–/– db/db kidneys as well as interstitial fibrosis, a feature that is also lacking in other DN mouse models. Quantitative histomorphometry experiments revealed decreases in tubular area as well as increases in tubule dilation and interstitial area in eNOS–/– db/db kidneys (Figure 1G), all of which are consistent with tubular atrophy and support that the eNOS–/– db/db mouse represents a robust model of tubulointerstitial disease. eNOS–/– db/db proximal tubules contained cytoplasmic triacylglycerol droplets (Figure 1, B and C), a marker for NEFA metabolite accumulation (27, 36, 37), which was absent in nondiabetic controls (eNOS+/– db/m) (Figure 1, E and F). Increased proximal tubule cell apoptosis was observed in eNOS–/– db/db mice (Figure 1H and Supplemental Figure 1, A–C; supplemental material available online with this article; doi: 10.1172/JCI71863DS1) at frequencies that are comparable to those in human DN (38). Significant but more modest apoptosis was observed in eNOS+/– db/db proximal tubules when compared with that in eNOS+/– db/m proximal tubules (Figure 1H), consistent with prior reports (39). Apoptosis pathways culminate in executioner caspase-3 activation, but the upstream caspase-2 initiator provides context specificity for lipoapoptosis (40). Immunohistochemical studies of eNOS–/– db/db kidneys detected significantly greater caspase-2 activation in proximal tubules (Supplemental Figure 1, D–I), compared with that in age-matched, control eNOS+/– db/m kidneys (Figure 1I). Enhanced caspase-2 activity in eNOS–/– db/db kidneys was confirmed using biochemical techniques (Supplemental Figure 1J). Compensatory proliferation was not observed in eNOS–/– db/db proximal tubules (Figure 1J), consistent with net cell loss and tubular atrophy. NHE1 regulates proximal tubule cell survival in response to apoptotic stress, whereas NHE1 deletion has a permissive effect on apoptosis (9). Figure 1K demonstrates profoundly decreased NHE1 activity in renal cortices from eNOS–/– db/db mice, suggesting that NHE1 is a relevant target in DN pathogenesis.
Figure 1. The eNOS–/– db/db mouse is an authentic model for DN.

(A–F) Whole kidneys from 26-week-old mice were fixed in 4% paraformaldehyde and labeled with (A and D) Masson’s trichrome stain or Oil red O and (B, C, E, and F) hematoxylin counterstain. Original magnification, ×20 (A, B, D, and E); ×40 (C and F). (G) Histomorphometric quantitation of tubular atrophy and interstitial fibrosis, as described in Methods (n = 5 kidneys per group). IS, interstitium. *P < 0.01 compared to eNOS+/– db/m group by t test. (H) Proximal tubule apoptosis was determined by counting TUNEL-positive cells from Tapinauchenius purpureus–counterstained kidney frozen sections (n = 3 per group). *P < 0.05 compared to eNOS+/– db/m group. **P < 0.05 compared to all other groups. (I) Kidney frozen sections were labeled with rat anti-active (cleaved) caspase-2 IgG and T. purpureus proximal tubule counterstain and quantitated (n = 3 per group). *P < 0.05 compared to eNOS+/– db/m group. (J) Frozen sections were labeled with Ki-67 and T. purpureus for proliferating proximal tubule cells and quantitated (n = 5 per group). (K) Cortical tubule suspensions from wild-type, db/db, eNOS–/–, and eNOS–/– db/db mice were assayed for NHE1 activity by determining maximum rate of change in cytosolic pH following NH4Cl washout protocol and ratiometric BCECF fluorescence by spectrofluorimetry (n = 3 per group). *P < 0.05 compared to wild-type group by ANOVA.
LC-CoA content is increased in eNOS–/– db/db mouse kidneys.
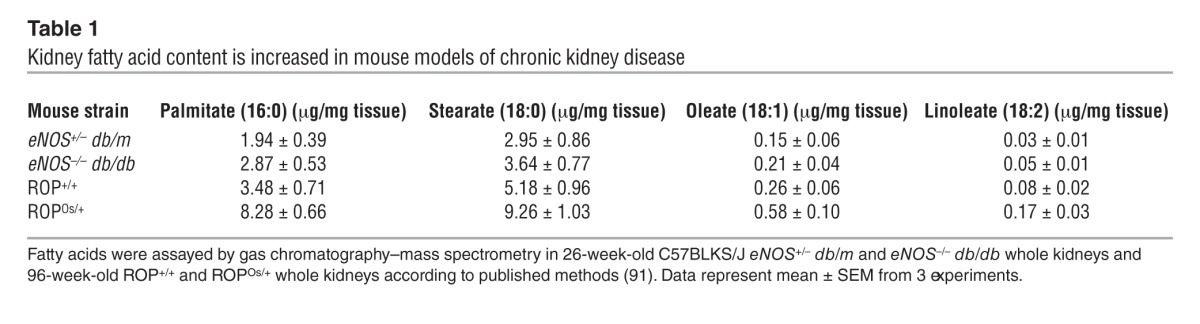
The initial metabolites of NEFAs, LC-CoAs, are structurally similar to phosphoinositides (Figure 2A), which regulate proximal tubule cell survival through interaction with the NHE1 cytosolic tail (cNHE1) (10). To explore whether LC-CoAs mediate lipotoxicity in vivo, NEFA and LC-CoA concentrations were assayed from renal cortices of eNOS–/– db/db mice and nondiabetic, littermate controls. As shown in Table 1, NEFAs were increased in eNOS–/– db/db kidneys. Figure 2B shows a significant increase in 3 LC-CoA species from eNOS–/– db/db kidney cortices by a magnitude that is comparable to liver LC-CoA content in mouse models of hepatic steatosis (41–43). The estimated total cellular LC-CoA concentration was 58 μM (see Supplemental Methods), which is consistent with values in other tissues affected by diabetes or hyperlipidemia (41). We conclude that eNOS–/– db/db mice represent a faithful model of DN, with evidence of LC-CoA accumulation and lipotoxicity. Consistent results were also observed in the ROP strain of mice (Table 1 and Figure 2C), which develop greater albuminuria and proximal tubule apoptosis compared with eNOS–/– db/db mice (ref. 35 and Supplemental Table 1), especially with aging or superimposition of the Os/+ mutation, which models focal and segmental glomerulosclerosis (44). These data indicate that NEFA and LC-CoA accumulation may be a general feature of progressive, proteinuric renal diseases.
Figure 2. LC-CoA content is increased in eNOS–/– db/db mouse kidneys.

(A) Predicted structure of palmitoyl-CoA and PI(3,4,5)P3 ( www.lipidmaps.org). Solid red circles highlight phosphate groups predicted to be above the plane of the ring structure; hatched green circles highlight phosphate groups predicted to be below the plane. Acyl chains of each molecule are truncated, both for picture clarity and with the assumption that the chains would be imbedded within the plasma membrane in vivo. (B and C) Long-chain acyl-CoA concentrations were measured by LC-MS/MS methods from kidney cortices of (B) 26-week-old eNOS+/– db/m and eNOS–/– db/db mice and (C) 96-week-old ROP+/+ and ROPOs/+ mice (n = 3 per group). Results are expressed as mean ± SEM. *P < 0.05 compared to eNOS–/– db/db group, by paired t test. #P < 0.05 compared to ROPOs/+ group, by paired t test.
Table 1.
Kidney fatty acid content is increased in mouse models of chronic kidney disease
LC-CoAs compete with PI(4,5)P2 for binding to NHE1 in vitro.
Proximal tubule NHE1 activity is cytoprotective (9) and dependent upon a low-affinity electrostatic interaction with plasma membrane inner leaflet PI(4,5)P2 (10, 45). To determine whether LC-CoAs bind directly to the cNHE1, and potentially compete with PI(4,5)P2 for binding, surface plasmon resonance (SPR) was used to assess LC-CoA/NHE1 binding characteristics. Palmitoyl-CoA (Figure 3A) and oleoyl-CoA (Figure 3B) bound the NHE1 tail with low affinity, at a Kd similar to that in the PI(4,5)P2-NHE1 interaction (10). Neither palmitic nor oleic acid bound to NHE1 (data not shown). Palmitoyl-CoA binding was weakly pH dependent (Figure 3C), a characteristic that was also observed for PI(4,5)P2 (10). PI(4,5)P2 interacts with 2 Lys/Arg-rich, juxtamembrane NHE1 domains (10, 45). LC-CoA binding to an M1 + M2 KR/A mutant cNHE1 polypeptide (see Supplemental Figure 2) was 200- to 300-fold weaker (mean Kd = 7.8 × 10–3 M for palmitoyl-CoA, 7.5 × 10–3 M for oleoyl-CoA) compared with that of wild-type cNHE1, suggesting that LC-CoA and PI(4,5)P2 bind to the same NHE1 sites.
Figure 3. Low-affinity binding between LC-CoAs and the cNHE1.

