

Abstract
Doublecortin-like kinase 1 protein (DCLK1) is a gastrointestinal tuft cell marker that has been proposed to identify quiescent and tumor growth–sustaining stem cells. DCLK1+ tuft cells are increased in inflammation-induced carcinogenesis; however, the role of these cells within the gastrointestinal epithelium and their potential as cancer-initiating cells are poorly understood. Here, using a BAC-CreERT–dependent genetic lineage–tracing strategy, we determined that a subpopulation of DCLK1+ cells is extremely long lived and possesses rare stem cell abilities. Moreover, genetic ablation of Dclk1 revealed that DCLK1+ tuft cells contribute to recovery following intestinal and colonic injury. Surprisingly, conditional knockdown of the Wnt regulator APC in DCLK1+ cells was not sufficient to drive colonic carcinogenesis under normal conditions; however, dextran sodium sulfate–induced (DSS-induced) colitis promoted the development of poorly differentiated colonic adenocarcinoma in mice lacking APC in DCLK1+ cells. Importantly, colonic tumor formation occurred even when colitis onset was delayed for up to 3 months after induced APC loss in DCLK1+ cells. Thus, our data define an intestinal DCLK1+ tuft cell population that is long lived, quiescent, and important for intestinal homeostasis and regeneration. Long-lived DCLK1+ cells maintain quiescence even following oncogenic mutation, but are activated by tissue injury and can serve to initiate colon cancer.
Introduction
Colorectal cancer arises as a result of a series of genetic changes that include activating mutations in oncogenes and inactivating mutations in tumor suppressor genes. The most common initial genetic event involves inactivation in the APC tumor suppressor gene, which leads to stabilization and nuclear translocation of β-catenin (1). For many years, this initiating event was thought to occur primarily, if not exclusively, in crypt stem cells in the colon. Indeed, activation of β-catenin in rapidly dividing crypt base columnar (CBC) cells has been shown to lead to intestinal neoplasia, while villous cells appear largely resistant to APC deletion (2).
However, more recent studies have suggested that intestinal tumors commonly arise from cellular compartments outside of CBC cells. First, many early adenomatous polyps are detected at the top of colonic glands without a clear connection to the stem cell region in the crypts, suggesting a “top-down model” for adenoma formation (3, 4). Second, recent genetic studies have revealed that the combination of β-catenin activation and NF-κB signaling can convert LGR5– cells into LGR5+ stem cells that give rise to intestinal neoplasms (5). This model of dedifferentiation or interconversion of postmitotic cells outside the stem cell compartment of the crypt into cancer-initiating cells is appealing. However, the study by Schwitalla et al. (5) did not answer the question as to whether all LGR5– cells, or only a subpopulation of these cells, are capable of such interconversion. In addition, the genetic models used required two simultaneous hits to produce this phenotype. For such a model to produce cancer-initiating cells in vivo, the cell in question would have to be very long lived, which is problematic, given that it is well established that most intestinal and colonic epithelial cells outside of the crypts turn over within 4 to 5 days (6), except for Paneth (7, 8) and enteroendocrine (9) cells, which can persist for up to 2 months.
The four well-studied intestinal cell types including goblet cells, Paneth cells, enterocytes, and enteroendocrine cells, have all been shown to be derived from actively cycling LGR5+ stem cells located in the crypt base (10). More recently, a fifth epithelial cell type, known as the tuft cell, has been recognized and shown to be LGR5 derived (11). This rare cell type, originally described in the rodent trachea (12) and stomach (13) more than 60 years ago, was subsequently found throughout the entire digestive and respiratory tracts (14). Tuft cells have been implicated in chemoreception (14–18) and express proteins of the eicosanoid pathway such as cyclo-oxygenase-1 and cyclo-oxygenase-2 (19). Recently, tuft cells have been shown to express the protein doublecortin-like kinase 1 protein (DCLK1, previously referred to as KIAA0369 or DCAMKL1), which encodes a microtubule-associated protein with a C-terminal serine-threonine kinase domain (20, 21). Jay and coworkers further classified DCLK1+ tuft cells as postmitotic and reported their dependence on the expression of the transcription factor ATOH1/MATH1, suggesting that tuft cells constitute a novel secretory lineage in the intestinal epithelium (11).
In addition to labeling intestinal tuft cells and embryonic neuronal stem cells (22), DCLK1 has also been proposed to be a marker of quiescent intestinal stem cells (23–25). This assumption was based on the expression of DCLK1 in rare +4-positioned cells within intestinal crypts. Upon isolation, intestinal DCLK1+ cells formed primitive epithelial spheres (24). Moreover, DCLK1+ tuft cells are substantially increased in a number of models of inflammation-induced carcinogenesis, arguing for a possible role in malignant transformation (26–28). Recently, Nakanishi and colleagues described the generation of a Dclk1-CreERT2 knockin mouse that allowed formal lineage tracing. The authors reported that DCLK1+ cells were short lived and rarely functioned as intestinal stem cells under physiologic and pathologic conditions, but functioned as cancer stem cells in established tumors of ApcMin/+ mice (29). Nevertheless, the role of DCLK1+ tuft cells and their contribution to the origin of cancer has not been studied using genetic fate-mapping methods.
Thus, to further study the DCLK1 lineage in homeostasis, regeneration, and carcinogenesis, we generated Dclk1-CreERT BAC transgenic mice that, in contrast to the previously reported Dclk1-CreERT2 knockin mouse (29), contained two intact endogenous Dclk1 loci. Here, we report that our Dclk1-CreERT BAC transgenic mouse faithfully labels intestinal tuft cells and identifies a small subset of DCLK1+ cells that is exceptionally long lived and quiescent. These cells require neuronal input for survival and are involved in the epithelial response to injury. More importantly, DCLK1+ cells remain quiescent and long lived, even upon loss of APC, but become powerful cancer-initiating cells in the setting of inflammatory insult.
Results
DCLK1 marks long-lived quiescent epithelial tuft cells.
To study DCLK1+ tuft cells in intestinal homeostasis and carcinogenesis, we established a genetic fate-mapping system to label DCLK1-expressing cells. Dclk1-BAC-Cre and Dclk1-BAC-CreERT transgenic mice were generated by pronuclear injection of the BAC clone RP23-283D6 that contained an approximately 50-kb 5′ sequence of the Dclk1 gene–coding region plus a CreTM-FrtNeoFrt cassette, inserted by homologous recombination directly upstream of the ATG in exon 2 of the Dclk1-coding region (Supplemental Figure 1A; supplemental material available online with this article; doi: 10.1172/JCI73434DS1). Dclk1-BAC-Cre and Dclk1-BAC-CreERT transgenic mice were then crossed with various reporter mice (Supplemental Table 2) to perform genetic lineage-tracing experiments (30) in homeostasis, inflammation, and cancer.
Twenty-four hours after tamoxifen administration, in four separate founders crossed with R26-LacZ or R26-TGFP reporter mice, recombination was evident in rare (0.5%–1.5% of all epithelial cells) cells (Supplemental Figure 1C) throughout the gastric, intestinal, and colonic mucosa (Figure 1A and Supplemental Figure 1B). The cells positive for the LacZ or GFP reporter were confirmed to be epithelial tuft cells by their morphology and expression of DCLK1+ protein as detected by DCLK1 immunofluorescence (Figure 1, A and B). To further characterize the labeled cells and validate the Cre expression pattern of our Dclk1-BAC-Cre and Dclk1-BAC-CreERT transgenic mice, we performed immunohistochemistry (IHC) for other tuft cell markers previously reported in the literature (19). DCLK1 protein expression colocalized with DCLK1 Cre expression approximately 95%–98% of the time, while α-gustducin and acetylated tubulin labeled the vast majority of the Dclk1-CreTM–expressing cells in the stomach, intestine, and colon 2 months after tamoxifen induction (Supplemental Table 1 and Supplemental Figure 2, A–C). We did not observe recombination without administration of tamoxifen in mice of both sexes up to 12 months of age (Supplemental Figure 1, C and D).
Figure 1. DCLK1 labels long-lived epithelial tuft cells.

(A) Representative LacZ staining of tuft cells in the gastrointestinal tract of Dclk1 R26LacZ mice 24 hours after tamoxifen administration. (B) Representative immunofluorescence staining for DCLK1 (red) in Dclk1-CreGFP mice. (C) Representative LacZ staining of tuft cells in the gastrointestinal tract of Dclk1 R26LacZ mice up to 18 months after tamoxifen administration. (D) Quantification of labeled tuft cells in Dclk1 R26LacZ mice at different time points after tamoxifen treatment (n >5). (E) Quantification of DCLK1+ cells in Dclk1 R26-TGFP × Dclk1flox/WT mice compared with controls 8 weeks after tamoxifen administration (n = 3). ***P < 0.001. (F and G) Representative immunofluorescence staining for DCLK1 (red) and BrdU (green) in the small intestine and colon. (H) Quantification of BrdU-labeled DCLK1+ tuft cells after 1, 3, and 6 months of continuous BrdU administration (n = 3). Scale bars: 10 μm (B), 20 μm (A and C), and 50 μm (C, top two right panels; F and G).
