

Abstract
Patients with Marfan syndrome (MFS), a multisystem disorder caused by mutations in the gene encoding the extracellular matrix (ECM) protein fibrillin 1, are unusually vulnerable to stress-induced cardiac dysfunction. The prevailing view is that MFS-associated cardiac dysfunction is the result of aortic and/or valvular disease. Here, we determined that dilated cardiomyopathy (DCM) in fibrillin 1–deficient mice is a primary manifestation resulting from ECM-induced abnormal mechanosignaling by cardiomyocytes. MFS mice displayed spontaneous emergence of an enlarged and dysfunctional heart, altered physical properties of myocardial tissue, and biochemical evidence of chronic mechanical stress, including increased angiotensin II type I receptor (AT1R) signaling and abated focal adhesion kinase (FAK) activity. Partial fibrillin 1 gene inactivation in cardiomyocytes was sufficient to precipitate DCM in otherwise phenotypically normal mice. Consistent with abnormal mechanosignaling, normal cardiac size and function were restored in MFS mice treated with an AT1R antagonist and in MFS mice lacking AT1R or β-arrestin 2, but not in MFS mice treated with an angiotensin-converting enzyme inhibitor or lacking angiotensinogen. Conversely, DCM associated with abnormal AT1R and FAK signaling was the sole abnormality in mice that were haploinsufficient for both fibrillin 1 and β1 integrin. Collectively, these findings implicate fibrillin 1 in the physiological adaptation of cardiac muscle to elevated workload.
Introduction
Heart function depends on the extracellular matrix (ECM) to transmit mechanical forces to cardiomyocytes, where they are converted into biochemical signals that regulate muscle activity to maintain homeostasis (1–3). Cardiomyocytes can also respond to elevated workload by augmenting contractility and increasing their mass (adaptive hypertrophy) in order to restore cardiac output to normal levels (3, 4). Cardiomyopathies are a clinically heterogeneous and often-lethal group of heart muscle diseases caused by genetic lesions or environmental insults that impair the adaptive response of cardiac muscle to hemodynamic overload (4). Chronically elevated blood pressure leads to hypertrophic cardiomyopathy, which is characterized by a significantly thicker heart wall and excessive deposition of an unorganized ECM (fibrosis) that further reduces muscle tissue compliance (3, 4). Sustained volume overload, on the other hand, promotes dilated cardiomyopathy (DCM), which is associated with moderate cardiac hypertrophy, little or no tissue fibrosis, and a substantially larger ventricular chamber diameter (3, 4). Studies of inherited forms of DCM have underscored the importance of sarcolemmal and intracellular components of cardiomyocytes that are implicated in sensing stretch signals and in generating and transmitting force from and to the sarcomere (4). By contrast, virtually nothing is known about the mechanism whereby the myocardial ECM transmits mechanical stimuli to resident cells so as to maintain muscle homeostasis (1, 5). This is a clinically important issue, as cardiac tissue degeneration is the histopathologic hallmark of acquired DCM.
Heart dysfunction is a controversial finding in Marfan syndrome (MFS), a relatively common disease of the connective tissue due to mutations that alter the structure or decrease the expression of the ECM protein and TGF-β regulator fibrillin 1 (6–8). While the prevailing view is that thoracic aortic aneurysm (TAA) and cardiac valve abnormalities overload the left ventricle (LV) by respectively stiffening the aortic wall and increasing valve regurgitation (6), several clinical studies of relatively small cohorts of MFS patients have reported LV pathology in the absence of severe valve regurgitation or aortic surgery, or that is out of proportion with aneurysm growth (9–20). These findings have been used to argue that mutations in fibrillin 1 may cause ventricular dysfunction by altering the structural properties of myocardial tissue and/or the local bioavailability of TGF-β signals (6, 18). Relevant to the former possibility, fibrillin 1 assemblies are broadly distributed in the myocardium, where they couple individual myocytes to the pericellular matrix, as basement membrane–associated (BM-associated) microfibrils, confer physical properties to cardiac tissue, as obligatory components of interstitial elastic fibers, and weave together adjoining clusters of myocytes, as molecular bridges between pericellular and interstitial matrices (5, 7). By analogy to the medial layer of the aortic wall (19), the interconnected meshwork of fibrillin 1 assemblies in the myocardium may therefore represent a key component of the structural scaffold believed to support proper muscle function (5).
Aside from endowing tissues with tensile strength and elasticity, fibrillin 1 assemblies also regulate cell behavior by interacting with integrin receptors and by modulating latent TGF-β bioavailability (7, 8). In accordance with the latter function, both the pan–TGF-β–neutralizing antibody 1D11 and the angiotensin II (AngII) type 1 receptor (AT1R) blocker (ARB) losartan, which also inhibits AngII-stimulated TGF-β synthesis (21, 22), have been shown to prevent aortic aneurysm formation in Fbn1C1039G/+ mice, an MFS model of nonlethal TAA (23, 24). This experimental evidence has resulted in several clinical trials assessing the efficacy of losartan therapy against TAA progression in MFS patients (25–29). In contrast to the wealth of new information about TAA pathogenesis, heart dysfunction has not been characterized in Fbn1 mutant mice, with negative implications for improving the management of cardiac complications in MFS patients, particularly in severely affected children or following prophylactic TAA repair (6, 30–32).
The present study was designed to address the aforementioned unresolved issues by characterizing the impact of fibrillin 1 deficiency on cardiac function using a combination of genetic and pharmacological approaches in mice. The results of these experiments refute the long-held belief that DCM is a secondary manifestation of MFS, in addition to revealing the unsuspected new function of fibrillin 1 as a force-transmitting molecule central to heart adaptation to increased workload. We believe that our findings significantly advance the current understanding of both cardiac pathogenesis in MFS and the role of the ECM in mechanotransduction.
Results
DCM is part of the cardiovascular phenotype of mice with severe MFS.
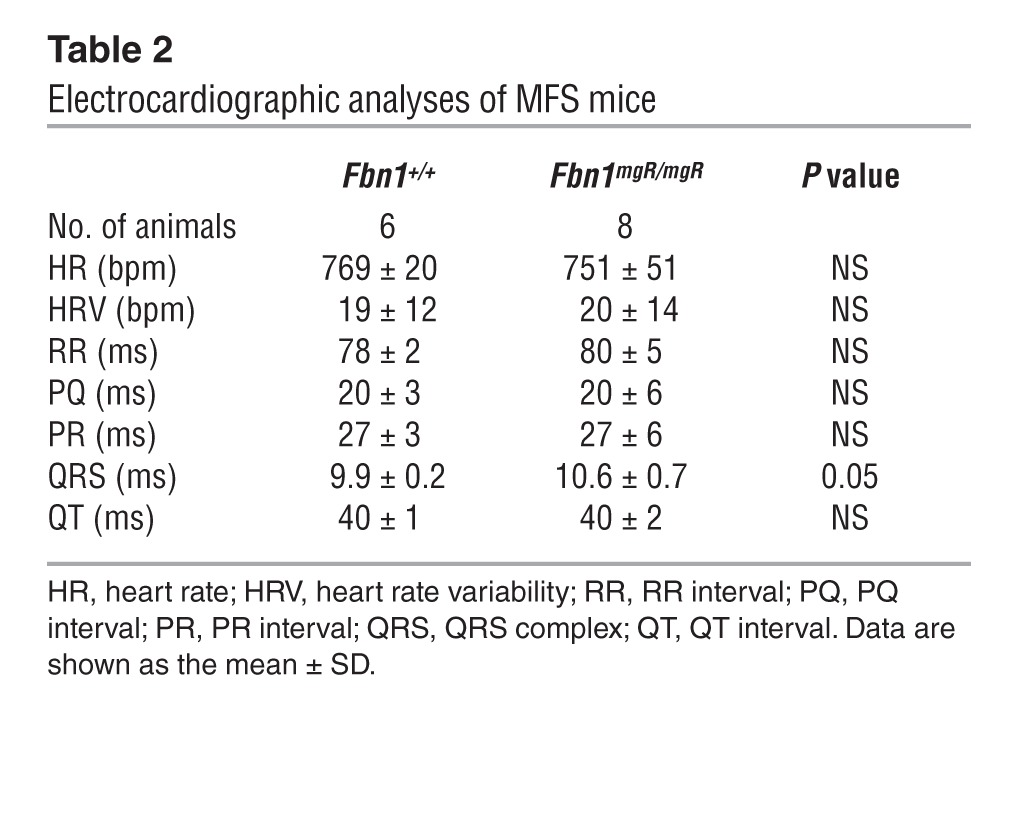
Cardiac function was monitored in Fbn1mgR/mgR mice, a validated animal model of progressively severe MFS (33). Fbn1mgR/mgR mice produce approximately 20% of the normal amount of fibrillin 1 and die from ruptured dissecting TAA during the first year of life (median survival 2.5 months) (33). Echocardiographic (echo) tests in 3-month-old Fbn1mgR/mgR mice confirmed significant dilation of the aortic root and proximal ascending aorta, as well as severe mitral and aortic regurgitation associated with increased atrial mass (data not shown and Supplemental Figure 1, A and B; supplemental material available online with this article; doi: 10.1172/JCI71059DS1). Higher pulse pressure secondary to decreased arterial diastolic pressure provided additional evidence of aortic regurgitation (Table 1). Importantly, echo findings of decreased fractional shortening and increased internal diameter of the LV indicated that cardiac dysfunction is part of the Fbn1mgR/mgR phenotype (Figure 1A). ECG measurements at 2.5 months of age showed evidence of abnormal conductance in Fbn1mgR/mgR relative to Fbn1+/+ mice, with increased delay in ventricular depolarization — i.e., QRS duration (Table 2).