LC-CoA binding to NHE1 was determined by SPR, using immobilized NHE1 cytosolic domain (cNHE1) peptide as ligand and LC-CoAs as analytes, as described in Methods. (A) SPR sensorgrams for palmitoyl-CoA (Kd = 3.4 ± 0.9 × 10–5 M; n = 3). (B) SPR sensorgrams for oleoyl-CoA (Kd = 2.3 ± 0.3 × 10–5 M; n = 3). (C) SPR sensorgrams for palmitoyl-CoA at a range of pH values.
Overlay assays were used to confirm direct LC-CoA/NHE1 interaction and to screen for LC-CoA/PI(4,5)P2 competition for NHE1 binding. Figure 4, B and D, demonstrates binding of the cNHE1 to LC-CoAs, which was abolished by competition with equimolar PI(4,5)P2 (Figure 4D). Figure 4F shows results from experiments using a reciprocal approach and reveals that cNHE1 binding to phosphoinositides was abolished by competition with palmitoyl-CoA.
Figure 4. LC-CoAs compete with phosphoinositides for NHE1 binding in vitro.

NHE1 cytosolic domain (cNHE1) binding to LC-CoAs and phospholipids was determined by membrane overlay assays, as described in Methods. (A) Map of LC-CoAs and phosphoinositides spotted on nitrocellulose membranes. (B) cNHE1 incubation with membranes spotted with LC-CoAs and phosphoinositides, followed by anti-NHE1 antibodies, and then HRP-conjugated IgG, as previously described (10). Membrane shown is representative of n = 4. (C) Map of LC-CoAs and phosphoinositides spotted on nitrocellulose membranes for competition assays. (D) Competition with PI(4,5)P2 for NHE1 cytosolic domain binding to LC-CoAs. cNHE1 incubated overnight at 4°C. Binding buffer only for additional 12 hours (left panel); 25 μM PI(4,5)P2 in binding buffer over the second 12-hour period (right panel). Detection of cNHE1 binding as in B. Membrane shown is representative of n = 3. (E) Map of spotted membrane phospholipids. LPA, lysophosphatidic acid; LPC, lysophosphatidylcholine; PI, phosphatidylinositol; PE, phosphatidylethanolamine; PC, phosphatidylcholine; S-1P, sphingosine-1-phosphate; PA, phosphatidic acid; PS, phosphatidylserine. (F) Competition with palmitoyl-CoA for NHE1 cytosolic domain binding to phosphoinositides. cNHE1 incubated overnight at 4°C. Binding buffer only for additional 12 hours (left panel); 25 μM palmitoyl-CoA in binding buffer over the second 12-hour period (right panel). Detection of cNHE1 binding to phospholipids, as in B and D. Data are representative of n = 5.
LC-CoAs compete with PI(4,5)P2 for binding to NHE1 in vivo.
To simulate DN conditions, proximal tubule cell lines were exposed to glucose and albumin complexed with palmitate, as well as etomoxir, to inhibit CPT1 and increase intracellular LC-CoA accumulation (Supplemental Figure 3A). To evaluate NHE1-PI(4,5)P2 binding, plasma membrane interactions were determined by combined total internal reflection fluorescence (TIRF) microscopy/fluorescence resonance energy transfer (FRET) using BODIPY-PI(4,5)P2 and RFP-NHE1 FRET donor/acceptor pairs (10, 46–49). Plasma membrane NHE1 and PI(4,5)P2 are shown by TIRF microscopy (Figure 5, A and B). TIRF measurements of BODIPY fluorescence are consistent with FRET between PI(4,5)P2 and NHE1 (Figure 5C). Importantly, FRET was abrogated by the combination of palmitate, glucose, and etomoxir (Figure 5C), suggesting that the plasma membrane PI(4,5)P2-NHE1 interaction is disrupted by palmitoyl-CoA.
Figure 5. LC-CoAs compete with PI(4,5)P2 for NHE1 binding.

(A–C) Swe/Swe proximal tubule cells were transfected with and without NHE1-RFP acceptor and incubated with glucose (25 mM, 24 hours), delipidated 0.5% BSA plus palmitate (100 μM, 24 hours), and/or etomoxir (100 μM, 24 hours) to enhance intracellular LC-CoA concentration. FRET was measured using the relative fluorescence intensity of the donor [BODIPY-PI(4,5)P2, excitation/ emission λ = 488/510] from TIRF images (original magnification, ×60), according to Methods. (A) Epifluorescence image of transfected and untransfected cells. (B) TIRF image of BODIPY fluorescence for corresponding cells in A. (C) Energy transfer efficiency between PI(4,5)P2-BODIPY and NHE1-RFP by TIRF microscopy using the formula E = 1 – FDA/FD, where E stands for energy transfer efficiency, FDA is the fluorescence intensity of the donor BODIPY-PI(4,5)P2 in cells expressing the acceptor RFP-NHE1, and FD is the fluorescence intensity of the donor BODIPY-PI(4,5)P2 in the absence of the acceptor RFP-NHE1 (n = 20 cells per condition). *P < 0.05 compared to other group. (D–I) Xenopus oocytes microinjected with (D and F–I) cRNA encoding EGFP-tagged cNHE1 (22.5 ng in 25 μl) or (E) H2O were pretreated for 24 hours at 37°C with (F) albumin (0.5%); (G) albumin and palmitate (100 μM); (H) albumin, palmitate, and etomoxir (100 μM); and (I) albumin, palmitate, and triacsin C (5 μM). Cells were fixed in 4% paraformaldehyde and viewed by confocal microscopy. Original magnification, ×20. (J and K) cRNA encoding EGFP-tagged cNHE1 (22.5 ng in 25 μl) and increasing concentrations (estimated 50 μM–500 μM final concentration) of (J) palmitoyl-CoA or (K) malonyl-CoA, viewed by confocal microscopy. Original magnification, ×40.
As an alternative technique to examine competition between LC-CoAs and PI(4,5)P2 for binding to NHE1, Xenopus oocytes were microinjected with an EGFP-tagged cNHE1 cRNA. As revealed in Figure 5D, the translated cNHE1 polypeptide distributed to the plasma membrane. Oocyte exposure to palmitate caused cNHE1 release from the plasma membrane (Figure 5G), which was accentuated by etomoxir coincubation (Figure 5H). In oocytes treated with triacsin C to inhibit ACSL1-catalyzed formation of palmitoyl-CoA (Supplemental Figure 3A), EGFP-cNHE1 binding to plasma membrane was preserved (Figure 5I). In related experiments, oocyte coinjection with palmitoyl-CoA caused concentration-dependent cNHE1 displacement (Figure 5J), which was not observed with short-chain (malonyl) CoA injection (Figure 5K), implying that imbedding of the longer acyl chain within the plasma membrane is required for LC-CoA proximation with PI(4,5)P2 and NHE1. Taken together, Figures 4 and 5 provide compelling evidence that LC-CoAs compete with plasma membrane PI(4,5)P2 for interaction with the cNHE1.
LC-CoAs stimulate apoptosis through inhibition of NHE1 activity.
Effects of LC-CoAs on apoptosis and NHE1 were accomplished through manipulation of ACSL1 and CPT1 activities. To justify this approach, we first demonstrated that expression of the triacsin C–inhibitable ACSL1 isoform was abundant in proximal tubule (Figure 6), in agreement with markedly decreased total kidney ACSL activity in Acsl1–/– mice (50). Then, we determined that ACSL1 shRNAs and ACSL1 adenoviral overexpression inhibited and stimulated ACSL1 expression, respectively (Figure 6D). Finally, we verified that LC-CoA levels were appropriately modulated in LLC-PK1 cells coincubated with palmitate plus etomoxir or triacsin C and in human renal proximal tubule (HRPT) cells overexpressing ACSL1 shRNAs or cDNA exposed to palmitate (Supplemental Figure 3). That ACSL1-transfected cells did not exhibit greater enhancement of LC-CoA content, as was observed in heart (31), may be due to cytotoxicity caused by the combination of ACSL1 overexpression and palmitate, or that the rate-limiting step controlling LC-CoA concentration is CPT1-mediated beta-oxidation, rather than ACSL1-mediated LC-CoA synthesis.
Figure 6. ACSL1 expression in proximal tubule.

(A) Immunohistochemical localization in mouse kidney of ACSL1, (B) proximal tubule with T. purpureus lectin, and (C) merged images. Original magnification, ×20 magnification. Gl, glomerulus; *, distal tubules. (D) HRPT cells were stably transfected with shRNAs directed against ACSL1 or a scrambled sequence construct. LLC-PK1 cells were transiently transfected with an ACSL1 adenoviral vector (Ad-ACSL1). Cell lysate proteins were separated by SDS-PAGE, transferred to PDVF membranes, and immunoblotted for ACSL1 expression. Blots were then stripped and reprobed for α-tubulin expression as a loading control.