Next, we performed lineage tracing in our Dclk1-BAC-CreERT transgenic mice and found that the majority of DCLK1+ cells seen 24 hours after tamoxifen induction disappeared within 10 to 14 days, a finding largely in accordance with previous reports (29). However, a small number (~5% of the cells initially labeled) of long-lived single LacZ+ DCLK1+ cells were easily detectable at later time points throughout the gastric, intestinal, and colonic epithelium, surviving for up to 18 months following induction with tamoxifen (Figure 1, C and D, and Supplemental Figure 1B). Importantly, these long-lived cells uniformly expressed the intestinal tuft cell–specific marker COX2 (Supplemental Figure 2D), proving that they were indeed tuft cells (19, 31). To confirm that our transgenic mice faithfully labeled COX2+ epithelial tuft cells, we performed COX2 staining on intestinal sections of Dclk1-BAC-CreERT × R26-tdTom mice and found that 91% of the DCLK1+ cells labeled in our mice stained positive for COX2 (Supplemental Figure 2, D and E).
To date, there remains controversy surrounding the lifespan of tuft cells in the gastrointestinal epithelium. Several studies have demonstrated that DCLK1+ cells only rarely take up BrdU after short-term labeling (23, 27, 32), while others have suggested that these cells cycle every 2 weeks (19, 29, 31). Using a Dclk1-CreERT knockin mouse that results in heterozygous deficiency of endogenous DCLK1, Nakanishi and colleagues reported a 2-week life span for DCLK1+ cells (29). These results are in sharp contrast to the population of long-lived DCLK1+ cells identified using our BAC transgenic mice. We hypothesized that loss of one allele of DCLK1 changes the behavior of tuft cells. To test this hypothesis, we took advantage of Dclk1floxed mice, which were crossed with Dclk1-BAC-CreERT × R26-TGFP mice. Eight weeks after induction with tamoxifen, recombined cells were easily identifiable in control (Dclk1-BAC-CreERT × R26-TGFP) mice, whereas heterozygous loss of DCLK1 led to a significant decrease in recombined cells (Figure 1E), providing a possible explanation for the differences between our results and previously published observations (29). To further confirm the relative quiescence and longevity of DCLK1+ cells, we performed long-term (1–6 months) continuous BrdU labeling experiments (Figure 1, F–H). In these studies, we found that only approximately 40% of DCLK1+ cells were positive for BrdU following 1 month of continuous labeling, despite greater than 95% of the remaining epithelium being labeled at this time point. This gradually increased to include the majority (>98% of cells) of DCLK1+ tuft cells over a period of 6 months of BrdU labeling. These results are also in keeping with a recent report (32) confirming that DCLK1+ cells have very low proliferating cell nuclear antigen (PCNA) and antigen Ki67 (Ki67) expression, indicating relative quiescence.
DCLK1+ cells are LGR5 derived and supported by neuronal signals.
To examine the origin of DCLK1+ cells in the epithelium, we performed lineage tracing in Lgr5-GFP-IRES-CreERT2 × R26-tdTom mice, which label the well-described LGR5+ stem cells of the gut (10). DCLK1 immunofluorescence revealed that the majority of DCLK1+ cells were indeed detected within the LGR5+ cell lineage (Figure 2, A and B).
Figure 2. DCLK1+ tuft cells are derived from LGR5 stem cells and require neural input for survival.

(A and B) Representative immunofluorescence for DCLK1 (green) in Lgr5-CreERT-IRES-GFP R26tdTom mice. Scale bars: 50 μm (A) and 20 μm (B). (C) Representative immunofluorescence for DCLK1 (red) in Lgr5-CreERT-IRES-GFP × Atohflox/flox mice. Scale bar: 10 μm. (D) Quantification of DCLK1+ tuft cells in GFP-positive crypts in Lgr5-CreERT-IRES-GFP Atohflox/flox mice 2 months after administration of tamoxifen (n = 3). (E and F) Representative images of intestinal organoids derived from Dclk1 R26tdTom mice 24 hours (E) and 7 days (F) after isolation. Scale bars: 20 μm (E) and 25 μm (F). (G) Quantification of recombined cells in intestinal organoids derived from Dclk1 R26tdTom mice (n = 3). (H) IHC for DCLK1 in paraffin-embedded intestinal organoids 14 days after isolation. Scale bar: 20 μm. (I) Reconstruction of cocultured colonic organoids (red) and primary neurons (green) imaged with 2-photon microscopy. (J and K) Colonic organoids derived from Dclk1 R26tdTom mice cultured in the absence (J) and presence (K) of GFP-positive neurons after 7 days in culture. Scale bars: 50 μm (I–K). (L) Quantification of recombined cells in colonic organoids derived from Dclk1 R26tdTom in the absence and presence of neurons 14 days after isolation. (M–P) Colonic organoids derived from Dclk1 R26-DTA mice cultured in the presence (N and P) and absence (M and O) of neurons. 4-OH-tamoxifen was added to induce the expression of DTA in DCLK+ cells (O and P). Scale bars: 100 μm (M–P). (Q) Quantification of colonic organoids in the presence (+ Neurons) and absence of neurons and after DCLK1+ cell ablation (+ Tam). Results are shown as percentages and normalized to control (– Tam). (R) Size of colonic organoids in the presence (+ Neurons) and absence of neurons and after DCLK1+ cell ablation (+ Tam) (n = 3). *P < 0.05; **P < 0.01; ***P < 0.001.
Tuft cells have previously been postulated to be part of the secretory lineage (which also includes goblet, enteroendocrine, and Paneth cells) of the intestinal epithelium (31). However, others have reported the presence of DCLK1+ cells in the setting of ATOH1 loss (33, 34). Thus, to assess the extent to which DCLK1+ tuft cells are a part of the ATOH1-dependent secretory lineage, we examined intestinal sections from Lgr5-GFP-IRES-CreERT2 mice crossed with Atoh1flox/flox mice (35, 36). When compared with WT control mice, Lgr5-GFP-IRES-CreERT2 × Atoh1flox/flox mice showed similar numbers of DCLK1+ tuft cells within LGR5-GFP+ crypts, even up to 2 to 3 months after tamoxifen administration (Figure 2, C and D). These data independently confirm previous reports (33, 34) that not all DCLK1+ tuft cells are part of the intestinal secretory lineage, as previously suggested (19).
Since DCLK1+ cells were derived from LGR5+ cells in vivo, we wondered whether LGR5+ stem cells were capable of generating DCLK1+ cells in in vitro. To examine this question, we studied gastrointestinal crypt 3D (organoid) cultures (37, 38) and were surprised to observe a rapid and complete loss of the DCLK1+ lineage over time, with no DCLK1+ cells detected by recombination or IHC after 1 week in culture (Figure 2, E–H). These results suggested that nonepithelial signals might be critical for tuft cell maintenance and longevity in organoid cultures. Given that previous reports suggested an association between epithelial tuft cells and neuronal structures (14, 18, 39), we hypothesized that neuronal input might support tuft cell survival in colonic, intestinal, and gastric organoid cultures. To test this hypothesis, we established a coculture system of primary neurons isolated from the spinal cord (40) and gastrointestinal organoids based on the protocols published by Clevers and coworkers (refs. 38, 41, 42, and Figure 2I). Upon coculture of organoids isolated from Dclk1-CreERT × R26-tdTom mice with primary neuronal cells, we observed that loss of DCLK1+ cells could be averted (Figure 2, J–L, Supplemental Figure 3, A and B, Supplemental Figure 4, A–E, and Supplemental Figure 5, A–F), suggesting that nerves can prolong the survival of DCLK1+ cells in vitro. Interestingly, the loss of DCLK1+ cells in vitro could also be prevented by the addition of the cholinergic agonist pilocarpine instead of primary neurons. This suggests that nerves may contribute to the maintenance of gastrointestinal tuft cells through cholinergic signaling (Supplemental Figure 5, G–I).