Table 1.
Blood pressure measurements in MFS mice
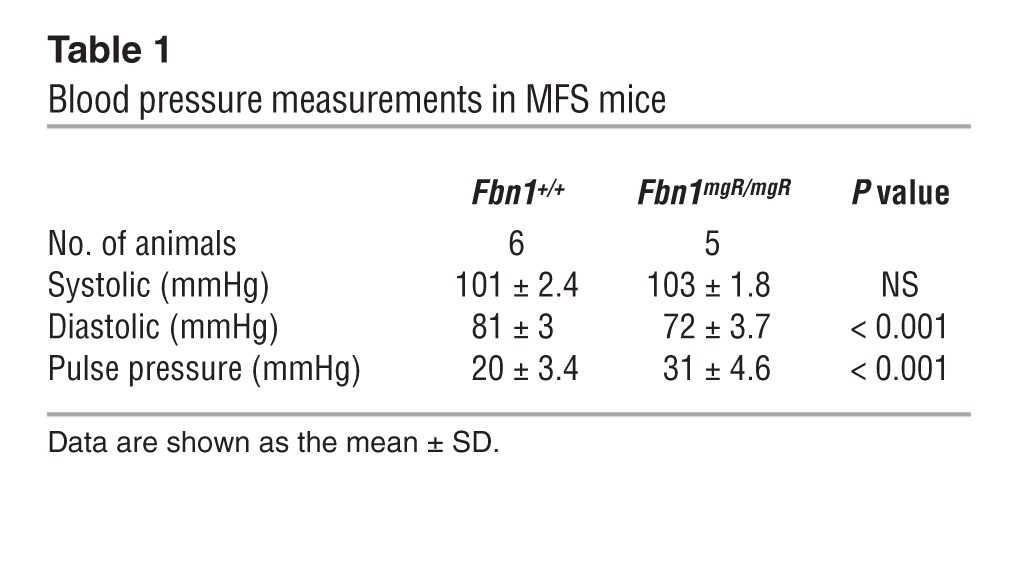
Figure 1. DCM detection in MFS mice.

(A) Top: representative echo M-mode images taken at the level of the mid-papillary muscle from 3-month-old Fbn1+/+ and Fbn1mgR/mgR mice; red and green double-headed arrows highlight LV internal diameter in diastole (LVIDd) and systole (LVIDs), respectively. Percentage of fractional shortening (FS) was calculated as (LVIDd – LVIDs) / LVIDd × 100. Bottom: Bar graphs summarize the LVIDd and percentage of FS data in Fbn1+/+ and Fbn1mgR/mgR mice (n >15 per genotype). (B) Representative in vivo PV loop tracings in Fbn1mgR/mgR (blue) compared with Fbn1+/+ (green) mice. PV loop quantification of cardiac function including EDV, ESV, ESPVR, PRSW, and dP/dT maximum and minimum values (n = 5 per genotype). *P < 0.05; bars indicate the mean ± SD.
Table 2.
Electrocardiographic analyses of MFS mice
Invasive hemodynamic measurements documented impaired contractile function in 3-month-old Fbn1mgR/mgR male mice compared with that found in sex-matched Fbn1+/+ littermates by showing: (a) rightward shift of the pressure-volume (PV) loop curve in association with a significant elevation of end-diastolic and end-systolic volumes (EDV and ESV, respectively); (b) lower slope of the end-systolic PV relationship (ESPVR); (c) lower preload recruitable stroke work (PRSW); and (d) reduced rates of maximum and minimum pressure changes in the LV (dP/dT) (Figure 1B). Heart dysfunction in Fbn1mgR/mgR mice was related to substantial cardiomegaly characterized by ventricular dilation, increased ventricular heart weight/body weight (HW/BW) ratio, and normal LV wall thickness (Figure 2A). Cardiomegaly was readily noticeable upon postmortem inspection of P16 Fbn1mgR/mgR mice and became gradually more pronounced with age (Figure 2A). In contrast to histological and molecular evidence of myocyte hypertrophy and reactivation of fetal myocardial genes (Figure 2B), we observed no appreciable signs of excessive collagen accumulation or upregulation of fibrogenic genes in the myocardium of 3- and 6-month-old Fbn1mgR/mgR mice compared with their WT littermates (Figure 2C and data not shown). Collectively, these findings constitute the first description of DCM in a mouse model of MFS.
Figure 2. DCM characterization in MFS mice.

(A) Representative Fbn1+/+ and Fbn1mgR/mgR hearts collected at the indicated stages of postnatal life with the respective ventricular HW/BW estimates shown by the bar graphs above them (n > 5 per genotype and time point). Scale bar: 5 mm. H&E staining of representative heart sections from 3-month-old Fbn1+/+ and Fbn1mgR/mgR mice are shown below. Scale bar: 1 mm. (B) Representative WGA-stained cross sections of Fbn1+/+ and Fbn1mgR/mgR hearts (white tracings highlight myocyte cross-sectional area [CSA]), with overall estimates of cardiac hypertrophy expressed as CSA. Levels of hypertrophy-related transcripts β-myosin heavy chain (Mhy7) and natriuretic peptide A (Nppa) are shown on the right. Scale bar: 100 μm. (C) Representative trichrome-stained cross sections of Fbn1+/+ and Fbn1mgR/mgR hearts (left) shown together with quantitative PCR estimates of fibrogenic transcripts periostin (Pstn) and collagen I (Col1) (right), and Smad3 immunoblots of total protein extracts (bottom) from 3-month-old Fbn1+/+ and Fbn1mgR/mgR myocardium. Scale bar: 100 μm. (D) Mean opening angle (degree) of the LV free wall (left) and elastic modulus (kPa) measurement (right) in the myocardium of 2-month-old Fbn1+/+ and Fbn1mgR/mgR hearts (n = 5 per genotype and assay). *P < 0.05; bars indicate the mean ± SD.
DCM is a primary cardiovascular manifestation of MFS mice.
Next, we used Fbn1 conditional null allele (Fbn1Lox) to address the long-standing controversy of cardiomyopathy origin in MFS by interrogating the potential contribution of the myocardial matrix to disease onset (34). Both collagen IV–producing (BM-producing) cardiomyocytes and collagen I–producing (interstitial matrix–producing) cardiac fibroblasts secrete fibrillin 1 and thus cooperate in assembling the interconnected meshwork of myocardial microfibrils (5, 7). Lacking suitable fibroblast-specific Cre-driving transgenes, we assessed the role of fibrillin 1 in the myocardium by selectively targeting Fbn1 gene expression in cardiomyocytes using the αMHC-Cre transgenic mouse (Supplemental Figure 2A and ref. 35). Negative controls included mice with Fbn1 inactivation in either the aortic arch or cardiac valves by the respective action of the Wnt1-Cre and Col2-Cre transgenes (Supplemental Figure 2A and refs. 36, 37). Since an aorta with no fibrillin 1 ruptures soon after birth (38), we interbred hypomorphic Fbn1Lox/mgR mice with Wnt1-Cre transgenic mice so as to decrease rather than eliminate fibrillin 1 deposition in the aortic media. Indeed, our quantitative estimates of Fbn1 transcripts in cardiomyocytes and cardiac fibroblasts of the various conditional mutant mice confirmed that cardiomyocytes from newborn Fbn1αMHC–/– mice are the only cells underexpressing Fbn1 (Supplemental Figure 2B). It should be noted that Fbn1 transcript levels in cells from conditional mutant mice are shown in Supplemental Figure 2B together with those from the respective parental mutant mice and are expressed for both groups relative to 100% Fbn1+/+ levels. Although higher than in the Fbn1mgR/mgR counterpart, we found that fibrillin 1 synthesis in the Fbn1αMHC–/– myocardium was sufficiently low to trigger disease; additionally, we found that Fbn1 underexpression by mutant myocyte cultures was consistent with the uneven distribution of X-gal–positive cells in the myocardium of αMHC-Cre;RosaLacZ transgenic mice (Supplemental Figure 2B).