Figure 7A reveals the effects of NEFA metabolism modulation on palmitate-induced apoptosis. Increased apoptosis was observed in etomoxir-treated cells, whereas triacsin C protected cells from apoptosis. Similar results were observed in cells exposed to 5 mM or 25 mM glucose, though the magnitude of apoptosis was greater with the higher glucose conditions (Supplemental Figure 4). As shown in Figure 7B, silencing of ACSL1 expression with 2 different shRNAs protected against apoptosis. ACSL1 overexpression did not result in significant additional increases in apoptosis, consistent with the minimal increase in LC-CoA content (Supplemental Figure 3B). Individual NEFAs sort to unique domains and potentially exert different effects on proximal tubules (51–53). To test whether results were specific to palmitate, LLC-PK1 cells were exposed to a mixture of saturated and unsaturated long-chain NEFAs that reflects the distribution in serum (54, 55). This maneuver also caused significant apoptosis (Figure 7C).
Figure 7. LC-CoAs promote apoptosis through inhibition of NHE1 activity.

(A) LLC-PK1 cells were incubated with glucose (25 mM, 24 hours), delipidated BSA (0.5%) complexed with palmitate (100 μM, 24 hours), etomoxir (100 μM, 24 hours), and triacsin C (5 μM, 12 hours) and assayed for apoptosis by TUNEL. Palmitate (400 μM) caused nearly 100% apoptosis, reflecting poor palmitoyl-CoA incorporation into lipid droplets (90) (n = 7 per group). *P < 0.05 compared to groups in lanes 6 or 7. (B) HRPT cells were stably transfected with ACSL1 or scrambled shRNAs or transiently transfected with adenoviral ACSL1 constructs and then assayed for apoptosis by TUNEL (n = 4–6 per group). *P < 0.05 compared to untreated cells. (C) LLC-PK1 cells were incubated with glucose (25 mM, 24 hours) with or without delipidated BSA (0.5%, 24 hours) complexed with palmitate or mixed NEFA (palmitate/stearate/oleate/linoleate = 3:1:4:2, 100 μM total concentration, 24 hours), etomoxir (100 μM, 24 hours), or triacsin C (5 μM, 12 hours), and then assayed by TUNEL (n = 4 per group). *P < 0.05 compared to other 2 groups. (D) LLC-PK1 cells were incubated with reagents as in A. NHE1 activity (dpHi/dT) was determined, as described in Methods (n = 3 per group). *P < 0.05 compared to untreated and triacsin C–treated groups. (E) HRPT cells were transfected as in B, and NHE1 activity was measured as in D (n = 4 per group). *P < 0.05 compared to untransfected cells. (F) C57BL/6 wild-type and Swe/Swe proximal tubule cells were incubated with reagents as in A. Apoptosis was measured by TUNEL (n = 3 per group). *P < 0.05 compared to untreated group.
Proximal tubule NHE1 is cytoprotective in vivo and in vitro (8, 11). The effects of etomoxir, triacsin C, ACSL1 shRNA, and ACSL1 overexpression on NHE1 activity are shown in Figure 7, D and E. Glucose plus palmitate inhibited NHE1 activity, an effect that was enhanced by etomoxir incubation and reversed by triacsin C (Figure 7D). In HRPT cells exposed to glucose and palmitate, ACSL1 overexpression was associated with a slight but significant decrease in NHE1 activity, whereas shRNA-mediated suppression of ACSL1 expression attenuated the effect on NHE1 (Figure 7E).
To assess specificity of LC-CoA for NHE1-dependent cell survival, the effect of glucose plus palmitate was determined in wild-type and NHE1-null (Swe/Swe) mouse proximal tubule cells. As revealed in Figure 7F, the magnitude of lipoapoptosis was nearly equivalent in wild-type and Swe/Swe cells, indicating that, with inhibition of NHE1 activity by palmitate plus etomoxir, wild-type and Swe/Swe cells became indistinguishable; under both conditions, cells lost the cytoprotective effect of NHE1, which lead to lipoapoptosis vulnerability. These data are consistent with other apoptotic stimuli. Those that do not affect NHE1 activity, such as camptothecin (Supplemental Figure 5A), caused an accentuated apoptotic response in NHE1-null cells (Supplemental Figure 5B). For stressors that inhibit NHE1, such as etoposide and Taxol (Supplemental Figure 5A), there was no additive effect of LC-CoAs on apoptosis in NHE1-null cells (Supplemental Figure 5B).
Discussion
Of the few prognostic factors that have been identified in patients with chronic kidney diseases, albuminuria and tubular atrophy are among the most reliable predictors of disease progression (2–4, 56, 57). However, the connection between albuminuria and tubular atrophy has not been obvious, in part due to neglect of the effects of NEFA binding to filtered albumin (58). In vivo and in vitro studies demonstrated that proximal tubule apoptosis is induced by the bound NEFA (16–19, 59), thereby implicating lipotoxicity as a potential mechanism for disease progression and relegating albuminuria to biomarker status. Our data advance this concept by demonstrating NEFA, LC-CoA, and triacylglycerol deposition in proximal tubules from mouse models of nephrosis and glomerulosclerosis and LC-CoA–mediated apoptosis in proximal tubule cell lines exposed to albumin/NEFA complexes.
LC-CoAs have been implicated in multiple diseases (29), though pathophysiologic mechanisms had not been established previously. Based upon our data, we envisioned a scenario for chronic kidney disease, as depicted in Figure 8. In proteinuric glomerular diseases, which are often accompanied by hyperlipidemia, NEFAs are heavily bound to albumin and filtered, thereby permitting proximal tubule luminal exposure and uptake of NEFAs in large quantities. The CD36 scavenger receptor has been suggested to be involved in proximal tubule NEFA transport in humans (60), but it is not expressed in the proximal tubules of wild-type or diabetic mice (Supplemental Figure 6 and ref. 60). Vectorial acylation (or metabolic channeling), by an enzyme complex that includes ACSL1 and fatty acid transport protein-2 (FATP2) (refs. 61, 62, and S. Khan and J.R. Schelling, unpublished observations), instead directs NEFAs for simultaneous uptake and metabolism (63, 64). Following esterification, LC-CoA intermediates are transported into mitochondria or peroxisomes by the rate-limiting enzyme, CPT1a, and oxidized to generate ATP (Figure 8, pathway 1). When energy needs are met, unmetabolized LC-CoAs are stored as cytoplasmic triacylglycerol-rich lipid droplets or cleaved by the Them/ACOT family of acyl-CoA thioesterases (65) to shield cells from lipotoxicity (Figure 8, pathway 2). In contrast to adipocytes, LC-CoA buffering capacity is limited in epithelial cells, resulting in lipoapoptosis through multiple biochemical mediators (Figure 8 and ref. 29). Our data highlight the importance of NHE1 as a final checkpoint for cell survival. We have shown previously that apoptotic stresses stimulate proximal tubule NHE1, which triggers generation of downstream cell survival signals (8–10). However, if LC-CoAs accumulate and uncouple PI(4,5)P2 binding from juxtamembrane NHE1 domains, NHE1 becomes disabled, which allows apoptosis to proceed (Figure 8, pathway 3).The functions of the NHE1 Na+/H+ exchanger can therefore now be expanded to include a metabolic sensor role.
Figure 8. Schematic diagram of NEFA uptake and metabolism in proteinuric states.

Numbers 1–3 indicate the sequential order of NEFA metabolism. Green arrows indicate stimulatory pathways; red arrows indicate inhibitory pathways. ACSL1, acyl-CoA synthetase-1; Alb, albumin; Amn, amnionless; CPT1a; carnitine palmitoyl transferase-1a; Cub, cubilin; DAG, diacylglycerol; Dgat2; diacylglycerol acyltransferase-2; FATP2, fatty acid transfer protein-2; NHE1, Na+/H+ exchanger-1; PI(4,5)P2, phosphatidylinositol-(4,5)-bisphosphate; TG, triacylglycerol; Them2, thioesterase superfamily member 2.
We showed previously that PI(4,5)P2, which resides in the inner leaflet plasma membrane at a concentration of 10 to 15 μM (66–68), binds the cNHE1 with Kd of 52 μM (10). LC-CoAs bound the cNHE1 with slightly greater (23–34 μM) affinity. In contrast to high-affinity binding, which tends to be irreversible, low-affinity interactions with NHE1 are likely to be physiologically relevant, by allowing rapid on-off binding of multiple molecules, resulting in fine modulation of NHE1 function (68). Importantly, we estimated that the increased intracellular LC-CoA concentration in eNOS–/– db/db kidneys approaches 60 μM, thereby enabling LC-CoA displacement of phosphoinositides from NHE1 binding sites.
Although our data are consistent with a model of lipoapoptosis due to LC-CoA competition with PI(4,5)P2 for NHE1 binding, we cannot exclude the possibility that intracellular LC-CoAs could also alter phosphoinositide fatty acid side chain composition (69) or affect the interaction with other phosphoinositide-binding, apoptosis-modulating proteins, such as Akt (70) and p90 ribosomal S6 kinases (71), which are associated with NHE1 regulation (8, 71–73), or KATP channels (32–34), which are expressed in the proximal tubule (74), and cause K+ efflux, a prominent apoptotic feature (75).