To test whether functional innervation was associated with tuft cell survival in vivo, we examined paired biopsies from intestinal transplant patients (n = 2) and found that while the transplanted graft (which was extrinsically denervated) lacked DCLK1+ cells, the endogenous intestine (which was normally innervated) had a normal complement of DCLK1+ cells (Supplemental Figure 3, A and B). Similarly, in Ret–/– mice lacking a functional enteric nervous system (refs. 43, 44, and Supplemental Figure 4C), embryonic intestinal tissues lacked intestinal COX2+ tuft cells when compared with those of control mice (Supplemental Figure 4, D and E). In these studies, epithelial COX2 (11, 19) was used to define intestinal tuft cells, since DCLK1 expression in the mouse is not detected until after birth (11). These findings suggested that DCLK1+ cells are dependent on proper neural innervation of the gut for their survival in vitro and in vivo.
Interestingly, the addition of nerves to in vitro cultures increased both the number and size of the resulting colonic organoids (Figure 2, M, N, Q, and R and Supplemental Figure 3, C, D, and G). To test whether this effect was at least partly mediated through DCLK1+ tuft cells, we ablated DCLK1+ cells in vitro using Cre-mediated expression of the diphtheria toxin subunit A (DTA) (45). Indeed, nerves failed to support organoid growth in the absence of DCLK1+ tuft cells (Figure 2, O, P, Q, and R and Supplemental Figure 3, E, F, and G), arguing that DCLK1+ cells are involved in the integration of neuronal signals into the epithelium.
DCLK1+ cells are involved in intestinal homeostasis and response to injury.
To examine the role of DCLK1+ cells in normal epithelial homeostasis, we treated Dclk1-CreERT mice crossed with R26-iDTR (46) mice, conditionally expressing the diphtheria toxin receptor, with a single dose of tamoxifen followed by diphtheria toxin (100 ng i.p. × 3 doses). This regimen caused a loss of DCLK1+ cells in the colon and small intestine and led to a significant reduction in epithelial Ki67+ cells compared with nondiphtheria toxin–treated controls (Figures 3, A–D), suggesting a role for DCLK1+ cells under homeostatic conditions.
Figure 3. Intestinal DCLK1+ cells are involved in tissue homeostasis and play a critical role in regeneration.

(A and C) IHC for DCLK1 and Ki67 in the small intestine (A) and colon (C) of control mice without diphtheria toxin (– DTX) and after ablation of DCLK1+ cells in Dclk1 R26-iDTR mice (+ DTX). (B and D) Quantification of Ki67 cells in the small intestine (A) and colon (C) of Dclk1 R26-iDTR after treatment with diphtheria toxin compared with controls (n = 3). (E) Overall survival of Dclk1 R26-DTA (n = 5) and control mice (n = 5) after whole-body irradiation. (F–H) Representative H&E-stained sections of the regenerating epithelium from control (F) and Dclk1 R26-DTA mice (G and H) 5–7 days after irradiation. (I) Overall survival of Dclk1 R26-DTA (n = 5) and control mice (n = 5) after DSS treatment. (J–L) Representative H&E-stained sections from control (J) and Dclk1 R26-DTA mice (K and L) 7 days after DSS treatment. *P < 0.05; **P < 0.01; ***P < 0.001. Scale bars: 20 μm (C). Original magnification, x100 (A, F–H, and J–L).
Surprisingly, the ablation of DCLK1+ cells using R26-iDTR mice was well tolerated, without clinical signs of gastrointestinal pathology, although we did observe a significant reduction of Ki67+ cells. This is in line with our in vitro findings, in so far that the presence of DCLK1+ cells was not critical to form and maintain gastrointestinal organoids (Figure 2, F, J, M, and R). Therefore, we investigated whether DCLK1+ cells were required in response to intestinal and colonic injury. In these studies, Dclk1-CreERT mice were crossed with R26-DTA mice (47), in which Cre recombination led to cell-specific expression of attenuated DTA. In these experiments, Cre-negative littermates (n = 5) served as controls. Following a single dose of tamoxifen, mice (n = 5) were either subjected to whole-body irradiation (1 × 12 Gy) or 3% dextran sodium sulfate (DSS) in the drinking water for 7 days. While all control Cre-negative animals survived for 7 days after irradiation or DSS treatment, Dclk1-CreERT × R26-DTA mice did not recover from irradiation or DSS injury and showed significant morbidity and mortality (Figure 3, E and I). Histological analysis of the small intestine of Dclk1-CreERT × R26 DTA (Dclk1 DTA) mice revealed impaired epithelial regeneration and crypt dropout (Figure 3, G and H). In the colon of Dclk1 R26-DTA mice, regeneration was greatly impaired, with marked crypt injury characterized by a denuded mucosa, epithelial cells showing scant cytoplasm, and occasional mucin vacuoles, rare retained goblet cells, and abundant apoptotic cells (Figure 3K). Furthermore, we noted areas of complete crypt dropout (Figure 3L). In contrast, control mice showed almost complete regeneration 7 days after DSS treatment (Figure 3J). From these studies, we conclude that DCLK+ cells are involved in the regulation of normal epithelial homeostasis and are especially important in response to epithelial injury.
DCLK1+ cells can serve as colon cancer–initiating cells.
DCLK1 has previously been proposed to mark quiescent stem cells in the gut (23, 24), although recent reports of lineage tracing have not supported this (29). To address this question, we examined hundreds of histologic sections from the stomach, intestine, and colon of Dclk1-CreERT × R26LacZ mice at different time points after tamoxifen treatment. In these experiments, we observed very rare lineage tracing of entire crypt-villus units in the intestine as well as complete glandular units in the stomach and colon (Figure 4, A and B). Given how infrequently this occurred, we postulated that if DCLK1 indeed marked a quiescent stem cell, tracing events should become more frequent after gastrointestinal injury. Surprisingly, we did not detect an increase in the frequency of lineage-tracing events following small intestinal radiation injury or DSS-induced colitis (data not shown), arguing against a prominent role for DCLK1+ tuft cells as a physiologically important reserve stem cell. These results are in keeping with the data published by Nakanishi and colleagues (29). Nevertheless, culture of single colonic and intestinal DCLK1+ cells and colonic and intestinal organoids in Wnt3a-conditioned medium (49) revealed the ability of DCLK1+ cells to give rise to and lineage trace entire organoids in vitro (Figures 4, C–E), indicating that in response to sufficient niche signals, DCLK1+ cells are able to overcome quiescence and become active progenitor cells.
Figure 4. Quiescent DCLK1+ cells serve as colon cancer–initiating cells.

(A) LacZ staining of Dclk1 R26LacZ mice showing traced crypts in the stomach, small intestine, and colon. Scale bars: 50 μm. (B) Quantification of traced crypts in Dclk1 R26LacZ mice. (C and D) Intestinal organoids derived from Dclk1 R26tdTom mice in the presence of Wnt3a at baseline (C) and after 2 weeks (D) in culture. Scale bars: 50 μm (C) and 25 μm (D). (E) DCLK1+ cells from the small intestine (upper panels) and colon (lower panels) of Dclk1 R26tdTom mice were sorted based on red fluorescent protein (RFP) expression (see also Supplemental Figure 1) and cultured in vitro for 7 days in the presence and absence of Wnt3a. Original magnification, x100. (F) Quantification of organoid formation in the presence and absence of Wnt3a (n = 3). (G) LacZ staining of the colon of a Dclk1 R26LacZ Apcflox/flox mouse 14 months after tamoxifen treatment. Scale bar: 25 μm. (H and I) IHC for β-gal (green) and β-catenin (red) in Dclk1 R26LacZ Apcflox/flox mice after tamoxifen treatment. (I) Representative image of a single recombined DCLK1+ cell without nuclear translocation of β-catenin. Scale bars: 20 μm (H) and 10 μm (I). (J) Experimental setup for DSS treatment in Dclk1 R26LacZ Apcflox/flox mice. (K) Gross pathology of resulting tumors in Dclk1 R26LacZ Apcflox/flox mice. Scale bar: 0.5 inches. (L) Quantification of tumor incidence in Dclk1 R26LacZ Apcflox/flox mice after treatment with tamoxifen, DSS only, and tamoxifen plus DSS (n ≥5 mice/group).
To test the hypothesis that an increase in Wnt signaling activates and transforms DCLK1+ cells, we crossed our Dclk1-CreERT × R26-LacZ mice with Apcflox/flox mice to conditionally knock out the Wnt regulator APC. To our surprise, we detected neither increased lineage tracing nor the formation of intestinal or colonic adenomas for up to 18 months after induction of APC loss (Figure 4G). Importantly, APC loss did not cause nuclear translocation of β-catenin (Figure 4, H and I) in DCLK1+ cells, a hallmark of pathological Wnt signaling (48). Thus, DCLK1+ tuft cells appeared to remain quiescent in the face of acute injury and for extended periods of time, even in the setting of loss of a key tumor suppressor gene.