In contrast to Fbn1Col2–/– and Fbn1Wnt1–/mgR mice, DCM was the sole abnormality in otherwise phenotypically normal Fbn1αMHC–/– mice (Figure 3A). As expected, Fbn1Col2–/– and Fbn1Wnt1–/mgR mice respectively displayed abnormal valves and increased atrial mass and TAA (Supplemental Figure 2C and data not shown). Lack of cardiomegaly in αMHC-Cre transgenic mice furthermore excluded a disease-causing effect of Cre recombinase expression (Supplemental Figure 2D). The latter finding, together with the aforementioned X-gal data (Supplemental Figure 2B), distinguish this particular αMHC-Cre transgenic mouse line from those in which excessive Cre expression has been reported to cause DCM (39). We determined that cardiac dysfunction in Fbn1αMHC–/– mice was correlated with a substantial reduction of immunoreactive fibrillin 1 in the matrix immediately around individual cardiomyocytes (BM-associated pericellular matrix; Figure 3B). Seemingly normal fibrillin 1 immunostaining in the interstitial matrix of the Fbn1αMHC–/– myocardium (but not of the Fbn1–/– myocardium) implied that cardiac fibroblasts are the main source of these microfibrils (Figure 3B). Consistent with this conclusion, vimentin-positive fibroblasts were largely distributed along the long cables of interstitial microfibrils (Figure 3C). Comparable collagen IV immunostaining in Fbn1+/+, Fbn1αMHC–/–, and Fbn1–/– myocardia excluded the possibility that fibrillin 1 deficiency interferes with BM organization (Figure 3B). Lack of appreciable ultrastructural differences in the BMs of Fbn1+/+ and Fbn1–/– myocardia strengthened this conclusion (Supplemental Figure 3).
Figure 3. Origin of MFS-related DCM.

(A) Representative hearts (left) collected from 6-week-old Fbn1+/+, Fbn1Col2–/–, Fbn1Wnt1–/mgR, and Fbn1αMHC–/– mice together with the indicated parameters of cardiac function or HW/BW. *P < 0.05 (n ≥ 5 per genotype) compared with control samples; bars indicate the mean ± SD; ANOVA P < 0.0001. Scale bar: 5 mm. (B) Collagen IV (Col IV, green) and fibrillin 1 (FBN1, red) immunofluorescence staining of neonatal myocardia of the indicated genotypes. Whenever detectable, immunostaining of interstitial and BM-containing pericellular matrices are highlighted by a yellow arrow and white arrowhead, respectively. Scale bar: 30 μm. Far left panels are higher-power images of Fbn1+/+ samples showing fibrillin 1 and collagen IV codistribution in the pericellular matrix. Scale bar: 5 μm. (C) Lower-power images of fibrillin 1 (FBN1, red) and vimentin (Vim, green) immunostaining of neonatal Fbn1+/+ myocardia that illustrate the almost exclusive distribution of cardiac fibroblasts along interstitial cables of fibrillin 1 microfibrils. Scale bar: 30 μm.
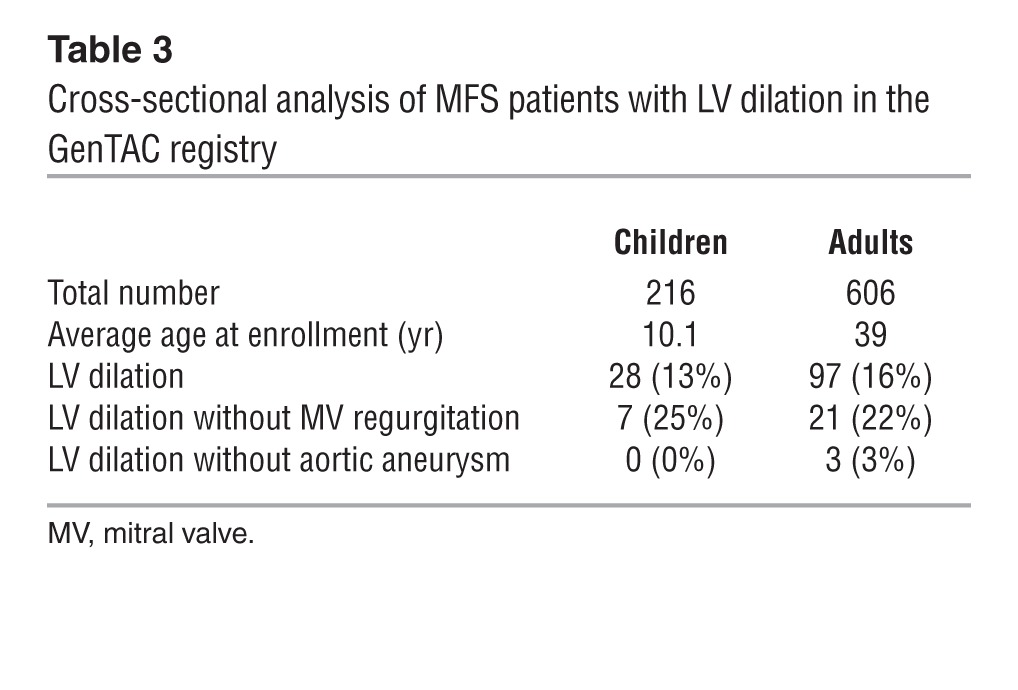
In light of the above mouse findings, we examined the clinical data regarding cardiovascular abnormalities in 822 MFS patients; these data have been collected over the past 5 years by the national registry of Genetically Triggered Thoracic Aortic Aneurysm and Cardiovascular Conditions (GenTAC) (40). This retrospective analysis revealed that approximately 23% of adult and pediatric patients display LV dilation without concomitant valvular disease and that approximately 3% of adult patients have ventricular, but not aortic, dilation (Table 3). It should be noted that the number of patients manifesting LV dilation in the absence of aortic disease is probably underestimated due to the use of aortic root enlargement as a major diagnostic criterion for MFS (6). Together, the mouse and human findings therefore imply that, contrary to the prevailing view, DCM is a primary MFS manifestation caused by a structurally deficient myocardial matrix.
Table 3.
Cross-sectional analysis of MFS patients with LV dilation in the GenTAC registry
Promiscuous TGF-β activity is not a prominent determinant of DCM in MFS mice.
Previous studies of Fbn1C1039G/+ mice have implicated promiscuous TGF-β signaling in MFS pathogenesis by documenting the ability of the neutralizing antibody 1D11 or losartan to mitigate aortic, valvular, and skeletal muscle abnormalities along with improper Smad2/3 activation (24, 41, 42). More recent findings in Fbn1C1039G/+ mice treated with the same agents or an MEK1/2 inhibitor have connected TAA progression with stimulation of the noncanonical (Smad-independent) ERK1/2 pathway (43, 44). We therefore explored the potential relationship between fibrillin 1 deficiency and improper activation of TGF-β signaling in myocardial tissue. In line with cardiac hypertrophy without significant fibrosis, we found that canonical (Smad-dependent) TGF-β signaling in the myocardium of 3-month-old Fbn1mgR/mgR mice was not appreciably different from that of WT littermates (Figure 2C). Additionally, systemic administration of 1D11–neutralizing antibody to Fbn1mgR/mgR or Fbn1αMHC–/– mice from P16 onward did not mitigate cardiac dysfunction (Figure 4B and Supplemental Figure 4). We excluded the formal possibility that the negative outcome of 1D11 treatment reflects poor penetration of the pan–TGF-β–neutralizing antibody by noting that 1D11 administration resulted in an appreciable — if not statistically significant — decrease in TGF-β signaling through the noncanonical ERK1/2 pathway in the myocardium of treated relative to untreated Fbn1αMHC–/– mice (Table 4). Hence, we concluded that, in contrast to other MFS manifestations (24, 42–44), promiscuous TGF-β signaling does not play a prominent role in DCM pathogenesis.
Figure 4. Molecular pathogenesis of MFS-related DCM.