In vivo validation of the role for LC-CoAs in the pathophysiology of kidney disease was achieved primarily in studies with the eNOS–/– db/db mouse, which was chosen because it is arguably the most reliable murine model of DN (76). A major advantage is the accelerated tempo of disease, with development of significant renal dysfunction by 26 weeks (35). We confirmed the glomerular histopathology as well as tubular atrophy and interstitial fibrosis, which are likely to contribute to the progression phenotype, since persistent proximal tubule apoptosis leads to glomerulosclerosis and kidney failure (77). In addition, we provide evidence that tubular atrophy is due to lipoapoptosis by demonstrating lipid accumulation and caspase-2 activation in proximal tubules. Although caspase-2 has been linked to multiple apoptotic stimuli, a recent report indicates that it is indispensable for lipoapoptosis (40).
One limitation of eNOS–/– db/db mice is that they represent a phenocopy for DN, since humans with diabetes harbor neither eNOS nor leptin receptor mutations. Baseline plasma NEFA concentrations are also lower in mice than in humans, though there is a similar 50% mean increase in plasma NEFAs in both db/db mice and humans with type 2 diabetes (78–80). Despite these interspecies differences, eNOS–/– db/db kidneys did contain lipid droplets and increased NEFA and LC-CoA concentrations, suggesting that eNOS–/– db/db mice reliably mimic human DN and lipotoxicity. Although we provide extensive in vitro data linking LC-CoA accumulation to apoptosis by NHE1 inhibition, an additional limitation of the experimental approach is that a direct cause-effect relationship could not be established using an in vivo model.
In most patients with chronic kidney disease, the mainstays of therapy are antihypertensive agents with dual antiproteinuric effects. Although this strategy would theoretically limit lipotoxicity by simultaneously diminishing NEFA exposure to the proximal tubule luminal transporters, our data suggest new targets that could be considered. One obvious possibility would be ACSL1 or FATP2 inhibitors, which inhibit vectorial acylation and/or limiting intracellular LC-CoA generation. LC-CoA must be esterified with carnitine to form acylcarnitine prior to CPT1a-dependent shuttling into mitochondria. L-carnitine supplementation might facilitate this process and thereby reduce cytosolic LC-CoA concentration (81). Dietary L-carnitine trials have achieved mixed results in dialysis patients, in an effort to improve multiple uremic symptoms, but the impact of carnitine on kidney disease progression has not been well studied (82). Preliminary trials have shown that PPARα agonists may represent an effective treatment for DN, though the mechanism of efficacy is unknown (83). Because PPARα agonists directly activate CPT1a (84), we speculate that a novel mechanism may be via CPT1a-directed acceleration of LC-CoA metabolism and abrogation of lipoapoptosis.
In summary, we describe a new biochemical mechanism, which is potentially applicable to multiple types of chronic kidney disease. Our data support a model in which diseased glomeruli with impaired permselectivity allow filtration and proximal tubule reabsorption of NEFA, which leads to accumulation of LC-CoAs, and competition with structurally similar phosphoinositides for binding to NHE1. In this capacity, the NHE1 Na+/H+ exchanger functions as a metabolic sensor for lipotoxicity, since LC-CoA–dependent NHE1 inactivation reduces the threshold for apoptosis, which culminates in tubular atrophy and renal failure.
Methods
Mice.
The generation and phenotype characteristics of C57BLKS/J eNOS–/– db/db (eNOS–/– db/db) and ROP/Le-Os Es1b/+ Es1a (ROPOs/+) mice have been described previously (35, 44, 85, 86). Mice were fed Prolabs Isopro RMH 3000 rat chow (LabDiet), containing 26% protein, 14% fat, and 60% carbohydrates.
Histology.
Kidneys fixed in 4% paraformaldehyde were imbedded in paraffin blocks, and 10-μm microtome sections were stained with Masson trichrome, according to the guidelines from Sigma-Aldrich (HT15), as previously described (87). To assess for tubular atrophy and interstitial fibrosis, quantitative histomorphometry was conducted on images at the magnification of ×40, which were overlaid with a 16 × 22 grid within Adobe Photoshop. Six sections per kidney were randomly selected for analysis. Coincidence of intersecting grid lines with tubule (nucleus, cytoplasm, or brush border), tubule lumen, or Masson’s trichrome–stained interstitium was counted, whereas glomeruli and blood vessels were omitted from calculations. The total of the 3 compartments was defined as 1.0, and the proportion of the total comprised by tubule cell, tubule lumen, or fibrosed interstitium was compared between eNOS+/– db/m and eNOS–/– db/db kidneys. Cytoplasmic lipid droplets were labeled with Oil red O (0.5%, 5 minutes, room temperature), followed by hematoxylin counterstaining. Stained sections were viewed at magnification of ×20 or ×40 by light microscopy. Apoptosis was assessed by TUNEL assays, and results were quantitated as previously described (11). In situ proximal tubule cell proliferation was determined by Ki-67 labeling, as previously described (87).
Immunohistochemistry.
Methods have been described previously in detail (87). Mouse kidney was frozen at –80°C. Samples were sectioned to 5 μm by cryostat, fixed in paraformaldehyde (4%, 10 minutes, 4°C), rinsed in PBS, and then permeabilized with Triton X-100 (Sigma-Aldrich; 0.2% in PBS, 10 minutes, 4°C). Sections were blocked with goat serum (5% in PBS, 1 hour, room temperature) and then incubated with anti-ACSL1 IgG (Cell Signaling; 1:25, overnight, 4°C) or anti-active caspase-2 (Abcam; 10 μg/ml, overnight, 4°C), followed by Alexa Fluor 488–conjugated goat anti-rabbit IgG (Invitrogen; 1:200, 1 hour, at room temperature). Proximal tubules were labeled with rabbit anti–aquaporin-1 IgG (1:200, 14 hours, 4°C) plus FITC-conjugated goat anti-rabbit IgG or Texas red– or FITC-conjugated Tetragonolobus purpureus lectin, as previously described (5). CD36 localization in kidney was determined by incubation with anti-CD36 IgG (Novus; 1:100, 14 hours, 4°C), followed by FITC- or Texas red–conjugated IgG. Sections were mounted in Aqueous Mounting Media (Vector) containing ToTo-3 nuclear counterstain (Invitrogen) and viewed by epifluorescence or confocal microscopy.
NHE1 function.
Mice were sacrificed at 12 weeks, kidneys were removed, and cortical suspensions were generated following serial sieving through mesh of progressively smaller diameter. Ultimately, fragments that flowed through 40-μm mesh, which excludes glomeruli and contains primarily proximal tubules, were harvested (88). Suspensions were loaded with BCECF-AM (1 μM, 30 minutes, 37°C), using 0.02% pluronic F127 to assist with uptake and dispersion. N-ethyl-N-isopropyl amiloride–sensitive (EIPA-sensitive; Sigma-Aldrich, 1 μM) Na+/H+ exchange was measured using NH4Cl washout acid load recovery protocol and BCECF fluorescence, which was assayed by spectrofluorimetry (89). In cultured cells, EIPA-sensitive Na+/H+ exchange was determined according to previously described methods (10). Similar results were observed for EIPA- and cariporide-inhibitable Na+/H+ exchange in cultured proximal tubule cells and kidney cortices ex vivo (Supplemental Figure 7), implying that the relevant Na+/H+ transporter is NHE1.
Cell culture.
C57BL/6 wild-type and NHE1-null C57BL/6Swe/Swe mouse proximal tubule cells were isolated by Percoll gradient centrifugation, maintained in primary culture in DMEM/F-12 (Invitrogen) plus 10% fetal bovine serum (HyClone) and 1% penicillin/streptomycin-fungizone (Sigma-Aldrich), and then immortalized by infection with temperature-sensitive SV40, as previously described (10). The HRPT cell line (gift from L. Racusen, Johns Hopkins University, Baltimore, Maryland, USA) was maintained in DMEM-F12 (Invitrogen) plus 10% fetal bovine serum (HyClone) and 1% penicillin/streptomycin-fungizone (Sigma-Aldrich), as described previously (11). LLC-PK1 cells (ATCC) were maintained in DMEM (Invitrogen) plus 10% fetal bovine serum (HyClone) and 1% penicillin/streptomycin-fungizone (Sigma-Aldrich), as described previously (10). For experiments testing the effects of high glucose and NEFA incubation, untreated control groups were maintained in DMEM with low glucose (5.56 mM) (Invitrogen).
Fatty acid analysis by gas chromatography–mass spectrometry.
The total concentrations of palmitate (16:0), stearate (18:0), oleate (18:1), and linoleate (18:2) were determined from tissues using gas chromatography–mass spectrometry (90). A known quantity of tissue was hydrolyzed and extracted after adding a known amount of heptadecanoic acid (17:0). Fatty acids were analyzed as their trimethylsilyl derivatives under electron impact ionization mode using an Agilent 5973N-MSD equipped with an Agilent 6890 GC system and a DB17-MS capillary column (30 m × 0.25-mm internal diameter × 0.25-μm film thickness).