Recently, it was proposed that intestinal organoid cultures simulate a state of injury or regeneration and promote regeneration from otherwise quiescent intestinal cells (8). Given the Wnt-dependent, robust lineage tracing of DCLK1+ cells in organoid cultures and the expansion of DCLK1+ cells in various settings of gastrointestinal injury and preneoplasia (Supplemental Figure 6), we decided to test the hypothesis that injury in the setting of APC loss overcomes DCLK1+ cell quiescence. Surprisingly, when DSS was administered to Dclk1-CreERT × ApcfLox/flox mice 2 weeks after tamoxifen induction (Figure 4J), we observed multiple colonic tumors in 100% of the mice (n >5; Figure 4, K and L). In fact, colonic tumor formation resulted in substantial mortality within 2 to 3 months of tamoxifen administration in the majority of mice examined (data not shown).
DCLK1+ cells give rise to poorly differentiated colorectal cancer.
Lineage-tracing studies of the colorectal tumors revealed that DCLK1+ cells indeed gave rise to the malignant epithelium (Figure 5, A and B). Thus, given the longevity of a subset of DCLK1+ cells (Figure 1), we tested whether these long-lived cells were equally effective in cancer initiation. Therefore, we extended the time between administration of tamoxifen and DSS challenge for up to 3 months (Figure 5C). Again, one hundred percent (100%) of the mice (n = 3) developed frank carcinoma of the colon (Figure 5D). To further characterize the resulting tumors, pathological examination of these tumors and comparison with other APC-driven models of murine colonic neoplasia (Supplemental Figure 7 and Table 1) were performed by a board-certified gastrointestinal pathologist (H. Remotti). We classified Dclk1-CreERT × Apcflox/flox tumors as invasive carcinomas with complex growth patterns, a prominent desmoplastic reaction, and a high number of Ki67+ cells (Figure 5, E and F, Supplemental Figure 7, and Table 1). Furthermore, we observed strong cytoplasmic and nuclear β-catenin (Figure 5H) staining when compared with unaffected areas of the colon (Figure 5G). Tumors were uniformly stained for pancytokeratin and showed frequent invasion into the surrounding mesenchyme (Figure 5, I and J). We observed that nuclear p53 staining, a predictor of p53 mutation (50), was sparse (Figure 5K) and argued against additional p53 mutations in Dclk1-CreERT × Apcflox/flox tumors. Finally, in keeping with previous reports of DCLK1 expression in neoplasia (29), we detected scattered DCLK1+ cells throughout these tumors (Figure 5L).
Figure 5. DCLK1+ cells give rise to poorly differentiated colorectal cancer.

(A) H&E staining of a colonic tumor in a Dclk1 Apcflox/flox mouse after DSS treatment. (B) LacZ staining of the tumor depicted in A. Original magnification, x40 (A and B). (C) Experimental setup for delayed DSS treatment studies. (D) Macroscopic appearance of resulting tumors after delayed administration of DSS. (E) H&E staining of resulting tumors in a Dclk1 Apcflox/flox mice. Scale bar: 50 μm. (F) IHC for Ki67 of the same tumor showing elevated numbers of Ki67+ cells. Scale bar: 100 μm. (G) IHC for β-catenin in an unaffected area of the colon. β-Catenin shows membrane association. Scale bar: 50 μm. (H) IHC for β-catenin in a resulting tumor showing strong cytoplasmic and nuclear staining. Scale bar: 50 μm. (I) IHC for pancytokeratin showing the complex architecture of the tumor. (J) IHC for pancytokeratin showing a malignant gland invading the surrounding stroma. Arrow shows budding tumor cells. Scale bar: 25 μm. (K) IHC for p53. Arrows show scattered cells with nuclear p53 staining. Scale bar: 50 μm. (L) IHC for DCLK1. Rare DCLK1+ cells (arrows) can be found in the tumor. Scale bar: 25 μm. (M) Proposed model for the role of DCLK1+ tuft cells in homeostasis and injury and as cancer-initiating cells. (N) In vitro lineage-tracing events in the presence of recombinant Wnt3a (red bar), macrophage-conditioned media supplemented with Wnt3a (blue bar), and medium supplemented with recombinant Wnt3a and IL-1β (yellow bar).
Table 1.
Pathological examination of murine models of colonic neoplasia
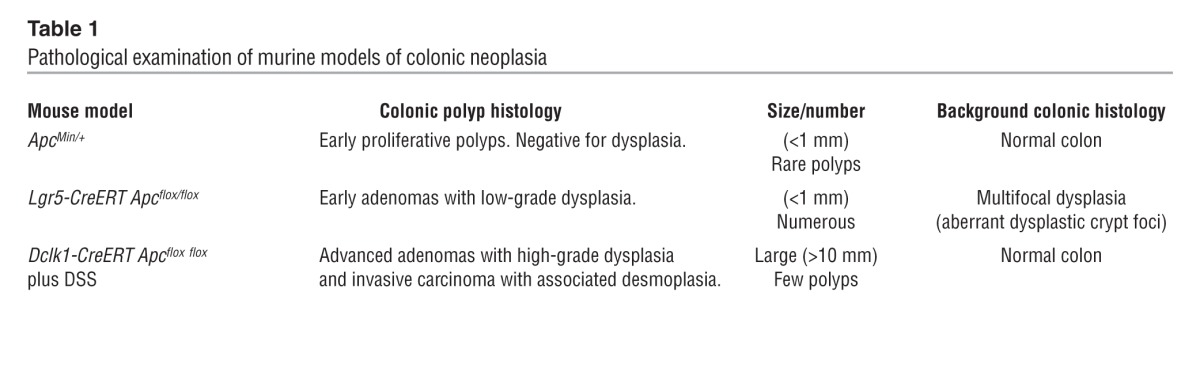
Based on these results, we propose a model (Figure 5M) in which DCLK1 marks a subset of long-lived, quiescent tuft cells that play a role in homeostasis and response to injury (Figure 3). Even the loss of a potent tumor suppressor is not sufficient to overcome the intrinsic quiescence of DCLK1+ cells (Figure 4, F and G, and Figure 5M). However, we found that the combination of APC loss and an injurious or inflammatory stimulus activates these cells and converts them into potent cancer-initiating cells (Figure 5M). To test this model, we cultured colonic organoids in the presence of recombinant Wnt3a and observed rare but robust lineage tracing. When we combined recombinant Wnt3a with macrophage-conditioned media or recombinant IL-1β, however, we observed an increase in lineage-tracing frequency, supporting our proposed model (Figure 5N).
Discussion
Using a novel Dclk1-BAC-CreERT transgenic mouse model, we demonstrate for what we believe to be the first time that a subpopulation of tuft cells represents a long-lived quiescent cell type with unexpected relevance to epithelial homeostasis that responds to gastrointestinal injury and carcinogenesis. Our data independently confirm the findings by Nakanishi et al. that most DCLK1+ tuft cells turn over within 1 to 2 weeks (29). However, we also show that, contrary to Nakanishi’s report, 5% of DCLK1+ tuft cells are remarkably long lived, representing the longest-living epithelial cell type in the gut (51). The discrepancy within the two models can likely be attributed to inactivation of one endogenous Dclk1 locus in the Dclk1-knockin mice, as heterozygous knockout of one Dclk1 allele using Dclk1flox/WT mice resulted in loss of the long-lived DCLK1 subpopulation. Furthermore, our findings also reconcile the disparate observations that most DCLK1+ tuft cells renew every 7 days (11, 29), yet DCLK1+ cells are often negative for proliferation markers such as Ki67 or BrdU (23, 24, 27). Indeed, we found that with continuous BrdU administration, the majority of short-lived DCLK1+ tuft cells were labeled within a few months, but a smaller subset of these cells remained slow cycling and could only be labeled if BrdU was administered for periods of 6 months or longer.
Interestingly, in vitro experiments using organoids from the gut revealed that under standard culture conditions, DCLK1+ cells do not survive in gut organoids. However, previous reports have suggested that tuft cells are often found in close proximity to neurons (14, 18, 39), and our collaborators have demonstrated that functional innervation regulates epithelial homeostasis and response to injury (52). Indeed, we found that DCLK1+ cells were sustained when organoids were cocultured with neurons and therefore represent, to the best of our knowledge, the first cell type that cannot be generated in organoid cultures in the absence of nonepithelial niche signals. Consistent with these observations, we found that DCLK1+ cells were largely absent in Ret–/– mice, which lack neuronal innervation of the gut, as well as in extrinsically denervated intestine following small intestinal transplantation. The interaction between epithelial cells and neurons may not be unique to DCLK1+ tuft cells and has, in fact, been suggested in other secretory and enteroendocrine cell types (53). However, a potential nerve/tuft cell axis also appears to be important in intestinal homeostasis, as intestinal proliferation, and thus the state of intestinal progenitors, was found to be highly dependent on neural innervation (52, 54). Our data indicate that the neural signal is at least partly mediated through DCLK1+ tuft cells, since genetic ablation of DCLK1+ cells markedly reduced epithelial proliferation. While DCLK1+ cell loss did not affect the growth and maintenance of intestinal organoids in vitro or cause a clinical phenotype under nonstressed conditions in vivo, we found that it resulted in reduced regeneration and decreased survival following tissue injury resulting from irradiation or DSS colitis, underscoring the importance of this cell during the coordinated response to injury.