(A) ERK1/2 immunoblots of total protein extracts from 3-month-old Fbn1+/+ and Fbn1mgR/mgR myocardia, with the numbers above each lane indicating ratios between phosphorylated and nonphosphorylated proteins. A statistical summary of these data is shown in Table 4. (B) Representative hearts collected from placebo-treated 2- to 3-month-old Fbn1+/+ and Fbn1mgR/mgR mice; Fbn1mgR/mgR mice treated with losartan (LOS), enalapril (ENA), propranolol (PPL), or pan–TGF-β–neutralizing antibody (1D11); or mice genetically deficient for AT1R (At1r–/–), β-arrestin 2 (Arrb2–/–), or angiotensinogen (Agt–/–). Scale bar: 5 mm. Below these images are evaluations of the same animals for the indicated parameters of cardiac function and size. Data relevant to Fbn1mgR/mgR;Agt–/– are shown separately because they are based on the analysis of 1-month-old control and experimental animals. *P < 0.05 (n ≥ 5 per genotype) relative to Fbn1+/+ mice, #P < 0.05 relative to untreated Fbn1mgR/mgR mice, ANOVA P < 0.0001; bars indicate the mean ± SD. (C) FAK immunoblots of total protein extracts from 2-month-old myocardia of Fbn1+/+ and mutant mice of the indicated genotypes. Numbers above each lane indicate ratios between phosphorylated and nonphosphorylated proteins, and bar graphs on the right summarize the data. *P < 0.05; n ≥ 5 per genotype.
Table 4.
Relative pERK1/2 levels in fibrillin 1–deficient myocardia

Fibrillin 1 deficiency stimulates abnormal mechanosignaling by cardiomyocytes.
Based on the above findings, we next investigated whether a structurally deficient matrix alters the mechanical properties of the myocardium and thereby influences mechanosignaling by cardiomyocytes. A reduced mean opening angle (a proxy for residual stress) of equatorial cross section rings from isolated Fbn1mgR/mgR ventricles suggested a reduction in passive mechanical tension of myocardial tissue (Figure 2D). Atomic force microscopy (AFM) measurements of whole hearts isolated from Fbn1mgR/mgR mice corroborated this observation by showing a significant decrease in tissue elastic modulus relative to Fbn1+/+ samples (Figure 2D). In line with these ex vivo observations, DCM in 3-month-old Fbn1mgR/mgR mice was associated with biochemical evidence of greater mechanical stress, such as elevated ERK signaling and abated focal adhesion kinase (FAK) activation (Figure 4A and Figure 5A). These molecular abnormalities paralleled DCM severity, since they were not noted in 1-month-old mutant hearts (data not shown).
Figure 5. Integrin involvement in MFS-related DCM.

(A) Illustrative immunoblots of pFAK and FAK proteins and β1 integrin in myocardial extracts from 3-month-old Fbn1+/+ and Fbn1mgR/mgR mice. Ponceau staining is shown below each immunoblot as an indication of total protein loading. (B) Cardiac function in 3-month-old mice of the indicated genotypes (cHI denotes compound haploinsufficient Fbn1+/–;Itgb1+/– mice), with the representative hearts shown below the assays. *P < 0.05 relative to Fbn1+/+ samples (n = 5 per genotype), and mean ± SD. ANOVA P < 0.05. Scale bar: 5 mm. (C) Illustrative immunoblots of FAK and ERK1/2 proteins in myocardial extracts from 3-month-old Fbn1+/+ and cHI mice (n = 5). In A and C, numbers above each lane indicate ratios between phosphorylated and nonphosphorylated proteins. FAK immunoblot in C includes lanes that were run on the same gel but were not contiguous.
AngII and mechanical stretch have been reported to activate AT1R-induced ERK1/2 signaling by cardiomyocytes through separate Gq and β-arrestin 2 pathways, respectively (45, 46). We used genetic and pharmacological approaches here to interrogate which one of these two pathways drives abnormally high phosphorylated ERK1/2 (pERK1/2) activity in the myocardium of MFS mice. Normal cardiac size and function in Fbn1mgR/mgR mice treated with the AT1R antagonist losartan or genetically deficient for AT1R confirmed cardiomyopathy in association with aberrant receptor signaling (Figure 4B). As expected, either treatment with losartan or genetic loss of AT1R eliminated the approximately 8-fold increase in pERK1/2 over ERK1/2 levels seen in the LV of Fbn1mgR/mgR mice relative to their Fbn1+/+ littermates (Table 4). Comparison of the pERK1/2 with ERK1/2 ratios in Fbn1mgR/mgR and Fbn1αMHC–/– myocardia implied that cardiomyocytes contribute to approximately 80% of the abnormal pERK1/2 increase in fibrillin 1–deficient myocardia (Table 4).
While the above findings implicated promiscuous AT1R activity in DCM pathogenesis, they did not discriminate between AngII- and stretch-induced signaling. To address this question, we first examined 3-month-old Fbn1mgR/mgR mice systemically treated with the angiotensin-converting enzyme enalapril at a dosage hemodynamically equivalent to that of losartan (24, 43, 44). The analyses revealed comparable myocardial dysfunction in treated and untreated Fbn1mgR/mgR mice (Figure 4B). Next, we generated Fbn1mgR/mgR mice genetically deficient for the precursor molecule angiotensinogen (Fbn1mgR/mgR;Agt–/– mice); however, compromised fitness of mice lacking angiotensinogen (47) restricted our analyses to measuring in 1-month-old Fbn1mgR/mgR;Agt–/– mice the percentage of fractional shortening and LV internal diameter in diastole. As with enalapril treatment of Fbn1mgR/mgR mice, the hearts of Fbn1mgR/mgR;Agt–/– mice displayed reduced muscle contractility and increased ventricle size (Figure 4B). Moreover, we found that cardiomegaly was also evident in the single Fbn1mgR/mgR;Agt–/– mouse that survived to approximately 3 months of age (Figure 4B). Together, these pharmacological and genetic experiments demonstrate ligand-independent activation of AT1R signaling in the myocardium of Fbn1mgR/mgR mice. We also found severe DCM in 3-month-old Fbn1mgR/mgR mice systemically treated with propranolol, a β adrenergic blocker currently prescribed as first-line therapy against TAA progression in MFS (Figure 4B and ref. 6). Like enalapril, propranolol was administered at a hemodynamically equivalent dosage to that of losartan (24, 43, 44). By contrast, we observed normal heart function associated with nearly normal pERK1/2 levels in Fbn1mgR/mgR mice deficient for the stretch mediator β-arrestin 2 (Figure 4B and Table 4). Together, these results corroborate our hypothesis that persistent mechanical stress causes spontaneous DCM in MFS mice.
Integrins are a class of cardiac mechanosensors that interact with the ECM to convert hemodynamic load into biochemical signals, such as FAK phosphorylation (48). We interpreted the barely detectable amounts of pFAK in the LV of 3-month-old Fbn1mgR/mgR mice as additional evidence that fibrillin 1 deficiency interferes with cardiac cell mechanosensing (Figure 5A). Moreover, normal amounts of β1 integrin excluded the possibility that depressed FAK activity is related to downregulation of α5β1, a major integrin of hypertrophic cardiomyocytes (Figure 5A and ref. 49). Based on these data, we examined cardiac size and function in mice haploinsufficient for both fibrillin 1 and β1 integrin under the assumption that reducing matrix integrity in the presence of fewer mechanosensors may phenocopy the myocardial abnormalities of MFS mice. Unlike Fbn1+/– or Itgb1+/– mice, compound haploinsufficiency for fibrillin 1 and β1 integrin resulted in an enlarged and dysfunctional heart (Figure 5B). Like Fbn1mgR/mgR mice, we found that DCM in Fbn1+/–;Itgb1+/– mice was associated with abated FAK activity and augmented ERK1/2 signaling (Figure 5C). Interestingly, normalization of both cardiac function and pFAK levels in Fbn1mgR/mgR;At1r–/– and Fbn1mgR/mgR;Arrb2–/– mice strongly suggested that stretch-induced AT1R signaling modulates integrin mechanosensing through a yet-to-be defined mechanism (Figure 4C). Altogether, our biomechanical, biochemical, genetic, and pharmacological experiments in Fbn1 mutant mice support a disease model in which a structurally deficient matrix compromises the physical properties of myocardial tissue, with the result of stimulating abnormal mechanosignaling by muscle cells and thereby ultimately causing spontaneous DCM.