LC-CoA analysis by liquid chromatography–mass spectrometry/MS.
In a known amount of tissue, acyl-CoAs were measured using liquid chromatography-mass spectrometry (LC-MS) methods, as previously described with slight modifications (91). Briefly, the tissue samples were spiked with an internal standard of [2H9]pentanoyl-CoA (0.4 nmol) and the supernatant fraction collected from tissue homogenate was loaded on to a preconditioned Supelco solid-phase extraction cartridge. Acyl-CoAs were eluted with 3 ml buffer (3:1 methanol/H2O with 50 mM ammonium formate) and 3 ml methanol. The eluent was evaporated under nitrogen gas and resuspended in the eluting buffer and then analyzed using a LC-MS/MS system. The LC was coupled with an API4000 Qtrap Mass Spectrometer (Applied Biosystems) and operated under positive ionization mode for the detection of acyl-CoA ions.
SPR.
Binding parameters for the interaction between LC-CoAs and the NHE1 cytosolic domain were measured using a Biacore 3000 SPR-based biosensor (Biacore AB), as previously described (10). Briefly, the wild-type rat cNHE1 or M1 + M2 mutant fusion proteins (Supplemental Figure 2; cDNAs were gifts from John Orlowksi, McGill University, Montreal, Quebec, Canada) were immobilized on a CM5 chip (Biacore) at a density of approximately 1,000 response units. Varying concentrations of palmitoyl-CoA or oleoyl-CoA analyte suspensions (Avanti Polar Lipids) in Biacore HBS-P buffer (10 mM HEPES [pH 7.4], 150 mM NaCl, 0.005% surfactant P20) were flowed at 5 μl per minute for 3 to 4 minutes to determine association rate constants, followed by a 4-minute perfusion with buffer only to determine dissociation rate constants. Data were analyzed using the BIAevaluation 3.1 program.
Membrane overlay assays.
The fusion protein corresponding to the entire rat cNHE1 (1 μg/ml, overnight, 4°C) was suspended in renaturation buffer and incubated with membrane phospholipids spotted on PIP strips (Echelon) or custom-made nitrocellulose strips spotted with palmitoyl-CoA, malonyl-CoA, or oleoyl-CoA (Sigma-Aldrich, 1 μl of 1 mM stock, 400 pmoles per spot) or C8-PI(4,5)P2 or C8-PI(3,4,5)P3 (Avanti, 1 μl of 1 mM, 400 pmoles per spot), which were then probed for cNHE1 binding with anti-NHE1 antibodies according to previously described methods (10).
TIRF microscopy and FRET.
FRET measurements were made between BODIPY-PI(4,5)P2 and N-terminal RFP-tagged NHE1 (gift from Jalal Khundmiri, University of Louisville, Louisville, Kentucky, USA) within TIRF microscopy fields according to previously described methods (10). RFP-tagged NHE1 retained Na+/H+ exchange activity (data not shown).
NHE1 localization in oocytes.
cRNA synthesis and oocyte microinjections were conducted as described previously (92). The cDNA encoding the entire rat cNHE1 (10) with a C-terminal EGFP tag was linearized with EcoRI and AgeI, purified using the QIAquick PCR Purification Kit (Qiagen), and subcloned into the Xenopus expression vector pGH19. Capped cRNA was transcribed from the linearized cDNA using the T7 mMessage mMachine Kit (Ambion). cRNA was purified and concentrated using the RNeasy MinElute RNA Cleanup Kit (Qiagen). Xenopus oocytes were surgically removed from anesthetized frog ovaries and separated by collagenase digestion. Stage V-VI oocytes were manually separated and stored at 18°C in oocyte recipe 3 (OR3) medium, supplemented with penicillin and streptomycin (500 U each). One day after isolation, oocytes were injected with 25 nl cRNA solution containing 900 ng/μl cRNA or 25 nl sterile water and then stored in OR3 medium at 18°C for 3 days prior to incubation with NEFA with or without chemical inhibitors or injection with acyl-CoAs. Upon experiment completion, oocytes were fixed in 4% paraformaldehyde, snap frozen in liquid nitrogen, cryostat sectioned, and viewed by confocal microscopy to assess EGFP plasma membrane localization.
Transfection with viral vectors.
To achieve ACSL1 overexpression, HRPT cells were infected with human ACSL1 adenoviral vector (Ad-ACSL1; gift from Jason Dyck, University of Alberta, Edmonton, Alberta, Canada). Ad-ACSL1 was propagated in E1A and E1B oncogene-transformed HEK 293A cells, and the virus supernatant was stored at –20°C. HRPT cells were plated on 10-cm dishes, grown to approximately 75% confluence, and infected with Ad-ACSL1 at a titer of 10 PFUs per cell. To achieve ACSL1 knockdown, HRPT cells were transfected with lentiviral shRNA vectors targeting human ACSL1 (RefSeq NM_001995.2). Oligonucleotides were purchased from the Mission shRNA Gene Set and cloned into pLKO.1-puro plasmid (Sigma-Aldrich), and lentiviral particles were produced in HEK 293T packaging cells using the ViraPower Expression System (Invitrogen). The viral supernatant was added to HRPT monolayers grown on 10-cm plates, and stably transduced cells were identified for further studies. Of the 5 constructs screened, 2 reliably silenced ACSL1 expression, as determined by immunoblotting (target coding sense sequences are underlined: ccggCCCTTGGTGTATTTCTATGATctcgAGATCATAGAAATACACCAAGGGtttttg [shRNA 520]; ccggGATGGTGATCGTTCCACTTTActcgAGTAAAGTGGAACGATCACCATCtttttg [shRNA 708]).
Immunoblotting.
Methods have been described previously (10). Briefly, cell monolayers were lysed and denatured in boiling SDS-PAGE buffer (125 mM Tris [pH 6.8], 2% SDS, 5% glycerol, 1% β-mercaptoethanol, 0.003% bromphenol blue) for 5 minutes. Samples (20 μg protein per lane) were resolved by SDS-PAGE and transferred to polyvinylidine difluoride membranes. Blots were blocked in 5% dried milk and probed with anti-ACSL1 IgG (Cell Signaling; 1:1,000 in 5% BSA, overnight, 4°C) and then HRP-conjugated IgG (1:10,000, 1 hour, room temperature). Band intensity was detected by enhanced chemiluminescence. Blots were exposed to stripping buffer (Thermo Scientific; 10 minutes, room temperature) and then reprobed with anti–α-tubulin IgG (Santa Cruz Biotechnology; 1:3,000, 1 hour, room temperature), followed by HRP-conjugated IgG (1:10,000, 1 hour, room temperature).
Immunoprecipitation.
Frozen mouse kidneys were pulverized in liquid nitrogen, and lysates (4 mg total protein per sample in RIPA buffer [20 mM Tris, pH 7.4, 20 mM NaF, 10% glycerol, 0.5% DCA, 125 mM NaCl, 0.1% SDS, 2 mM EDTA, 1% Triton X-100 plus protease inhibitor cocktail]) were immunoprecipitated with rabbit polyclonal anti-active caspase-2 (Abcam, ab2251, 1 μg, overnight, 4°C). On the following day, GammaBind Sepharose beads (GE Healthcare) were added (40 μl, blocked with 5% bovine serum albumin, 90 minutes, 4°C). Then, washed beads were incubated with SDS-PAGE buffer, to which 100 mM DTT was added (10 minutes, 70°C). Precipitated proteins were resolved by SDS-PAGE on an 18% gel and transferred to polyvinylidine difluoride membranes, which were blocked in 5% nonfat dried milk. Membranes were probed with anti-active caspase-2 IgG (1 μg/ml, overnight at 4°C), followed by HRP-conjugated protein A (1:4,000, 1 hour, room temperature), and band intensity was detected by enhanced chemiluminescence.
Statistics.
All experiments were conducted a minimum of 3 times. Results are expressed as mean ± SEM. Two-tailed paired t tests were used for statistical analysis between 2 groups. Two-way ANOVA was used for comparisons among more than 2 groups. Statistical significance was defined as P < 0.05.
Study approval.
All protocols were approved by the Institutional Animal Care and Use Committees at Vanderbilt and Case Western Reserve Universities.
Supplementary Material
Acknowledgments
This work was supported by NIH grants R01 DK067528 and R01 DK072348 (to J.R. Schelling), R01 DK30344 (to M.D. Parker), R01 HL097355 (to W.P. Schilling), and U24 DK76174 (to M.A. Puchowicz) as well as American Heart Association fellowship grant 0825593D (to B.G. Abu Jawdeh) and The Thailand Research Fund and Commission on Higher Education (TRF-CHE) Research Grant for New Scholar MRG5580125 (to K. Shinlapawittayatorn). We are grateful to John Orlowski (McGill University), who supplied the NHE1 cDNAs; to Jalal Khundmiri (University of Louisville), who supplied the RFP-NHE1 cDNA; and to Jason Dyck (University of Alberta), who supplied the adenoviral ACSL1 construct.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2014;124(3):1057–1068. doi:10.1172/JCI71863.