Several reports have previously raised the possibility that DCLK1+ tuft cells represent unique stem cells within the intestinal epithelium (23–25). We found that long-term lineage tracing in the presence of two intact endogenous Dclk1+ loci revealed a distinct subset of DCLK1+ tuft cells that remain quiescent and long lived, but only very rarely lineage trace, even in the setting of acute intestinal or colonic injury. These results are in line with previous reports (29) and argue against a prominent role of DCLK1+ cells as quiescent or reserve stem cells. Interestingly, while the number of DCLK1+ cells did not increase with acute injury, we observed an expansion of these cells in the setting of chronic injury, inflammation, and preneoplasia, including that associated with chronic infection with B. fragilis (55) and H. hepaticus (56, 57). These findings are in keeping with previous reports showing an expansion of DCLK1+ cells in several preneoplastic states (24, 26, 27, 58, 59). Nakanshi et al. reported that DCLK1 serves as a marker for cancer stem cells in established ApcMin/+ adenomas (29). Interestingly, we did not observe lineage tracing of tumors in ApcMin/+ mice or in mice treated with AOM/DSS (data not shown) using either our Dclk1-BAC-CreERT or Dclk1-BAC-Cre mice. Thus, the presence or absence of two intact Dclk1+ loci might explain the differences between Nakanishi’s (29) results and the data presented here.
Most interestingly, DCLK1+ tuft cells remained quiescent and resistant to transformation, even after genetically induced loss of APC. Surprisingly, this phenotype was extraordinarily stable, with an absence in any detectable tumors extending out as long as 18 months after APC loss. Remarkably, loss of APC in DCLK1+ cells did not cause nuclear translocation of β-catenin, normally a hallmark of Wnt activation in colorectal cancer (60). Nevertheless, we found that DCLK1+ cells readily gave rise to advanced colorectal cancer when APC loss was followed by an inflammatory stimulus such as DSS colitis. Importantly, it did not matter whether the inflammatory stimulus was given immediately after tamoxifen induction or 3 months later, indicating that a quiescent cell expressing truncated APC could remain quiescent for a prolonged period until activated by an inflammatory stimulus. Recently, it has been suggested that 3D organoid cultures resemble a state of injury and regeneration (8). Accordingly, in 3D cultures, we found that DCLK1+ cells were responsive to ligand-induced Wnt signaling and showed robust lineage tracing. More importantly, inflammatory factors such as IL-1β, when combined with external Wnt3a, further increased tracing from DCLK1+ cells, thus recapitulating our in vivo findings.
Taken together, we have established that long-lived colonic tuft cells, normally resistant to proliferation, can serve as a potent cell originator for colon cancer. To the best of our knowledge, this is the first demonstration using a genetic model showing that oncogenic mutation (such as loss of APC) can be acquired in a long-lived cell type without phenotypic consequences over the lifetime of a mouse. In this unique cell lineage, APC mutations are insufficient to drive abnormal Wnt signaling and are thus inadequate for the transformation of these cells. However, the combination of APC loss with an inflammatory insult alters the surrounding niche by ultimately activating proliferation and converting these cells into bona fide cancer-initiating cells. This model of quiescent cells harboring oncogenic mutations that are possibly acquired early in life and later become activated by additional hits or inflammatory insults could considerably alter our understanding of the timeline for cancer progression and our strategy for cancer screening and surveillance.
While active stem cells display potent DNA repair mechanisms (61), mutations in long-lived quiescent cells can remain undetected, and thus unrepaired, for prolonged periods. Notably, Greten and colleagues recently proposed that the mechanism for top-down development of colorectal cancer may, in part, occur through the reacquisition of stem cell properties and the malignant transformation of more differentiated cells (3, 4). Importantly, this phenotype was only achieved upon activation of pathological Wnt signaling combined with an inflammatory stimulus (5). This finding is consistent with our observations that transformation of DCLK1+ tuft cells requires not only loss of APC but also an additional insult. Thus, it is tempting to speculate that some of the lesions in the Schwitalla et al. study (5) in fact arose from reactivated DCLK1+ cells.
Of note, colitis-associated colorectal cancers tend to be more poorly differentiated (62, 63), as we observed in the tumors in our mouse model. Therefore, an alternative explanation for our observations could be that DCLK1+ cells represent a cellular origin of colitis-associated colorectal cancer. Importantly, the concept that different colonic (stem) cell types give rise to different subtypes of colorectal cancer has been proposed before (64).
In summary, we define a subset of DCLK1+ tuft cells as a unique, long-lived, quiescent epithelial cell type that is involved in tissue homeostasis and that is critical in the response to epithelial injury. Although resistant to proliferation upon APC loss, DCLK1+ tuft cells lacking APC can be effectively reactivated following inflammatory injury and represent a novel cellular origin for colorectal cancer. We believe that the data presented here fundamentally change our understanding of the early steps of colon carcinogenesis and the plasticity of presumably differentiated cell types.
Methods
Generation of Dclk1-CreERT and Dclk1-CreGFP transgenic mice.
BAC recombination was performed using the CreTM-FrtNeoFrt or the EGFPcre-Frt-kan-Frt (pIGCN21) cassette, as described previously (65). Briefly, the CreTM-FrtNeoFrt cassette was ligated into a p451 plasmid before generating a probe containing a 40-bp sequence homologous to the BAC sequence directly upstream of the ATG in exon 2 (Supplemental Figure 1). A BAC clone RP23-283D6 containing an approximately 50-kb 5′ sequence of the Dclk1 gene–coding region (CHORI) was isolated and transferred into SW105-competent cells. The correct sequence was confirmed by using restriction enzyme digestion and PCR in the region of interest. The purified CreTM-FrtNeoFrt with the 40-bp BAC homolog on both ends was electroporated into SW105 Dclk1-BAC–containing cells. BAC DNA was isolated, linearized with PI-SceI, and then microinjected into the pronucleus of fertilized CBA × C57BL/6J oocytes at the Columbia University Transgenic Animal Core facility. Four positive founders were identified and backcrossed to C57BL/6J mice (The Jackson Laboratory).
Animal studies.
For a detailed list of all mouse strains used in these studies, see Supplemental Table 2. Dclk1-CreERT and Dclk1-CreGFP mice were crossed with Rosa26LacZ (R26LacZ), Rosa26-mTomato/mGFP (R26-TGFP), and Rosa26tdTomato (R26Tom) reporter strains obtained from The Jackson Laboratory. Mice expressing GFP under the human ubiquitin C promoter (UBC-GFP), Rosa26DTA (R26-DTA) (47), Rosa26iDTR (R26-iDTR) (46), and Dclk1flox/flox mice were purchased from The Jackson Laboratory. Apcflox/flox mice were obtained from the National Cancer Institute (NCI). Tamoxifen (1 or 6 mg) was administered by oral gavage to mice over 6 weeks of age once or every other day for 6 days. The four founder lines showed a similar expression pattern, but with a difference in expression intensity after tamoxifen induction. For lineage-tracing experiments, at least 20 mice of both sexes were analyzed at any given time point.
Determination of β-gal activity.
After intracardiac perfusion with a fixative-containing solution (4% PFA and 2% glutaraldehyde in 0.1 M Sorensen’s phosphate buffer [pH 7.4] with 2 mM MgCl2 and 5 mM EGTA), tissue was post-fixed with 4% formaldehyde (from PFA) at 4°C for 4 hours. Specimens were then cryopreserved in 30% sucrose, embedded in OCT compound, and sectioned on a cryostat microtome. Frozen sections (5 μm) were washed (0.01% sodium deoxycholate and 0.02% Nonidet P-40) and incubated for 4 hours at 37°C in 0.1% X-gal solution [4% 4-chloro-5-bromo-3-indolyl-D-galactopyranoside (X-gal) dissolved in dimethylformamide, 5 mM K3Fe(CN)6, 5 mM K4Fe(CN)6.6H2O] and counterstained with nuclear fast red.
Histology, IHC, immunofluorescence, and microscopy.