Discussion
Cardiac dysfunction is a common finding in severely affected MFS infants that is often accompanied by congestive heart failure (12, 30, 32). Subclinical cardiomyopathy is also a risk factor for adolescent and adult MFS patients, as surgical TAA repair can lead to an acute rise in LV afterload due to the low compliance of Dacron implants (12, 31). For example, a retrospective study of 143 MFS patients identified that the long-term mortality rate associated with aortic repair was 20% and that congestive heart failure was almost as great a risk factor as rupture/dissection of a secondary aneurysm (19% vs. 23%, respectively) (31). Since first described in a 1985 report (9), cardiomyopathy in MFS has been evaluated in several clinical studies totaling over 800 patients and collectively suggesting an average disease prevalence of approximately 20% with cohort-specific ranges from 0% to 68% (10–20). Different factors are likely to have contributed to this extreme variance; among others, they include the exclusion criteria used in the studies, the size and age of the cohorts examined, the medication status of individual patients, the parameters used to normalize cardiac measurements, and the accuracy and resolution across different imaging modalities. A recent study has yielded more reliable information than before by using cardiovascular MRI to evaluate myocardial performance in a relatively large cohort of adult MFS patients (18). This retrospective analysis has revealed mild to asymptomatic ventricular enlargement and dysfunction in approximately 25% of afflicted individuals with no cardiovascular surgery or severe valvular regurgitation. While our cross-sectional analysis of 822 pediatric and adult MFS patients identified a comparable frequency of LV dilation without concurrent valvular disease and similarly underestimated its incidence in the absence of aortic disease, genetic compartmentalization of cardiovascular manifestations in Fbn1 conditional null mice provided the strongest evidence for the primary origin of myocardial dysfunction in MFS.
First, we demonstrated that reduced fibrillin 1 production by cardiomyocytes is sufficient to precipitate DCM in otherwise normal mice. This result does not necessarily exclude participation of cardiac fibroblasts (i.e.: interstitial matrix–producing cells) in disease pathogenesis, as both cell types contribute to the formation of the complex meshwork of fibrillin 1 assemblies that support muscle function (5, 7). Indeed, it is entirely possible that reducing the amount of pericellular fibrillin 1 deposited by cardiomyocytes negatively impacts the assembly of interstitial microfibrils as well. Second, we established a causal relationship between a structurally deficient ECM, a mechanically impaired muscle tissue, and abnormal mechanosignaling by cardiomyocytes. Supporting evidence included: (a) a significant reduction of passive mechanical tension and elastic modulus myocardial tissue deficient for fibrillin 1; (b) AngII-independent activation of abnormal ERK1/2 signaling in Fbn1mgR/mgR ventricles through the stretch-stimulated AT1R/β-arrestin 2 pathway; (c) comparable levels of augmented ERK1/2 activity in the myocardia of mice with either germline or cardiomyocyte-specific Fbn1 hypomorphism (Fbn1mgR/mgR and Fbn1αMHC–/– mice, respectively); and (d) equivalent anatomical, physiological, and signaling abnormalities of the hearts of mice underexpressing fibrillin 1 (Fbn1mgR/mgR mice) or haploinsufficient for both fibrillin 1 and β1 integrin (Fbn1+/–;Itgb1+/– mice).
While the role of AT1R in the development of load-induced cardiac hypertrophy has long been established (2), the underlying molecular mechanism has only recently begun to emerge. Yasuda et al. (50) were the first to show that mechanical stress–induced AT1R activation involves a change in receptor conformation. They furthermore argued that angiotensin receptor blockers (ARBs) prevent stretch-induced AT1R activation by locking the receptor into an inactive conformation, thereby functioning as inverse agonists rather than antagonists (50). Our finding that losartan treatment or AT1R loss can prevent DCM in MFS mice is consistent with this molecular model of ARB action. Rakesh et al. (46) subsequently demonstrated that mechanical stretch also promotes an AT1R-mediated conformational change in β-arrestin 2, which in turn results in receptor signaling independent of AngII or G protein activation. The normal heart of Fbn1mgR/mgR;Arrb2–/– mice is independent validation of the authors’ main conclusion that β-arrestin 2–biased AT1R agonism is the major driver of load-induced cardiac hypertrophy (46). Our data also suggest a hierarchical interaction between AT1R and integrins in their response to mechanical stress, since normal pFAK levels were restored in Fbn1mgR/mgR mice genetically deficient for AT1R or β-arrestin 2. Ongoing investigations are further exploring the mechanistic significance of this intriguing observation. This point notwithstanding, we believe that the dramatically low levels of pFAK in Fbn1mgR/mgR ventricles more likely reflect the broad impact of altered tissue properties on cell mechanosensors than loss of integrin binding to fibrillin 1 (7).
The demonstration that extracellular microfibrils are intimately involved in transmitting mechanical signals to intracellular compartments complements previous genetic studies of inherited DCM that have identified mutations in cellular proteins that participate in force generation and transmission from the sarcomere to the surrounding matrix (4). Accordingly, MFS-related DCM should be viewed as an integral component of the clinical spectrum of heart diseases associated with mutations in the ECM/sarcomere multiprotein complex that supports cardiomyocyte activity. Cingolani et al. (51) recently reported that mice lacking the matricellular protein thrombospondin 4 (TSP4) have normal cardiac function at baseline, but do not properly respond to acute pressure overload, allegedly as the result of improper integrin binding. Based on the present findings and preliminary evidence from transaortic constriction experiments in asymptomatic Fbn1+/– mice, we argue that fibrillin 1 supports cardiac baseline function and adaptation to increased workload largely by conferring key physical properties to myocardial tissue rather than by binding to integrin receptors on cardiac cells.
Recent studies of MFS mice with nonlethal TAA have suggested that anti–TGF-β therapy is a general strategy to ameliorate the pleiotropic manifestations of this heritable connective tissue disease (8, 24, 43, 44). Our data indicate that cardiac dysfunction, a clinically important MFS manifestation, is an exception to the broad use of anti–TGF-β therapy. We believe that stress signals in response to a fibrillin 1–deficient ECM should also be considered potential targets of pharmacological intervention, particularly for tissues subjected to cyclic mechanical load, such as the myocardium and ascending aorta. Ongoing investigations are addressing the latter possibility in our validated mouse model of severe MFS. The outcome of this work notwithstanding, the present study, to the best of our knowledge, is the first demonstration that fibrillin 1, a structural component of the architectural ECM, is a force-transmitting molecule that protects the mammalian heart against cardiomyopathy. This information has important implications for both the clinical management of cardiac complications in MFS and the understanding of degenerative processes in acquired DCM.
Methods
Mice.
Fbn1 mutant mice have been previously described (33, 34, 38). At1ar (referred to in the text as At1r) and Agt mutant mice and αMHC-Cre transgenic mice were purchased from the Jackson Laboratory mouse repository (stock nos. 002682, 002681, and 011038). Itgb1 conditional null mice were also purchased from the Jackson Laboratory mouse repository (stock no. 004605), and the conditional null allele was excised by crossing these mice with CMV-Cre transgenic mice (stock no. 006054) prior to their mating with Fbn1+/– mice. Col2-Cre and Arrb2 mutant mice were gifts of V. Lefebvre (Cleveland Clinic Lerner Research Institute, Cleveland, Ohio, USA) and R. Lefkowitz (Duke University Medical Center, Durham, North Carolina, USA). Wnt1-Cre transgenic mice and Rosa-LacZ reporter mice were provided by P. Soriano (Icahn School of Medicine at Mount Sinai). All analyses were performed on male mutant mice and sex-matched littermates.
Physiological analyses.