Bassam G. Abu Jawdeh’s present address is: Department of Medicine, University of Cincinnati, Cincinnati, Ohio, USA.
Krekwit Shinlapawittayatorn’s present address is: Cardiac Electrophysiology Research and Training Center, Department of Physiology, Faculty of Medicine, Chiang Mai University, Thailand.
References
- 1.Coresh J, et al. Prevalence of chronic kidney disease in the United States. JAMA. 2007;298(17):2038–2047. doi: 10.1001/jama.298.17.2038. [DOI] [PubMed] [Google Scholar]
- 2.Risdon RA, Sloper JC, De Wardener HE. Relationship between renal function and histological changes found in renal-biopsy specimens from patients with persistent glomerular nephritis. Lancet. 1968;2(7564):363–366. doi: 10.1016/s0140-6736(68)90589-8. [DOI] [PubMed] [Google Scholar]
- 3.Schainuck LI, Striker GE, Cutler RE, Benditt EP. Structural-functional correlations in renal disease. Hum Pathol. 1970;1(4):631–641. doi: 10.1016/S0046-8177(70)80061-2. [DOI] [PubMed] [Google Scholar]
- 4.Bohle A, Muller GA, Wehrmann M, Mackensen-Haen S, Xiao J-C. Pathogenesis of chronic renal failure in the primary glomerulopathies, renal vasculopathies, and chronic interstitial nephritides. Kidney Int Suppl. 1996;54:S2–S9. doi: 10.1046/j.1523-1755.1998.06802.x. [DOI] [PubMed] [Google Scholar]
- 5.Schelling JR, Nkemere N, Kopp JB, Cleveland RP. Fas-dependent fratricidal apoptosis is a mechanism of tubular epithelial cell deletion in chronic renal failure. Lab Invest. 1998;78(7):813–824. [PubMed] [Google Scholar]
- 6.Khan S, Cleveland RP, Koch CJ, Schelling JR. Hypoxia induces renal tubular epithelial cell apoptosis in chronic renal disease. Lab Invest. 1999;79(9):1089–1099. [PubMed] [Google Scholar]
- 7.Schelling JR, Cleveland RP. Involvement of Fas-dependent apoptosis in renal tubular epithelial cell deletion in chronic renal failure. Kidney Int. 1999;56(4):1313–1316. doi: 10.1046/j.1523-1755.1999.00684.x. [DOI] [PubMed] [Google Scholar]
- 8.Wu KL, et al. The NHE1 Na+/H+ exchanger recruits ezrin/radixin/moesin proteins to regulate Akt-dependent cell survival. J Biol Chem. 2004;279(25):26280–26286. doi: 10.1074/jbc.M400814200. [DOI] [PubMed] [Google Scholar]
- 9.Schelling JR, Abu Jawdeh BG. Regulation of cell survival by Na+/H+ exchanger-1 (NHE1). Am J Physiol Renal Physiol. 2008;295(3):F625–F632. doi: 10.1152/ajprenal.90212.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abu Jawdeh BG, et al. Phosphoinositide binding differentially regulates NHE1 Na+/H+ exchanger-dependent proximal tubule cell survival. J Biol Chem. 2011;286(49):42435–42445. doi: 10.1074/jbc.M110.212845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu KL, et al. Renal tubular epithelial cell apoptosis is associated with caspase cleavage of the NHE1 Na+/H+ exchanger. Am J Physiol Renal Physiol. 2003;284(4):F829–F839. doi: 10.1152/ajprenal.00314.2002. [DOI] [PubMed] [Google Scholar]
- 12.Weinberg JM. Lipotoxicity. Kidney Int. 2006;70(9):1560–1566. doi: 10.1038/sj.ki.5001834. [DOI] [PubMed] [Google Scholar]
- 13.Ruan XZ, Varghese Z, Moorhead JF. An update on the lipid nephrotoxicity hypothesis. Nat Rev Nephrol. 2009;5(12):713–721. doi: 10.1038/nrneph.2009.184. [DOI] [PubMed] [Google Scholar]
- 14.Sun L, Halaihel N, Zhang W, Rogers T, Levi M. Role of sterol regulatory element-binding protein 1 in regulation of renal lipid metabolism and glomerulosclerosis in diabetes mellitus. J Biol Chem. 2002;277(21):18919–18927. doi: 10.1074/jbc.M110650200. [DOI] [PubMed] [Google Scholar]
- 15.Proctor G, Jiang T, Iwahashi M, Wang Z, Li J, Levi M. Regulation of renal fatty acid and cholesterol metabolism, inflammation, and fibrosis in Akita and OVE26 mice with type 1 diabetes. Diabetes. 2006;55(9):2502–2509. doi: 10.2337/db05-0603. [DOI] [PubMed] [Google Scholar]
- 16.Thomas ME, Harris KPG, Walls J, Furness PN, Brunskill NJ. Fatty acids exacerbate tubulointerstitial injury in protein-overload proteinuria. Am J Physiol. 2002;283(4):F640–F647. doi: 10.1152/ajprenal.00001.2002. [DOI] [PubMed] [Google Scholar]
- 17.Kamijo A, et al. Urinary free fatty acids bound to albumin aggravate tubulointerstitial damage. Kidney Int. 2002;62(5):1628–1637. doi: 10.1046/j.1523-1755.2002.00618.x. [DOI] [PubMed] [Google Scholar]
- 18.van Timmeren MM, Bakker SJ, Stegeman CA, Gans RO, van GH. Addition of oleic acid to delipidated bovine serum albumin aggravates renal damage in experimental protein-overload nephrosis. Nephrol Dial Transplant. 2005;20(11):2349–2357. doi: 10.1093/ndt/gfh964. [DOI] [PubMed] [Google Scholar]
- 19.Arici M, Chana R, Lewington A, Brown J, Brunskill NJ. Stimulation of proximal tubular cell apoptosis by albumin-bound fatty acids mediated by peroxisome proliferator activated receptor-γ. J Am Soc Nephrol. 2003;14(1):17–27. doi: 10.1097/01.ASN.0000042167.66685.EA. [DOI] [PubMed] [Google Scholar]
- 20.Suzuki H, et al. Structure and regulation of rat long-chain acyl-CoA synthetase. J Biol Chem. 1990;265(15):8681–8685. [PubMed] [Google Scholar]
- 21.Achouri Y, et al. Long chain fatty acyl-CoA synthetase 5 expression is induced by insulin and glucose: involvement of sterol regulatory element-binding protein-1c. Biochimie. 2005;87(12):1149–1155. doi: 10.1016/j.biochi.2005.04.015. [DOI] [PubMed] [Google Scholar]
- 22.Li LO, Klett EL, Coleman RA. Acyl-CoA synthesis, lipid metabolism and lipotoxicity. Biochim Biophys Acta. 2010;1801(3):246–251. doi: 10.1016/j.bbalip.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mishra R, Emancipator SN, Miller C, Kern T, Simonson MS. Adipose differentiation-related protein and regulators of lipid homeostasis identified by gene expression profiling in the murine db/db diabetic kidney. Am J Physiol Renal Physiol. 2004;286(5):F913–F921. doi: 10.1152/ajprenal.00323.2003. [DOI] [PubMed] [Google Scholar]
- 24.Schmid H, et al. Modular activation of nuclear factor-κB transcriptional programs in human diabetic nephropathy. Diabetes. 2006;55(11):2993–3003. doi: 10.2337/db06-0477. [DOI] [PubMed] [Google Scholar]
- 25.Woroniecka KI, Park AS, Mohtat D, Thomas DB, Pullman JM, Susztak K. Transcriptome analysis of human diabetic kidney disease. Diabetes. 2011;60(9):2354–2369. doi: 10.2337/db10-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garbarino J, et al. Sterol and diacylglycerol acyltransferase deficiency triggers fatty acid mediated cell death. J Biol Chem. 2009;284(45):30994–31005. doi: 10.1074/jbc.M109.050443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Urahama Y, et al. Lipid droplet-associated proteins protect renal tubular cells from fatty acid-induced apoptosis. Am J Pathol. 2008;173(5):1286–1294. doi: 10.2353/ajpath.2008.080137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Greenberg AS, et al. The role of lipid droplets in metabolic disease in rodents and humans. J Clin Invest. 2011;121(6):2102–2110. doi: 10.1172/JCI46069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schaffer JE. Lipotoxicity: when tissues overeat. Curr Opin Lipidol. 2003;14(3):281–287. doi: 10.1097/00041433-200306000-00008. [DOI] [PubMed] [Google Scholar]
- 30.Riedel MJ, Light PE. Saturated and cis/trans unsaturated acyl CoA esters differentially regulate wild-type and polymorphic beta-cell ATP-sensitive K+ channels. Diabetes. 2005;54(7):2070–2079. doi: 10.2337/diabetes.54.7.2070. [DOI] [PubMed] [Google Scholar]