Paraffin-embedded or PFA-fixed frozen sections (5 μm) were prepared for IHC and immunofluorescence, respectively. For immunofluorescence, slides were washed with 1% Triton X-100 in PBS. then rinsed and blocked for 30 minutes with 2% BSA (Sigma-Aldrich). Primary antibodies and fluorophore-conjugated secondary antibodies were diluted in 2% BSA and incubated for 1 hour each at room temperature. Slides were counterstained with DAPI and mounted in Vectashield medium (Vector Laboratories). See Supplemental Table 3 for a complete list of antibodies used in this study. Confocal fluorescence microscopy was performed on a Zeiss LSM 510 NLO multiphoton confocal microscope system based on an Axioskop 2 FS MOT microscope stand. For IHC staining, slides were deparaffinized in xylene, and endogenous peroxidase was blocked by incubation with 3% hydrogen peroxide in methanol for visualization of the peroxidase reaction. Alternatively, for visualization of the alkaline phosphatase reaction, slides were incubated with 20% acetic acid in methanol for 2 minutes. Antigen retrieval was performed by boiling the slides in citrate buffer (10 mM, pH 6.0) in a water bath for 20 minutes. Slides were rinsed in PBS Tween 0.05% and blocked for 30 minutes with 2% BSA. Primary antibodies and biotinylated secondary antibodies (Jackson ImmunoResearch Laboratories) were diluted in 2% BSA and incubated for 1 hour each at room temperature. Subsequently, slides were incubated with alkaline phosphatase or peroxidase-conjugated streptavidin (Dako) and either VECTOR Red substrate (Vector Laboratories) or 3,3′-diaminobenzidine (Sigma-Aldrich) as chromogens, respectively. Slides were counterstained with hematoxylin and mounted for viewing. Bright-field and fluorescence images were acquired using an Eclipse TU2000-U microscope (Nikon) connected to a cooled color CCD camera (Diagnostic Instruments) using SPOT software.
Small intestinal radiation injury.
To examine the role of DCLK1+ tuft cells in the epithelial response to small intestinal or colonic injury, Dclk1-CreERT mice were crossed with R26-DTA mice. For assessment of responses to small intestinal irradiation, Dclk1-CreERT;R26-DTA mice were dosed with tamoxifen (2 mg by oral gavage) and then exposed to 12 Gy whole-body irradiation 2 days after tamoxifen treatment. Mortality and, whenever possible, small intestinal epithelial responses were then assessed by histological examination of H&E-stained intestine. Comparisons were made with similarly treated control Cre-negative R26-DTA mice. Similarly, the colonic epithelial response to DSS-induced colitis was assessed by treating Dclk1-CreERT;R26-DTA mice with 3% DSS in the drinking water for 5 days starting 2 days after tamoxifen treatment. Comparisons were again made with similarly treated control Cre-negative R26-DTA mice with respect to mortality and histology.
DSS-induced colitis model.
According to the protocol previously described, 3-month-old mice were treated with 3% (wt/vol) DSS (molecular weight 36,000–50,000; MP Biomedicals) given in the drinking water for 5 days. The control mice received distilled water. The mice were monitored closely during the treatment and recovery periods using body weight and established criteria (66, 67) to assess animal well-being. For regeneration studies, the mice were euthanized when they reached the endpoint criteria (>20% loss of body weight and a hunching score of 4) or after a recovery period of 7 days following DSS treatment. A detailed experimental setup for the treatment of Dclk1-CreERT × Apcflox/flox mice is depicted in Figure 4J and Figure 5C. Briefly, mice were given three 6-mg doses of tamoxifen. After washout, 2-week-old (Figure 4I) to 3-month-old (Figure 5C) mice were treated with DSS as detailed above. Mice were analyzed at various time points (for 30 to 150 days) after DSS colitis or when they became moribund. The tissue was processed as described above.
Long-term BrdU treatment.
Eight-week-old C57BL/6 mice were administered BrdU (1 mg/ml in the drinking water) continuously for 1 to 6 months. Mice were then sacrificed at various time points following BrdU treatment, and stomach, small intestine, and colonic tissues were taken for histological assessment. Paraffin sections were obtained and double stained for BrdU and DCLK1. Quantification was done by counting the number of BrdU+/ DCLK1+ double-positive cells versus all DCLK1+ cells. At each time point, at least 3 animals were included in the analysis, and 100 crypts were counted per mouse.
In vitro culture system.
Crypt and single-cell isolation and cultures were performed as described previously, with minor modifications (38, 41, 42). Small intestine, colon, or antrum was removed from Dclk1-CreERT mice crossed with R26-Tom, R26-TGFP, or R26-DTA mice or their littermate controls. Villi were scraped off using a razor blade, and the tissue was chopped into approximately 5-mm pieces. Afterward, the tissue was washed with cold PBS and incubated in 2.5 mM EDTA in PBS for 60 minutes on ice. Tissue fragments were suspended vigorously with a 10-ml pipette in cold PBS containing 10% FBS, yielding supernatants enriched in crypts. Crypt fractions were centrifuged and passed through 100-μm filters (BD Biosciences). Afterward, the crypt fractions were centrifuged at 150 to 200 g for 5 minutes, and the crypts were embedded in extracellular matrix (provided by the NCI), seeded on prewarmed 24-well or 48-well plates, and overlaid with culture medium. The number of organoids per well was counted on microscopic images. Organoid size was analyzed using ImageJ software (NIH). Analysis was done in a blinded fashion. Single cells were isolated and cultured as described previously (38). Briefly, crypts were dissociated with TrypLE Express (Invitrogen) including 1 mg/ml DNase I (Roche Applied Science) for 10 minutes at 37°C. Dissociated cells were passed through a 20-μm cell strainer, washed with 2% FBS/PBS, and sorted by flow cytometry. Viable cells were collected, pelleted, and embedded in ECM, followed by seeding on a 48-well plate (100–3,000 singlets per well). In some experiments, 100 ng/ml Wnt3a (Peprotech), Wnt3A-conditioned medium (1:1 concentration), macrophage-conditioned medium (1:2 concentration), or 100 ng/ml murine IL-1β (PeproTech) was added to the culture medium. Wnt3a-conditioned medium was prepared as previously described (49). For macrophage-conditioned medium, murine macrophages were cultured from the BM, as described previously (68). After differentiation, day-6 BM-derived macrophages were incubated in serum-free DMEM for 24 hours, and conditioned medium was harvested. Murine primary neuronal cells from UBC-GFP mice were prepared as previously described (40), with minor modifications. After the back skin, muscle, and roof of the vertebral canal were removed, the thoracic and lumbar spinal cord was isolated. Tissues were cut into small pieces, washed with PBS, and incubated with 5 mg/ml Dispase type II (Roche Applied Science) at 37°C for 15 minutes. Cells were centrifuged at 200 g for 2 minutes and washed with PBS. Cells were then collected and mixed with extracted gastrointestinal crypts in the extracellular matrix at a 1:5 crypt/neuron ratio. In coculture experiments, murine NGF (20 ng/ml; PeproTech) and cholera toxin (100 ng/ml) (69) were added to the culture medium. The organoid images were acquired using fluorescence microscopy (TE2000-U; Nikon).
Analysis of Ret–/– mice.
Fixed embryonic tissue from E17 embryos of Ret–/– and control mice were a gift from Robert O. Heuckeroth (Department of Pediatrics, Washington University, St. Louis, Missouri, USA). Intestine and colon were cut into small pieces, embedded, and cut to obtain luminal sections. Sections were stained as detailed above, and at least five complete luminal sections per embryo were analyzed. Each group (knockout versus control) consisted of at least three animals.
Analysis of mice infected with B. fragilis and H. hepaticus.
Paraffin-embedded sections (three sections per animal) from B6 mice infected with B. fragilis (n = 3–8 months of age, infected at 3 weeks of age) were a gift from Cynthia Sears (Center for Global Health, Johns Hopkins University, Baltimore, Maryland, USA). Frozen sections from ApcMin/+ mice treated with H. hepaticus (n = 3–15 weeks of age, infected at 5 weeks of age) were a gift from James Fox (Institute for Comparative Medicine, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA). Uninfected ApcMin/+ mice served as controls. DCLK1+ cells and total crypts were counted in five random high-power fields on each section, and the results are indicated as the number of DCLK1+ cells per crypt.
Analysis of Lgr5-CreERT.
Frozen sections (n = 3) from Lgr5-GFP-IRES-CreERT2 mice crossed with Atoh1flox/flox mice were a gift from Ramesh Shivdasani (Department of Oncology, Dana Faber Institute, Boston, Massachusetts, USA). DCLK1+ cells were counted only in LGR5+ crypts (indicated by GFP expression) and compared with Lgr5-GFP-IRES-CreERT2 mice that had intact Atoh1 loci (n = 3).
Human samples.