Echo was performed at designated time points in male mice under sedation by i.p. injections of up to 80 mg/kg of ketamine. The degree of sedation was optimized to maintain a heart rate greater than 400 beats per minute, and Nair was used to remove hair from the anterior chest wall. Short-axis parasternal 2D views of the LV at the mid-papillary level and long-axis parasternal views of the LV were acquired using a GE Vivid echocardiography apparatus with a 13- to 14-MHz linear array probe (GE Healthcare). Aortic diameters were measured from a parasternal long-axis view as the maximal internal dimension at the level of the root and ascending aorta. M-mode measurements of the size of the LV wall and cavity were obtained by 2D guidance from the short-axis view of the LV, as recommended by the American Society of Echocardiography. For increased resolution, a Vevo 2100 ultrasound system with a 550-MHz linear array probe (Visual Sonics) was used for identification of valvular regurgitation by long-axis views of the mitral and aortic valves in mice sedated with isofluorane. PV loop analysis was performed as previously described (52). Briefly, mice were i.p. injected with urethane (1 g/kg), etomidate (10 mg/kg), and morphine (1 mg/kg) and mechanically ventilated with 7 μl/g stroke volume at 125 respirations/min–1. The upper abdomen and diaphragm were opened, and a 1.2 French mouse P-V catheter (Scisense) was inserted into the LV apex through an apical stab performed with a 25-gauge needle. To determine the absolute ventricular volumes via admittance technology, myocardial and blood conductances were obtained prior to placement in the LV (53). Hemodynamic recordings were performed after 5 minutes of a stable heart rate. The inferior vena cava was transiently occluded to reduce ventricular preload in order to obtain load-independent pressure-volume relationships. Hemodynamic measurements were acquired and analyzed using IOX software (EMKAtech). Linear fits were obtained for the end-systolic pressure-volume relationship (ESPVR) and preload recruitable stroke work slope (PRSW). In all PV tracings, the end-systolic pressure (ESP) and ESV were determined at the end of the systolic ejection phase. Blood pressure measurements were acquired in 3-month-old conscious animals using the tail cuff method (CODA-2; Kent Scientific). Mice were habituated to the tail cuff pressure system over the course of 3 days with subsequent recording over the course of 2 days. The average systolic, diastolic, and pulse pressure measured in more than 60 blood pressure recordings was used for each animal. Noninvasive ECG recordings were conducted in 2.5-month-old conscious mice via the ECGenie (Mouse Specifics).
Pharmacological treatments.
Fbn1+/+ and Fbn1 mutant mice received i.p. injections of pan–TGF-β–neutralizing antibody 1D11 (R&D Systems) diluted in PBS (pH 7.4) and administered at a dose of 10 mg/kg body weight 3 times per week starting at P16 (24). Equivalent reduction of blood pressure was achieved by administering propranolol (0.5 g/l), losartan (0.6 g/l), or enalapril (0.15 g/l) via the drinking water (24, 43, 44). In all instances, oral therapy was carried out from P16 until 3 months of age, when the mice were sacrificed to be analyzed as described in the text.
Histology, biomechanics, and microscopy.
Hearts were injected with 1 M KCl prior to overnight fixation in formalin and embedded in paraffin as previously described (54). Six-micrometer paraffin cardiac cross sections were stained with Masson trichrome or H&E to evaluate collagen accumulation and aortic tissue morphology, respectively. Frozen sections (6-μm thick) from the LV midportion were stained with Texas Red-X–conjugated wheat germ agglutinin (WGA) or DAPI (Invitrogen) to respectively visualize cell membranes and nuclei. The myocyte cross-sectional area was measured from digital microscopy images using ImageJ software, version 1.45 (NIH) to trace the outline of 100 to 200 myocytes. X-gal staining of whole-mount and tissue section preparations of control and experimental hearts was performed as described (37). The following antibodies were used for immunofluorescence staining of myocardial tissue: mouse fibrillin 1 (a gift of L. Sakai, Shriners Hospital for Children, Portland, Oregon, USA), mouse collagen IV (1:1,000 dilution; Rockland Immunochemicals), and mouse vimentin (1:100 dilution; GenScript USA). The mean opening angle was assessed in hearts injected with an arresting solution containing NaCl (4 g/l), KCl (4.4 g/l), NaHCO3 (1 g/l), glucose (2 g/l), 2,3-butanedione (3 g/l), and EGTA (3.8 g/l) (55). A 2-mm-thick cross-sectional equatorial slice was submerged in arresting solution in a Petri dish and photographed 2 minutes after death. The LV free wall was radially cut opposite the center of the right ventricle to relieve the internal residual stress, and the heart was imaged 30 seconds after the cut. Images were imported into ImageJ software to measure the resulting mean opening angle. For AFM measurements, intact hearts were from 2-month-old Fbn1+/+ and Fbn1mgR/mgR mice, potted in 3% agarose in a 60-mm tissue culture dish with the exposed LV free wall facing up, immersed in physiologic Tyrode’s solution supplemented with blebbistatin (25 μm) to achieve a stable passive configuration, and equilibrated to 37°C for testing, which was initiated within 10 minutes of isolating the heart. A 5 × 5 array of 25 indentations, covering an 80 × 80-μm region of tissue, was performed at 3 to 5 sites per heart using a pyramidal AFM probe with a spring constant of 0.07 N/m. AFM experiments were completed within 15 minutes, followed by Hertz contact analysis to determine the apparent elastic modulus of the tissue (56), which yielded a single average modulus value for each heart. For electron microscopy, 16-day-old Fbn1–/– and Fbn1–/– mice were euthanized with CO2 and the hearts immediately perfused with saline followed by fixation with 0.5% gluteraldehyde and 0.2% tannic acid in PBS. After fixation, a portion of the LV free wall was removed and transferred to modified Karnovsky’s fixative (4% formaldehyde and 2.5% gluteraldehyde containing 8 mM CaCl2 in 0.1 M sodium cacodylate buffer, pH 7.4) and postfixed in 1% osmium tetroxide for 1 hour (57). After fixation, the tissue was dehydrated through ethanol and embedded in Epon/SPURR resin (EM Science). Semi-thin (1 μm) and thin sections (~90 nm) were cut with a diamond knife on a Leica model EMUC6 ultramicrotome, stained with 1% methylene blue in 1% sodium borate, and imaged with a Nikon Eclipse50 microscope. Thin sections were stained with saturated uranyl acetate followed by 0.2% lead citrate and imaged with a Philips CM-12 transmission electron microscope fitted with a digital camera (Hamamatsu ORCA-HR) controlled by AMT Image Capture software.
Protein analyses.
Protein extracts were prepared from frozen tissues using a plastic pestle in the presence of RIPA buffer with the addition of Complete Mini EDTA-free protease inhibitor cocktail (Roche Applied Sciences) and the Phosphatase Inhibitor Cocktail Set II (Calbiochem EMD-Serono) (58). Protein concentration was evaluated using a BCA Protein Assay Kit (Thermo Scientific). Antibodies against Smad2/3 (Millipore) and pSmad2/3 (Invitrogen), ERK1/2 and pERK1/2, FAK and p(Y397)-FAK (Cell Signaling Technology), and integrin β1 (EMD Millipore) were diluted at 1:1,000 in Tris-buffered saline, pH 7.4, and 0.1% (v/v) Tween 20 in the presence of 5% BSA and left on the membrane for 12 hours at 4°C. In all cases, Ponceau S Solution staining (Sigma-Aldrich) of the transfer membrane was used as a loading control. Protein extracts (50 μg per lane) were electrophoresed in a 10% (w/v) polyacrylamide gel and electroblotted onto an Immobilon-P membrane (Millipore). Membranes were incubated for 1 hour at room temperature with a peroxidase-labeled anti-rabbit antibody (1:50,000; Jackson ImmunoResearch Laboratories) in the presence of 5% (w/v) skimmed milk in Tris-buffered saline (pH 7.4) and 0.05% (v/v) Tween 20. Immunoreactive products were visualized by chemiluminescence using Clarity Western ECL substrate (Bio-Rad), and their relative intensity was evaluated using Adobe Photoshop.
RNA analyses.
Total RNA was purified from the heart or cardiac cells of Fbn1+/+ and Fbn1 mutant mice using the Prep Ease kit (Affymetrix) (58). Cardiac cells were isolated from newborn hearts and processed using a modification of the published protocol (59). Briefly, experimental and control myocardia were digested in a buffer containing 430 mg/l of collagenase type II (Worthington Biochemical) and 0.05% Trypsin (Invitrogen). Purified cardiac cells were cultured for 2 hours on plastic dishes to isolate adhering cardiac fibroblasts, whereas cells still in suspension were seeded on gelatin-coated plates to enable adhesion of cardiomyocytes. Cardiac fibroblasts were subsequently cultured in DMEM containing 10% FBS and antibiotic-antimycotic solution (Corning Cellgro). Cardiomyocytes were cultured for 24 hours in DMEM containing 10% FBS and 0.1 mmol/l bromodeoxyuridine and in DMEM containing 0.1% insulin-transferrin-selenium (Invitrogen) until collection. For all experiments, cells were cultured at 5 × 104 cells/cm2 unless otherwise stated. Purified RNA (100 ng) was transcribed with a First-Strand cDNA Synthesis Kit (Affymetrix) and PCR amplified in Maxima SYBR Green/ROX Q Master Mix (Thermo Scientific) using forward and reverse primers specific for Col1a1 (forward: 5′-AACCCTGGAAACAGACGAAC-3′ and reverse: 5′-CACGTTCAGTTGGTCAAAGG-3′; accession number NM_007742), Vim (forward: 5′-GATTTCTCTGCCTCTGCCAAC-3′ and reverse: 5′-CAACCAGAGGAAGTGACTCCA-3′; accession number NM_011701.4), and Pstn, Nppa, and Mhy7 (SABiosciences, QIAGEN). Each sample was run in triplicate with an initial 10-minute reaction at 95°C for reverse transcription followed by 40 cycles at 95°C for 30 seconds, 60°C for 30 seconds, and 72°C for 1 minute. Amplification data were collected using the Mastercycler EP Realplex system and analyzed with Realplex 2.0 software (Eppendorf). RNA concentrations were calculated from the threshold cycle (Ct) at which fluorescence is first detected using the 2-ΔΔCt comparative method. Vimentin transcript levels were used to normalize cardiomyocyte preparations for contaminating fibroblasts.