- 31.Riedel MJ, et al. Metabolic regulation of sodium-calcium exchange by intracellular acyl CoAs. EMBO J. 2006;25(19):4605–4614. doi: 10.1038/sj.emboj.7601321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Larsson O, Deeney JT, Branstrom R, Berggren PO, Corkey BE. Activation of the ATP-sensitive K+ channel by long chain acyl-CoA. A role in modulation of pancreatic β-cell glucose sensitivity. J Biol Chem. 1996;271(18):10623–10626. doi: 10.1074/jbc.271.18.10623. [DOI] [PubMed] [Google Scholar]
- 33.Rohacs T, Lopes CM, Jin T, Ramdya PP, Molnar Z, Logothetis DE. Specificity of activation by phosphoinositides determines lipid regulation of Kir channels. Proc Natl Acad Sci U S A. 2003;100(2):745–750. doi: 10.1073/pnas.0236364100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rapedius M, et al. Long chain CoA esters as competitive antagonists of phosphatidylinositol 4,5-bisphosphate activation in Kir channels. J Biol Chem. 2005;280(35):30760–30767. doi: 10.1074/jbc.M503503200. [DOI] [PubMed] [Google Scholar]
- 35.Zhao HJ, et al. Endothelial nitric oxide synthase deficiency produces accelerated nephropathy in diabetic mice. J Am Soc Nephrol. 2006;17(10):2664–2669. doi: 10.1681/ASN.2006070798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chander PN, et al. Nephropathy in Zucker diabetic fat rat is associated with oxidative and nitrosative stress: prevention by chronic therapy with a peroxynitrite scavenger ebselen. J Am Soc Nephrol. 2004;15(9):2391–2403. doi: 10.1097/01.ASN.0000135971.88164.2C. [DOI] [PubMed] [Google Scholar]
- 37.Wang Z, et al. Regulation of renal lipid metabolism, lipid accumulation, and glomerulosclerosis in FVBdb/db mice with type 2 diabetes. Diabetes. 2005;54(8):2328–2335. doi: 10.2337/diabetes.54.8.2328. [DOI] [PubMed] [Google Scholar]
- 38.Kumar D, Robertson S, Burns KD. Evidence of apoptosis in human diabetic kidney. Mol Cell Biochem. 2004;259(1–2):67–70. doi: 10.1023/B:MCBI.0000021346.03260.7e. [DOI] [PubMed] [Google Scholar]
- 39.Brezniceanu ML, et al. Attenuation of interstitial fibrosis and tubular apoptosis in db/db transgenic mice overexpressing catalase in renal proximal tubular cells. Diabetes. 2008;57(2):451–459. doi: 10.2337/db07-0013. [DOI] [PubMed] [Google Scholar]
- 40.Johnson ES, et al. Metabolomic profiling reveals a role for caspase-2 in lipoapoptosis. J Biol Chem. 2013;288(20):14463–14475. doi: 10.1074/jbc.M112.437210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Knudsen J, Neergaard TB, Gaigg B, Jensen MV, Hansen JK. Role of acyl-CoA binding protein in acyl-CoA metabolism and acyl-CoA-mediated cell signaling. J Nutr. 2000;130(2S suppl):294S–298S. doi: 10.1093/jn/130.2.294S. [DOI] [PubMed] [Google Scholar]
- 42.Neschen S, et al. Prevention of hepatic steatosis and hepatic insulin resistance in mitochondrial acyl-CoA:glycerol-sn-3-phosphate acyltransferase 1 knockout mice. Cell Metab. 2005;2(1):55–65. doi: 10.1016/j.cmet.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 43.Monetti M, et al. Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metab. 2007;6(1):69–78. doi: 10.1016/j.cmet.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 44.He CJ, et al. Dissociation of glomerular hypertrophy, cell proliferation, and glomerulosclerosis in mouse strains heterozygous for a mutation (Os) which induces a 50% reduction in nephron number. J Clin Invest. 1996;97(5):1242–1249. doi: 10.1172/JCI118539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aharonovitz O, Zaun HC, Balla T, York JD, Orlowski J, Grinstein S. Intracellular pH regulation by Na+/H+ exchange requires phosphatidylinositol 4,5-bisphosphate. J Cell Biol. 2000;150(1):213–224. doi: 10.1083/jcb.150.1.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Epand RM, Vuong P, Yip CM, Maekawa S, Epand RF. Cholesterol-dependent partitioning of PtdIns(4,5)P2 into membrane domains by the N-terminal fragment of NAP-22 (neuronal axonal myristoylated membrane protein of 22 kDa). Biochem J. 2004;379(pt 3):527–532. doi: 10.1042/BJ20040204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang J, Chen H, Vlahov IR, Cheng JX, Low PS. Evaluation of disulfide reduction during receptor-mediated endocytosis by using FRET imaging. Proc Natl Acad Sci U S A. 2006;103(37):13872–13877. doi: 10.1073/pnas.0601455103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sweeney G, et al. Intracellular delivery of phosphatidylinositol (3,4,5)-trisphosphate causes incorporation of glucose transporter 4 into the plasma membrane of muscle and fat cells without increasing glucose uptake. J Biol Chem. 2004;279(31):32233–32242. doi: 10.1074/jbc.M402897200. [DOI] [PubMed] [Google Scholar]
- 49.Casas J, Gijon MA, Vigo AG, Crespo MS, Balsinde J, Balboa MA. Phosphatidylinositol 4,5-bisphosphate anchors cytosolic group IVA phospholipase A2 to perinuclear membranes and decreases its calcium requirement for translocation in live cells. Mol Biol Cell. 2006;17(1):155–162. doi: 10.1091/mbc.E05-06-0545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ellis JM, et al. Mouse cardiac acyl-CoA synthetase-1 deficiency impairs fatty acid oxidation and induces cardiac hypertrophy. Mol Cell Biol. 2011;31(6):1252–1262. doi: 10.1128/MCB.01085-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thomas ME, Morrison AR, Schreiner GF. Metabolic effects of fatty acid-bearing albumin on a proximal tubule cell line. Am J Physiol. 1995;268(6 pt 2):F1177–F1184. doi: 10.1152/ajprenal.1995.268.6.F1177. [DOI] [PubMed] [Google Scholar]
- 52.Feldkamp T, Kribben A, Roeser NF, Ostrowski T, Weinberg JM. Alleviation of fatty acid and hypoxia-reoxygenation-induced proximal tubule deenergization by ADP/ATP carrier inhibition and glutamate. Am J Physiol Renal Physiol. 2007;292(5):F1606–F1616. doi: 10.1152/ajprenal.00476.2006. [DOI] [PubMed] [Google Scholar]
- 53.Soumura M, et al. Oleate and eicosapentaenoic acid attenuate palmitate-induced inflammation and apoptosis in renal proximal tubular cell. Biochem Biophys Res Commun. 2010;402(2):265–271. doi: 10.1016/j.bbrc.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 54.Fraser DA, Thoen J, Rustan AC, Forre O, Kjeldsen-Kragh J. Changes in plasma free fatty acid concentrations in rheumatoid arthritis patients during fasting and their effects upon T-lymphocyte proliferation. Rheumatology. 1999;38(10):948–952. doi: 10.1093/rheumatology/38.10.948. [DOI] [PubMed] [Google Scholar]
- 55.Artwohl M, et al. Fatty acids induce apoptosis in human smooth muscle cells depending on chain length, saturation, and duration of exposure. Atherosclerosis. 2009;202(2):351–362. doi: 10.1016/j.atherosclerosis.2008.05.030. [DOI] [PubMed] [Google Scholar]
- 56.De Zeeuw D, et al. Proteinuria, a target for renoprotection in patients with type 2 diabetic nephropathy: Lessons from RENAAL. Kidney Int. 2004;65(6):2309–2320. doi: 10.1111/j.1523-1755.2004.00653.x. [DOI] [PubMed] [Google Scholar]
- 57.Peterson JC, et al. Blood pressure control, proteinuria, and the progression of renal disease. The Modification of Diet in Renal Disease Study. Ann Intern Med. 1995;123(10):754–762. doi: 10.7326/0003-4819-123-10-199511150-00003. [DOI] [PubMed] [Google Scholar]
- 58.Remuzzi G, Bertani T. Pathophysiology of progressive nephropathies. N Engl J Med. 1998;339(20):1448–1456. doi: 10.1056/NEJM199811123392007. [DOI] [PubMed] [Google Scholar]
- 59.Ghiggeri GM, et al. Characterization of cationic albumin in minimal change nephropathy. Kidney Int. 1987;32(4):547–553. doi: 10.1038/ki.1987.243. [DOI] [PubMed] [Google Scholar]