Anonymized paraffin-embedded paired biopsies from patients (n = 2) after small intestinal transplantation and control tissue were received from the Department of Pathology of Columbia University Medical Center.
Statistics.
Statistical analyses were performed using 2-tailed Student’s t tests. Error bars denote the mean ± SEM. A P value of less than 0.05 was considered statistically significant. *P < 0.05; **P < 0.01; ***P < 0.001.
Study approval.
All experiments involving mice were performed in compliance with federal laws and institutional guidelines and approved by the IACUC of the Columbia University Animal Care Facility (protocol AC-AAAD7600).
Supplementary Material
Acknowledgments
The authors thank Yagnesh Tailor, Karan Nagar, and Chintan Kapadia for their help with animal welfare. B. fragilis samples were provided by Cynthia Sears (Johns Hopkins University). H. hepaticus samples were provided by James G. Fox (Massachusetts Institute of Technology). Lgr5-CreERT × Atohflox/flox samples were provided by Ramesh A. Shivdasani (Harvard Medical School). T. C. Wang received grants from the NIH (RO1 DK097016) and the Pancreatic Cancer Action Network - ACCR (Innovative Grant). Y. Hayakawa was supported by a grant from the Mitsukoshi Health and Welfare Foundation and JSPS Postdoctoral Fellowships for Research Abroad. C.B. Westphalen was supported by grants from the German Research Foundation (Deutsche Forschungsgemeinschaft). S. Asfaha was supported by a Canadian Institute for Health Research Clinician Scientist Phase I Award. M. Quante was supported by a grant from the Max Eder Program of the Deutsche Krebshilfe (109789).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2014;124(3):1283–1295. doi:10.1172/JCI73434.
References
- 1.Saif MW, Chu E. Biology of colorectal cancer. Cancer J. 2010;16(3):196–201. doi: 10.1097/PPO.0b013e3181e076af. [DOI] [PubMed] [Google Scholar]
- 2.Barker N, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457(7229):608–611. doi: 10.1038/nature07602. [DOI] [PubMed] [Google Scholar]
- 3.Leedham SJ, Wright NA. Expansion of a mutated clone: from stem cell to tumour. J Clin Pathol. 2008;61(2):164–171. doi: 10.1136/jcp.2006.044610. [DOI] [PubMed] [Google Scholar]
- 4.Shih IM, et al. Top-down morphogenesis of colorectal tumors. Proc Natl Acad Sci U S A. 2001;98(5):2640–2645. doi: 10.1073/pnas.051629398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwitalla S, et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell. 2013;152(1–2):25–38. doi: 10.1016/j.cell.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 6.Barker N, Bartfeld S, Clevers H. Tissue-resident adult stem cell populations of rapidly self-renewing organs. Cell Stem Cell. 2010;7(6):656–670. doi: 10.1016/j.stem.2010.11.016. [DOI] [PubMed] [Google Scholar]
- 7.Clevers H. Stem cells: A unifying theory for the crypt. Nature. 2013;495(7439):53–54. doi: 10.1038/nature11958. [DOI] [PubMed] [Google Scholar]
- 8.Buczacki SJ, et al. Intestinal label-retaining cells are secretory precursors expressing Lgr5. Nature. 2013;495(7439):65–69. doi: 10.1038/nature11965. [DOI] [PubMed] [Google Scholar]
- 9.Tsubouchi S, Leblond CP. Migration and turnover of entero-endocrine and caveolated cells in the epithelium of the descending colon, as shown by radioautography after continuous infusion of 3H-thymidine into mice. Am J Anat. 1979;156(4):431–451. doi: 10.1002/aja.1001560403. [DOI] [PubMed] [Google Scholar]
- 10.Barker N, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449(7165):1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 11.Gerbe F, et al. Distinct ATOH1 and Neurog3 requirements define tuft cells as a new secretory cell type in the intestinal epithelium. J Cell Biol. 2011;192(5):767–780. doi: 10.1083/jcb.201010127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rhodin J, Dalhamn T. Electron microscopy of the tracheal ciliated mucosa in rat. Z Zellforsch Mikrosk Anat. 1956;44(4):345–412. doi: 10.1007/BF00345847. [DOI] [PubMed] [Google Scholar]
- 13.Jarvi O, Keyrilainen O. On the cellular structures of the epithelial invasions in the glandular stomach of mice caused by intramural application of 20-methylcholantren. Acta Pathol Microbiol Scand Suppl. 1956;39(suppl 111):72–73. [PubMed] [Google Scholar]
- 14.Sato A. Tuft cells. Anat Sci Int. 2007;82(4):187–199. doi: 10.1111/j.1447-073X.2007.00188.x. [DOI] [PubMed] [Google Scholar]
- 15.Hofer D, Drenckhahn D. Identification of the taste cell G-protein, α-gustducin, in brush cells of the rat pancreatic duct system. Histochem Cell Biol. 1998;110(3):303–309. doi: 10.1007/s004180050292. [DOI] [PubMed] [Google Scholar]
- 16.Hofer D, Puschel B, Drenckhahn D. Taste receptor-like cells in the rat gut identified by expression of α-gustducin. Proc Natl Acad Sci U S A. 1996;93(13):6631–6634. doi: 10.1073/pnas.93.13.6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sato A, Miyoshi S. Fine structure of tuft cells of the main excretory duct epithelium in the rat submandibular gland. Anat Rec. 1997;248(3):325–331. doi: 10.1002/(SICI)1097-0185(199707)248:3<325::AID-AR4>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 18.Morroni M, Cangiotti AM, Cinti S. Brush cells in the human duodenojejunal junction: an ultrastructural study. J Anat. 2007;211(1):125–131. doi: 10.1111/j.1469-7580.2007.00738.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gerbe F, Legraverend C, Jay P. The intestinal epithelium tuft cells: specification and function. Cell Mol Life Sci. 2012;69(17):2907–2917. doi: 10.1007/s00018-012-0984-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin PT, Gleeson JG, Corbo JC, Flanagan L, Walsh CA. DCAMKL1 encodes a protein kinase with homology to doublecortin that regulates microtubule polymerization. J Neurosci. 2000;20(24):9152–9161. doi: 10.1523/JNEUROSCI.20-24-09152.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gerbe F, Brulin B, Makrini L, Legraverend C, Jay P. DCAMKL-1 expression identifies Tuft cells rather than stem cells in the adult mouse intestinal epithelium. Gastroenterology. 2009;137(6):2179–2180. doi: 10.1053/j.gastro.2009.06.072. [DOI] [PubMed] [Google Scholar]
- 22.Walker TL, Yasuda T, Adams DJ, Bartlett PF. The doublecortin-expressing population in the developing and adult brain contains multipotential precursors in addition to neuronal-lineage cells. J Neurosci. 2007;27(14):3734–3742. doi: 10.1523/JNEUROSCI.5060-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.May R, et al. Doublecortin and CaM kinase-like-1 and leucine-rich-repeat-containing G-protein-coupled receptor mark quiescent and cycling intestinal stem cells, respectively. Stem Cells. 2009;27(10):2571–2579. doi: 10.1002/stem.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.May R, Riehl TE, Hunt C, Sureban SM, Anant S, Houchen CW. Identification of a novel putative gastrointestinal stem cell and adenoma stem cell marker, doublecortin and CaM kinase-like-1, following radiation injury and in adenomatous polyposis coli/multiple intestinal neoplasia mice. Stem Cells. 2008;26(3):630–637. doi: 10.1634/stemcells.2007-0621. [DOI] [PubMed] [Google Scholar]
- 25.Gagliardi G, Bellows CF. DCLK1 expression in gastrointestinal stem cells and neoplasia. J Cancer Res Ther. 2012;1:12. [Google Scholar]
- 26.Vega KJ, et al. Identification of the putative intestinal stem cell marker doublecortin and CaM kinase-like-1 in Barrett’s esophagus and esophageal adenocarcinoma. J Gastroenterol Hepatol. 2012;27(4):773–780. doi: 10.1111/j.1440-1746.2011.06928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saqui-Salces M, et al. Gastric tuft cells express DCLK1 and are expanded in hyperplasia. Histochem Cell Biol. 2011;136(2):191–204. doi: 10.1007/s00418-011-0831-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quante M, et al. Bile acid and inflammation activate gastric cardia stem cells in a mouse model of Barrett-like metaplasia. Cancer Cell. 2012;21(1):36–51. doi: 10.1016/j.ccr.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakanishi Y, et al. Dclk1 distinguishes between tumor and normal stem cells in the intestine. Nat Genet. 2013;45(1):98–103. doi: 10.1038/ng.2481. [DOI] [PubMed] [Google Scholar]