Clinical studies.
The design of the GenTAC registry has been described (40). Eight hundred and twenty-two patients with a diagnosis of MFS were enrolled in the longitudinal cohort study between November 1, 2009, and April 1, 2013.
Statistics.
Unpaired 2-tailed Student’s t tests were used to determine the significance between two groups, assuming significance at P < 0.05. Analyses between multiple groups used 1-way ANOVA, with P < 0.05 considered statistically significant. All values are expressed as the mean ± SD.
Study approval.
Mouse studies were approved by the IACUC of the Icahn School of Medicine at Mount Sinai. Data mined from the GenTAC national registry include patients originally recruited at five regional clinical centers, with written informed consent obtained from all patients at the time of enrollment and approval given by the IRBs of the participating centers (40).
Supplementary Material
Acknowledgments
We are indebted to D. Rifkin (NYU Langone School of Medicine, New York, New York, USA) for critical evaluation of the study as well as to our colleagues S. Smaldone and V. Todorovic for helpful suggestions. We thank J.J. Lee and K. Johnson for technical assistance and help in organizing the manuscript, respectively. This work was supported by grants from the NIH (to P. Yurchenco, R.J. Hajjar, F. Ramirez, and the GenTAC Consortium) and by a Research Award from the National Marfan Foundation (to F. Ramirez). J.R. Cook, E.R. Chemaly, and S.K. Rao were respectively supported by pre- and postdoctoral training grants from the NIH. The GenTAC Registry has been supported by US Federal Government contracts HHSN268200648199C and HHSN268201000048C from the National Heart, Lung, and Blood Institute (NHLBI) and the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS). Please see Supplemental Acknowledgments for the list of GenTAC Registry Consortium participating centers and investigators. The Microscopy Shared Research Facility, the Mouse Genetics and Gene Targeting Shared Research Facility, and the Atomic Force Microscopy Departmental Core Facility at the Icahn School of Medicine at Mount Sinai provided support for this work.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2014;124(3):1329–1339. doi:10.1172/JCI71059.
References
- 1.Harvey PA, Leinwand LA. The cell biology of disease: cellular mechanisms of cardiomyopathy. J Cell Biol. 2011;194(3):355–365. doi: 10.1083/jcb.201101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sheehy SP, Grosberg A, Parker KK. The contribution of cellular mechanotransduction to cardiomyocyte form and function. Biomech Model Mechanobiol. 2012;11(8):1227–1239. doi: 10.1007/s10237-012-0419-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Berlo JH, Maillet M, Molkentin JD. Signaling effectors underlying pathologic growth and remodeling of the heart. J Clin Invest. 2013;123(1):37–45. doi: 10.1172/JCI62839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seidman JG, Seidman C. The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell. 2001;104(4):557–567. doi: 10.1016/S0092-8674(01)00242-2. [DOI] [PubMed] [Google Scholar]
- 5.Parker KK, Ingber DE. Extracellular matrix, mechanotransduction and structural hierarchies in heart tissue engineering. Philos Trans R Soc Lond B Biol Sci. 2007;362(1484):1267–1279. doi: 10.1098/rstb.2007.2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pyeritz PE. The Marfan syndrome. Annu Rev Med. 2000;51:481–510. doi: 10.1146/annurev.med.51.1.481. [DOI] [PubMed] [Google Scholar]
- 7.Ramirez F, Sakai LY. Biogenesis and function of fibrillin assemblies. Cell Tissue Res. 2010;339(1):71–82. doi: 10.1007/s00441-009-0822-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramirez F, Rifkin DB. Extracellular microfibrils: contextual platforms for TGFβ and BMP signaling. Curr Opin Cell Biol. 2009;21(5):616–622. doi: 10.1016/j.ceb.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fujseki Y, Okuno K, Tanaka M, Shimada M, Takahashi M, Kawanishi K. Myocardial involvement in the Marfan syndrome. Jpn Heart J. 1985;26(6):1043–1050. doi: 10.1536/ihj.26.1043. [DOI] [PubMed] [Google Scholar]
- 10.Savolainen A, et al. Left ventricular function in children with the Marfan syndrome. Eur Heart J. 1994;15(5):625–630. doi: 10.1093/oxfordjournals.eurheartj.a060558. [DOI] [PubMed] [Google Scholar]
- 11.Chatrath R, Beauchesne LM, Connolly HM, Michels VV, Driscoll DJ. Left ventricular function in the Marfan syndrome without significant valvular regurgitation. Am J Cardiol. 2003;91(7):914–916. doi: 10.1016/S0002-9149(03)00039-0. [DOI] [PubMed] [Google Scholar]
- 12.Yetman AT, Bornemeier RA, McCrindle BW. Long-term outcome in patients with Marfan syndrome: is aortic dissection the only cause of sudden death? J Am Coll Cardiol. 2003;41(2):329–332. doi: 10.1016/s0735-1097(02)02699-2. [DOI] [PubMed] [Google Scholar]
- 13.Meijboom LJ, et al. Evaluation of left ventricular dimensions and function in Marfan’s syndrome without significant valvular regurgitation. Am J Cardiol. 2005;95(6):795–797. doi: 10.1016/j.amjcard.2004.11.042. [DOI] [PubMed] [Google Scholar]
- 14.Das BB, Taylor AL, Yetman AT. Left ventricular diastolic dysfunction in children and young adults with Marfan syndrome. Pediatr Cardiol. 2006;27(2):256–258. doi: 10.1007/s00246-005-1139-5. [DOI] [PubMed] [Google Scholar]
- 15.DeBacker JF, et al. Primary impairment of left ventricular function in Marfan syndrome. Int J Cardiol. 2006;112(3):353–358. doi: 10.1016/j.ijcard.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 16.Rybczynski M, et al. Tissue Doppler imaging identifies myocardial dysfunction in adults with Marfan syndrome. Clin Cardiol. 2007;30(1):19–24. doi: 10.1002/clc.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kiotsekoglou A, et al. Early impairment of left ventricular long-axis systolic function demonstrated by reduced atrioventricular plane displacement in patients with Marfan syndrome. Eur J Echocardiogr. 2008;9(5):605–613. doi: 10.1093/ejechocard/jen003. [DOI] [PubMed] [Google Scholar]
- 18.Alpendurada F, et al. Evidence for Marfan cardiomyopathy. Eur J Heart Fail. 2010;12(10):1085–1091. doi: 10.1093/eurjhf/hfq127. [DOI] [PubMed] [Google Scholar]
- 19.Kahveci G, Erkol A, Yilmaz F. Dilated cardiomyopathy in a patient with Marfan syndrome accompanied by chronic type A aortic dissection and right atrial thrombus. Intern Med. 2010;49(23):2583–2586. doi: 10.2169/internalmedicine.49.3880. [DOI] [PubMed] [Google Scholar]
- 20.de Witte P, et al. Intrinsic biventricular dysfunction in Marfan syndrome. Heart. 2011;97(24):2063–2068. doi: 10.1136/heartjnl-2011-300169. [DOI] [PubMed] [Google Scholar]
- 21.Lim DS, et al. Angiotensin II blockade reverses myocardial fibrosis in a transgenic mouse model of human hypertrophic cardiomyopathy. Circulation. 2001;103(6):789–791. doi: 10.1161/01.CIR.103.6.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schultz Jel J, et al. TGF-β1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J Clin Invest. 2002;109(6):787–796. doi: 10.1172/JCI14190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Judge DP, et al. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J Clin Invest. 2004;114(2):172–181. doi: 10.1172/JCI200420641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Habashi JP, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312(5770):117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lacro RV, et al. Rationale and design of a randomized clinical trial of β-blocker therapy (atenolol) versus angiotensin II receptor blocker therapy (losartan) in individuals with Marfan syndrome. Am Heart J. 2007;154(4):624–631. doi: 10.1016/j.ahj.2007.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gambarin FI, et al. Rationale and design of a trial evaluating the effects of losartan vs. nebivolol vs. the association of both on the progression of aortic root dilation in Marfan syndrome with FBN1 gene mutations. J Cardiovasc Med (Hagerstown). 2009;10(4):354–362. doi: 10.2459/JCM.0b013e3283232a45. [DOI] [PubMed] [Google Scholar]