- 60.Susztak K, Ciccone E, McCue P, Sharma K, Bottinger EP. Multiple metabolic hits converge on CD36 as novel mediator of tubular epithelial apoptosis in diabetic nephropathy. PLoS Med. 2005;2(2):e45. doi: 10.1371/journal.pmed.0020045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hirsch D, Stahl A, Lodish HF. A family of fatty acid transporters conserved from mycobacterium to man. Proc Natl Acad Sci U S A. 1998;95(15):8625–8629. doi: 10.1073/pnas.95.15.8625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johnson AC, Stahl A, Zager RA. Triglyceride accumulation in injured renal tubular cells: alterations in both synthetic and catabolic pathways. Kidney Int. 2005;67(6):2196–2209. doi: 10.1111/j.1523-1755.2005.00325.x. [DOI] [PubMed] [Google Scholar]
- 63.Richards MR, Harp JD, Ory DS, Schaffer JE. Fatty acid transport protein 1 and long-chain acyl coenzyme A synthetase 1 interact in adipocytes. J Lipid Res. 2006;47(3):665–672. doi: 10.1194/jlr.M500514-JLR200. [DOI] [PubMed] [Google Scholar]
- 64.Digel M, Ehehalt R, Stremmel W, Fullekrug J. Acyl-CoA synthetases: fatty acid uptake and metabolic channeling. Mol Cell Biochem. 2009;326(1–2):23–28. doi: 10.1007/s11010-008-0003-3. [DOI] [PubMed] [Google Scholar]
- 65.Hunt MC, Rautanen A, Westin MA, Svensson LT, Alexson SE. Analysis of the mouse and human acyl-CoA thioesterase (ACOT) gene clusters shows that convergent, functional evolution results in a reduced number of human peroxisomal ACOTs. FASEB J. 2006;20(11):1855–1864. doi: 10.1096/fj.06-6042com. [DOI] [PubMed] [Google Scholar]
- 66.McLaughlin S, Wang J, Gambhir A, Murray D. PIP(2) and proteins: interactions, organization, and information flow. Annu Rev Biophys Biomol Struct. 2002;31:151–175. doi: 10.1146/annurev.biophys.31.082901.134259. [DOI] [PubMed] [Google Scholar]
- 67.Liu AP, Fletcher DA. Actin polymerization serves as a membrane domain switch in model lipid bilayers. Biophys J. 2006;91(11):4064–4070. doi: 10.1529/biophysj.106.090852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gamper N, Shapiro MS. Regulation of ion transport proteins by membrane phosphoinositides. Nat Rev Neurosci. 2007;8(12):921–934. doi: 10.1038/nrn2257. [DOI] [PubMed] [Google Scholar]
- 69.Gu Z, et al. Polyunsaturated fatty acids affect the localization and signaling of PIP3/AKT in prostate cancer cells. Carcinogenesis. 2013;34(9):1968–1975. doi: 10.1093/carcin/bgt147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296(5573):1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 71.Takahashi E, et al. p90(RSK) is a serum-stimulated Na+/H+ exchanger isoform-1 kinase. Regulatory phosphorylation of serine 703 of Na+/H+ exchanger isoform-1. J Biol Chem. 1999;274(29):20206–20214. doi: 10.1074/jbc.274.29.20206. [DOI] [PubMed] [Google Scholar]
- 72.Snabaitis AK, Cuello F, Avkiran M. Protein kinase B/Akt phosphorylates and inhibits the cardiac Na+/H+ exchanger NHE1. Circ Res. 2008;103(8):881–890. doi: 10.1161/CIRCRESAHA.108.175877. [DOI] [PubMed] [Google Scholar]
- 73.Meima ME, Webb BA, Witkowska HE, Barber DL. The sodium-hydrogen exchanger NHE1 is an Akt substrate necessary for actin filament reorganization by growth factors. J Biol Chem. 2009;284(39):26666–26675. doi: 10.1074/jbc.M109.019448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brochiero E, Wallendorf B, Gagnon D, Laprade R, Lapointe JY. Cloning of rabbit Kir6.1, SUR2A, and SUR2B: possible candidates for a renal K(ATP) channel. Am J Physiol Renal Physiol. 2002;282(2):F289–F300. doi: 10.1152/ajprenal.00063.2001. [DOI] [PubMed] [Google Scholar]
- 75.Bortner CD, Hughes FM, Hughes FM, Jr, Cidlowski JA. A primary role for K+ and Na+ efflux in the activation of apoptosis. J Biol Chem. 1997;272(51):32436–32442. doi: 10.1074/jbc.272.51.32436. [DOI] [PubMed] [Google Scholar]
- 76.Brosius FC 3rd, et al. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2009;20(12):2503–2512. doi: 10.1681/ASN.2009070721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Grgic I, et al. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int. 2012;82(2):172–183. doi: 10.1038/ki.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yajima K, et al. Combination therapy with PPARγ and PPARα agonists increases glucose-stimulated insulin secretion in db/db mice. Am J Physiol Endocrinol Metab. 2003;284(5):E966–E971. doi: 10.1152/ajpendo.00149.2002. [DOI] [PubMed] [Google Scholar]
- 79.Fraze E, Donner CC, Swislocki AL, Chiou YA, Chen YD, Reaven GM. Ambient plasma free fatty acid concentrations in noninsulin-dependent diabetes mellitus: evidence for insulin resistance. J Clin Endocrinol Metab. 1985;61(5):807–811. doi: 10.1210/jcem-61-5-807. [DOI] [PubMed] [Google Scholar]
- 80.Reaven GM, Hollenbeck C, Jeng CY, Wu MS, Chen YD. Measurement of plasma glucose, free fatty acid, lactate, and insulin for 24 h in patients with NIDDM. Diabetes. 1988;37(8):1020–1024. doi: 10.2337/diab.37.8.1020. [DOI] [PubMed] [Google Scholar]
- 81.Hoppel C. The role of carnitine in normal and altered fatty acid metabolism. Am J Kidney Dis. 2003;41(4 suppl 4):S4–S12. doi: 10.1016/s0272-6386(03)00112-4. [DOI] [PubMed] [Google Scholar]
- 82.Calo LA, Vertolli U, Davis PA, Savica V. L-carnitine in hemodialysis patients. Hemodial Int. 2012;16(3):428–434. doi: 10.1111/j.1542-4758.2012.00679.x. [DOI] [PubMed] [Google Scholar]
- 83.Kouroumichakis I, Papanas N, Zarogoulidis P, Liakopoulos V, Maltezos E, Mikhailidis DP. Fibrates: therapeutic potential for diabetic nephropathy? Eur J Intern Med. 2012;23(4):309–316. doi: 10.1016/j.ejim.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 84.Rakhshandehroo M, Knoch B, Muller M, Kersten S. Peroxisome proliferator-activated receptor alpha target genes. PPAR Res. 2010;2010:612089. doi: 10.1155/2010/612089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gruneburg H. Genetical studies on the skeleton of the mouse. XVIII. Three gene for syndactylism. J Genet. 1956;54:113–145. doi: 10.1007/BF02981706. [DOI] [Google Scholar]
- 86.El-Meanawy MA, et al. Use of serial analysis of gene expression to generate kidney expression libraries. Am J Physiol Renal Physiol. 2000;279(2):F383–F392. doi: 10.1152/ajprenal.2000.279.2.F383. [DOI] [PubMed] [Google Scholar]
- 87.Khan S, et al. Mesangial cell integrin αvβ8 provides glomerular endothelial cell cytoprotection by sequestering TGF-β and regulating PECAM-1. Am J Pathol. 2011;178(2):609–620. doi: 10.1016/j.ajpath.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Terryn S, et al. A primary culture of mouse proximal tubular cells, established on collagen-coated membranes. Am J Physiol Renal Physiol. 2007;293(2):F476–F485. doi: 10.1152/ajprenal.00363.2006. [DOI] [PubMed] [Google Scholar]
- 89.Chambrey R, Achard JM, St John PL, Abrahamson DR, Warnock DG. Evidence for an amiloride-insensitive Na+/H+ exchanger in rat renal cortical tubules. Am J Physiol. 1997;273(3 pt 1):C1064–C1074. doi: 10.1152/ajpcell.1997.273.3.C1064. [DOI] [PubMed] [Google Scholar]
- 90.Brunengraber DZ, McCabe BJ, Kasumov T, Alexander JC, Chandramouli V, Previs SF. Influence of diet on the modeling of adipose tissue triglycerides during growth. Am J Physiol Endocrinol Metab. 2003;285(4):E917–E925. doi: 10.1152/ajpendo.00128.2003. [DOI] [PubMed] [Google Scholar]
- 91.Zhang GF, et al. Catabolism of 4-hydroxyacids and 4-hydroxynonenal via 4-hydroxy-4-phosphoacyl-CoAs. J Biol Chem. 2009;284(48):33521–33534. doi: 10.1074/jbc.M109.055665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Parker MD, Musa-Aziz R, Rojas JD, Choi I, Daly CM, Boron WF. Characterization of human SLC4A10 as an electroneutral Na/HCO3 cotransporter (NBCn2) with Cl- self-exchange activity. J Biol Chem. 2008;283(19):12777–12788. doi: 10.1074/jbc.M707829200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.