- 30.Kretzschmar K, Watt FM. Lineage tracing. Cell. 2012;148(1–2):33–45. doi: 10.1016/j.cell.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 31.Gerbe F, et al. Distinct ATOH1 and Neurog3 requirements define tuft cells as a new secretory cell type in the intestinal epithelium. J Cell Biol. 2011;192(5):767–780. doi: 10.1083/jcb.201010127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Itzkovitz S, et al. Single-molecule transcript counting of stem-cell markers in the mouse intestine. Nat Cell Biol. 2011;14(1):106–114. doi: 10.1038/ncb2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bjerknes M, et al. Origin of the brush cell lineage in the mouse intestinal epithelium. Dev Biol. 2012;362(2):194–218. doi: 10.1016/j.ydbio.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.VanDussen KL, et al. Notch signaling modulates proliferation and differentiation of intestinal crypt base columnar stem cells. Development. 2012;139(3):488–497. doi: 10.1242/dev.070763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Durand A, et al. Functional intestinal stem cells after Paneth cell ablation induced by the loss of transcription factor Math1 (Atoh1). Proc Natl Acad Sci U S A. 2012;109(23):8965–8970. doi: 10.1073/pnas.1201652109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim TH, Escudero S, Shivdasani RA. Intact function of Lgr5 receptor-expressing intestinal stem cells in the absence of Paneth cells. Proc Natl Acad Sci U S A. 2012;109(10):3932–3937. doi: 10.1073/pnas.1113890109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barker N, et al. Lgr5(+ve) stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell. 2010;6(1):25–36. doi: 10.1016/j.stem.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 38.Sato T, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459(7244):262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 39.Breer H, Eberle J, Frick C, Haid D, Widmayer P. Gastrointestinal chemosensation: chemosensory cells in the alimentary tract. Histochem Cell Biol. 2012;138(1):13–24. doi: 10.1007/s00418-012-0954-z. [DOI] [PubMed] [Google Scholar]
- 40.Malin SA, Davis BM, Molliver DC. Production of dissociated sensory neuron cultures and considerations for their use in studying neuronal function and plasticity. Nat Protoc. 2007;2(1):152–160. doi: 10.1038/nprot.2006.461. [DOI] [PubMed] [Google Scholar]
- 41.Yui S, et al. Functional engraftment of colon epithelium expanded in vitro from a single adult Lgr5(+) stem cell. Nat Med. 2012;18(4):618–623. doi: 10.1038/nm.2695. [DOI] [PubMed] [Google Scholar]
- 42.Sato T, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology. 2011;141(5):1762–1772. doi: 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
- 43.Schuchardt A, D’Agati V, Larsson-Blomberg L, Costantini F, Pachnis V. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature. 1994;367(6461):380–383. doi: 10.1038/367380a0. [DOI] [PubMed] [Google Scholar]
- 44.Heanue TA, Pachnis V. Enteric nervous system development and Hirschsprung’s disease: advances in genetic and stem cell studies. Nat Rev Neurosci. 2007;8(6):466–479. doi: 10.1038/nrn2137. [DOI] [PubMed] [Google Scholar]
- 45.Pappenheimer AM, Pappenheimer AM., Jr Diphtheria toxin. Annu Rev Biochem. 1977;46:69–94. doi: 10.1146/annurev.bi.46.070177.000441. [DOI] [PubMed] [Google Scholar]
- 46.Buch T, et al. A Cre-inducible diphtheria toxin receptor mediates cell lineage ablation after toxin administration. Nat Methods. 2005;2(6):419–426. doi: 10.1038/nmeth762. [DOI] [PubMed] [Google Scholar]
- 47.Wu S, Wu Y, Capecchi MR. Motoneurons and oligodendrocytes are sequentially generated from neural stem cells but do not appear to share common lineage-restricted progenitors in vivo. Development. 2006;133(4):581–590. doi: 10.1242/dev.02236. [DOI] [PubMed] [Google Scholar]
- 48.Schepers A, Clevers H. Wnt signaling, stem cells, and cancer of the gastrointestinal tract. Cold Spring Harb Perspect Biol. 2012;4(4):a007989. doi: 10.1101/cshperspect.a007989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Willert K, et al. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature. 2003;423(6938):448–452. doi: 10.1038/nature01611. [DOI] [PubMed] [Google Scholar]
- 50.Kaserer K, et al. Staining patterns of p53 immunohistochemistry and their biological significance in colorectal cancer. J Pathol. 2000;190(4):450–456. doi: 10.1002/(SICI)1096-9896(200003)190:4<450::AID-PATH545>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 51.van der Flier LG, Clevers H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol. 2009;71:241–260. doi: 10.1146/annurev.physiol.010908.163145. [DOI] [PubMed] [Google Scholar]
- 52.Gross ER, Gershon MD, Margolis KG, Gertsberg ZV, Li Z, Cowles RA. Neuronal serotonin regulates growth of the intestinal mucosa in mice. Gastroenterology. 2012;143(2):408–417. doi: 10.1053/j.gastro.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tolhurst G, Reimann F, Gribble FM. Intestinal sensing of nutrients. Handb Exp. 2012;(209):309–335. doi: 10.1007/978-3-642-24716-3_14. [DOI] [PubMed] [Google Scholar]
- 54.Margolis KG, et al. Enteric neuronal density contributes to the severity of intestinal inflammation. Gastroenterology. 2011;141(2):588–598. doi: 10.1053/j.gastro.2011.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu S, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med. 2009;15(9):1016–1022. doi: 10.1038/nm.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nagamine CM, Sohn JJ, Rickman BH, Rogers AB, Fox JG, Schauer DB. Helicobacter hepaticus infection promotes colon tumorigenesis in the BALB/c-Rag2(–/–) Apc(Min/+) mouse. Infect Immun. 2008;76(6):2758–2766. doi: 10.1128/IAI.01604-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nagamine CM, Rogers AB, Fox JG, Schauer DB. Helicobacter hepaticus promotes azoxymethane-initiated colon tumorigenesis in BALB/c-IL10-deficient mice. Int J Cancer. 2008;122(4):832–838. doi: 10.1002/ijc.23175. [DOI] [PubMed] [Google Scholar]
- 58.Quante M, et al. Bile acid and inflammation activate gastric cardia stem cells in a mouse model of Barrett-like metaplasia. Cancer Cell. 2012;21(1):36–51. doi: 10.1016/j.ccr.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tu S, et al. IFN-γ inhibits gastric carcinogenesis by inducing epithelial cell autophagy and T-cell apoptosis. Cancer Res. 2011;71(12):4247–4259. doi: 10.1158/0008-5472.CAN-10-4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012;149(6):1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 61.Hua G, et al. Crypt base columnar stem cells in small intestines of mice are radioresistant. Gastroenterology. 2012;143(5):1266–1276. doi: 10.1053/j.gastro.2012.07.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Potack J, Itzkowitz SH. Colorectal cancer in inflammatory bowel disease. Gut Liver. 2008;2(2):61–73. doi: 10.5009/gnl.2008.2.2.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Heimann TM, et al. Colorectal carcinoma associated with ulcerative colitis: a study of prognostic indicators. Am J Surg. 1992;164(1):13–17. doi: 10.1016/S0002-9610(05)80638-5. [DOI] [PubMed] [Google Scholar]
- 64.Sadanandam A, et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med. 2013;19(5):619–625. doi: 10.1038/nm.3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Quante M, Marrache F, Goldenring JR, Wang TC. TFF2 mRNA transcript expression marks a gland progenitor cell of the gastric oxyntic mucosa. Gastroenterology. 2010;139(6):2018–2027. doi: 10.1053/j.gastro.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wesselmann U, Czakanski PP, Affaitati G, Giamberardino MA. Uterine inflammation as a noxious visceral stimulus: behavioral characterization in the rat. Neurosci Lett. 1998;246(2):73–76. doi: 10.1016/S0304-3940(98)00234-1. [DOI] [PubMed] [Google Scholar]
- 67.Sevcik MA, et al. Endogenous opioids inhibit early-stage pancreatic pain in a mouse model of pancreatic cancer. Gastroenterology. 2006;131(3):900–910. doi: 10.1053/j.gastro.2006.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Maeda S, et al. Nod2 mutation in Crohn’s disease potentiates NF-κB activity and IL-1β processing. Science. 2005;307(5710):734–738. doi: 10.1126/science.1103685. [DOI] [PubMed] [Google Scholar]
- 69.McKeehan WL, Adams PS, Rosser MP. Modified nutrient medium MCDB 151, defined growth factors, cholera toxin, pituitary factors, and horse serum support epithelial cell and suppress fibroblast proliferation in primary cultures of rat ventral prostate cells. In Vitro. 1982;18(2):87–91. doi: 10.1007/BF02796399. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.