- 27.Detaint D, et al. Rationale and design of a randomized clinical trial (Marfan Sartan) of angiotensin II receptor blocker therapy versus placebo in individuals with Marfan syndrome. Arch Cardiovasc Dis. 2010;103(5):317–325. doi: 10.1016/j.acvd.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 28.Radonic T, et al. Losartan therapy in adults with Marfan syndrome: study protocol of the multi-center randomized controlled COMPARE trial. Trials. 2010;11:3. doi: 10.1186/1745-6215-11-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Groenink M. et al. Losartan reduces aortic dilatation rate in adults with Marfan syndrome: a randomized controlled trial. Eur Heart J. 2013;34(45):3491–3500. doi: 10.1093/eurheartj/eht334. [DOI] [PubMed] [Google Scholar]
- 30.Geva T, Sanders SP, Diogenes MS, Rockenmacher S, Praagh RV. Two-dimensional and Doppler echocardiographic and pathologic characteristics of the infantile Marfan syndrome. Am J Cardiol. 1990;65(18):1230–1237. doi: 10.1016/0002-9149(90)90979-b. [DOI] [PubMed] [Google Scholar]
- 31.Finkbohner R, Johnston D, Crawford ES, Coselli J, Milewicz DM. Marfan syndrome. Long-term survival and complications after aortic aneurysm repair. Circulation. 1995;91(3):728–733. doi: 10.1161/01.CIR.91.3.728. [DOI] [PubMed] [Google Scholar]
- 32.Booms P, et al. Novel exon skipping mutation in the fibrillin-1 gene: Two ‘hot spots’ for the neonatal Marfan syndrome. Clin Genet. 1999;55(2):110–117. doi: 10.1034/j.1399-0004.1999.550207.x. [DOI] [PubMed] [Google Scholar]
- 33.Pereira L, et al. Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1. Proc Natl Acad Sci U S A. 1999;96(7):3819–3823. doi: 10.1073/pnas.96.7.3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cook JR, et al. Generation of Fbn1 conditional null mice implicates microfibrils in osteoprogenitor recruitment. . Genesis. 2012;50(8):635–641. doi: 10.1002/dvg.22022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gaussin V, et al. Endocardial cushion and myocardial defects after cardiac myocyte-specific conditional deletion of the bone morphogenetic protein receptor ALK3. Proc Natl Acad Sci U S A. 2002;99(5):2878–2883. doi: 10.1073/pnas.042390499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peacock JD, Levay AK, Gillaspie DB, Tao G, Lincoln J. Reduced sox9 function promotes heart valve calcification phenotypes in vivo. Circ Res. 2010;106(4):712–719. doi: 10.1161/CIRCRESAHA.109.213702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chai Y, et al. Fate of the mammalian cranial neural crest during tooth and mandibular morphogenesis. Development. 2000;127(8):1671–1679. doi: 10.1242/dev.127.8.1671. [DOI] [PubMed] [Google Scholar]
- 38.Carta L, et al. Fibrillins 1 and 2 perform partially overlapping functions during aortic development. J Biol Chem. 2006;281(12):8016–8023. doi: 10.1074/jbc.M511599200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buerger A, et al. Dilated cardiomyopathy resulting from high-level myocardial expression of Cre-recombinase. J Card Fail. 2006;12(5):392–398. doi: 10.1016/j.cardfail.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 40.Eagle KA, GenTAC Consortium. Rationale and design of the National Registry of Genetically Triggered Thoracic Aortic Aneurysms and Cardiovascular Conditions (GenTAC). Am Heart J. 2009;157(2):319–326. doi: 10.1016/j.ahj.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ng CM, et al. TGF-β-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J Clin Invest. 2004;114(11):1586–1592. doi: 10.1172/JCI200422715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cohn RD, et al. Angiotensin II type 1 receptor blockade attenuates TGF-β-induced failure of muscle regeneration in multiple myopathic states. Nat Med. 2007;13(2):204–210. doi: 10.1038/nm1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Holm TM, et al. Noncanonical TGF-β signaling contributes to aortic aneurysm preogression in Marfan syndrome mice. Science. 2011;332(6027):358–361. doi: 10.1126/science.1192149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Habashi JP, et al. Angiotensin II Type 2 receptor signaling attenuates aortic aneurysm through ERK antagonism. Science. 2011;332(6027):361–365. doi: 10.1126/science.1192152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wei H, et al. Independent β-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc Natl Acad Sci U S A. 2003;100(19):10782–10787. doi: 10.1073/pnas.1834556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rakesh K, Yoo B, Kim IM, Salazar N, Kim KS, Rockman HA. β-Arrestin-biased agonism of the angiotensin receptor induced by mechanical stress. Sci Signal. 2010;3(125):ra46. doi: 10.1126/scisignal.2000769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim HS, et al. Genetic control of blood pressure and the angiotensinogen locus. Proc Natl Acad Sci U S A. 1995;92(7):2735–2739. doi: 10.1073/pnas.92.7.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lal H, et al. Integrins and proximal signaling mechanisms in cardiovascular disease. Front Biosci (Landmark Ed). 2009;14:2307–2334. doi: 10.2741/3381. [DOI] [PubMed] [Google Scholar]
- 49.Babbitt CJ, Shai SY, Harpf AE, Pham CG, Ross RS. Modulation of integrins and integrin signaling molecules in the pressure-loaded murine ventricle. Histochem Cell Biol. 2002;118(6):431–439. doi: 10.1007/s00418-002-0476-1. [DOI] [PubMed] [Google Scholar]
- 50.Yasuda N, et al. Conformational switch of angiotensin II type I receptor underlying mechanical stress-induced activation. EMBO Rep. 2008;9(2):179–186. doi: 10.1038/sj.embor.7401157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cingolani OH, et al. Thrombospondin-4 is required for stretch-mediated contractility augmentation in cardiac muscle. Circ Res. 2011;109(12):1410–1414. doi: 10.1161/CIRCRESAHA.111.256743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pacher P, Nagayama T, Mukhopadhyay P, Batkai S, Kass DA. Measurement of cardiac function using pressure-volume conductance catheter technique in mice and rats. Nat Protoc. 2008;3(9):1422–1434. doi: 10.1038/nprot.2008.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Porterfield JE, et al. Dynamic correction for parallel conductance, GP, and gain factor, α, in invasive murine left ventricular volume measurements. J Appl Physiol (1985). 2009;107(6):1693–1703. doi: 10.1152/japplphysiol.91322.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Redfern CH, et al. Conditional expression of a Gi-coupled receptor causes ventricular conduction delay and a lethal cardiomyopathy. Proc Natl Acad Sci U S A. 2000;97(9):4826–4831. doi: 10.1073/pnas.97.9.4826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Omens JH, Fung YC. Residual strain in rat left ventricle. Circ Res. 1990;66(1):37–45. doi: 10.1161/01.RES.66.1.37. [DOI] [PubMed] [Google Scholar]
- 56.Azeloglu EU, Costa KD. Cross-bridge cycling gives rise to spatiotemporal heterogeneity of dynamic subcellular mechanics in cardiac myocytes probed with atomic force microscopy. Am J Physiol Heart Circ Physiol. 2010;298(3):H853–H860. doi: 10.1152/ajpheart.00427.2009. [DOI] [PubMed] [Google Scholar]
- 57.Tsiper MV, Yurchenco PD. Laminin assembles into separate basement membrane and fibrillar matrices in Schwann cells. J Cell Sci. 2002;115(pt 5):1005–1015. doi: 10.1242/jcs.115.5.1005. [DOI] [PubMed] [Google Scholar]
- 58.Carta L, et al. p38 MAPK is an early determinant of promiscuous Smad2/3 signaling in the aortas of fibrillin-1 (Fbn1) null mice. J Biol Chem. 2009;284(9):5630–5636. doi: 10.1074/jbc.M806962200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Koitabashi N, et al. Increased connective tissue growth factor relative to brain natriuretic peptide as a determinant of myocardial fibrosis. Hypertension. 2007;49(5):1120–1127. doi: 10.1161/HYPERTENSIONAHA.106.077537. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.